
Anthraquinones, Diphenyl Ethers, and Their Derivatives from the Culture of the Marine Sponge-Associated Fungus Neosartorya spinosa KUFA 1047 †

 , ,
, ,  ,
,  , , ,
, , ,  and
and 
Abstract
:
1. Introduction
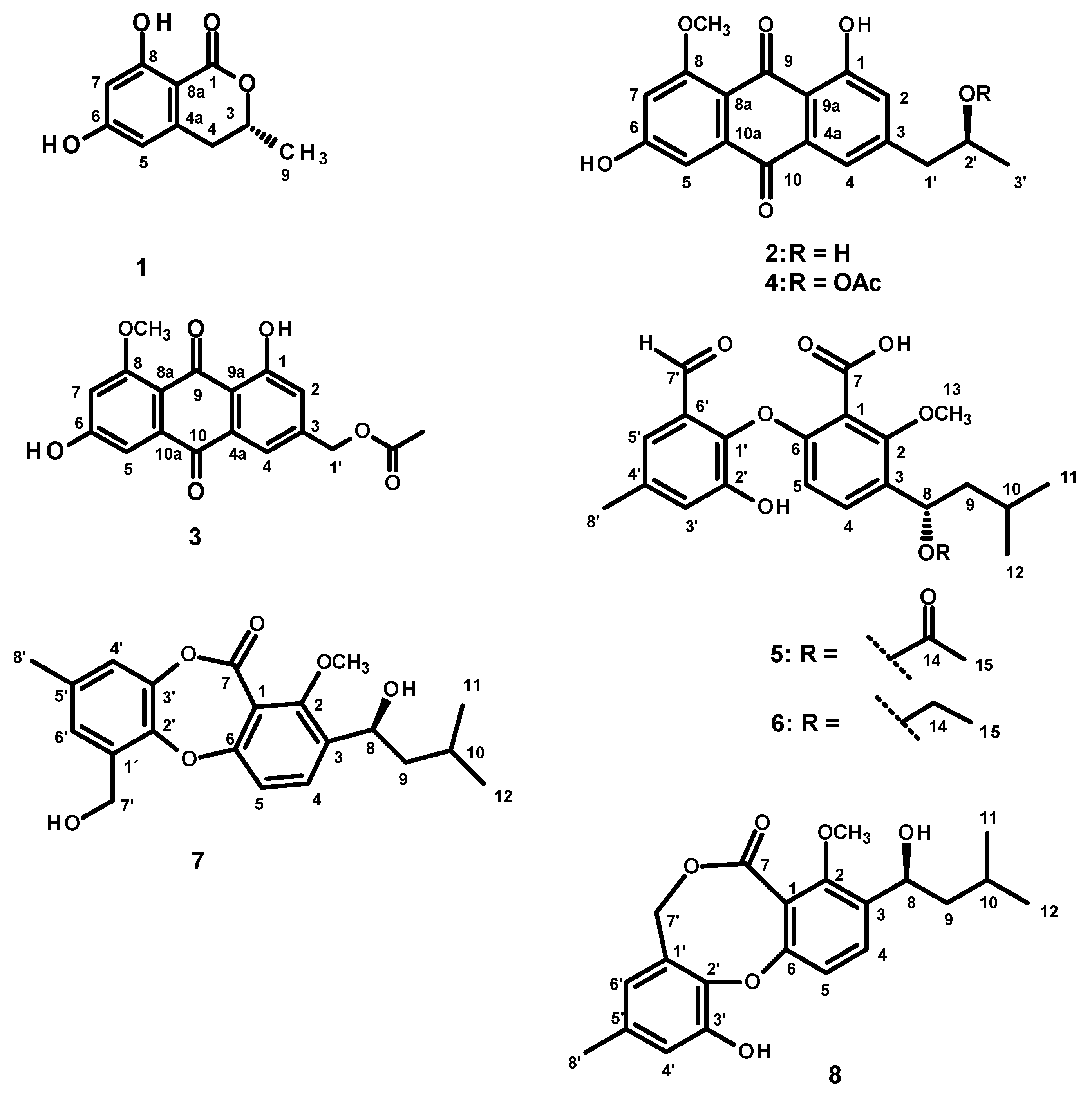
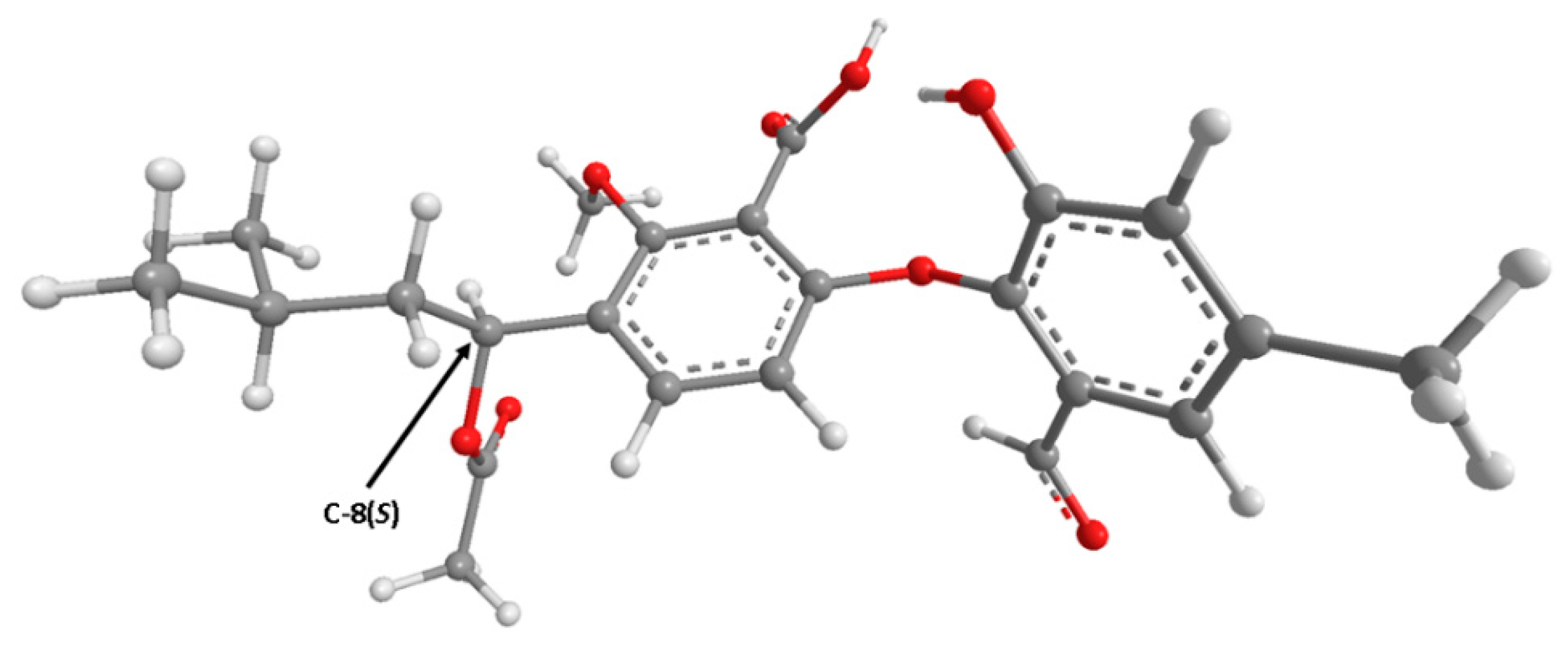
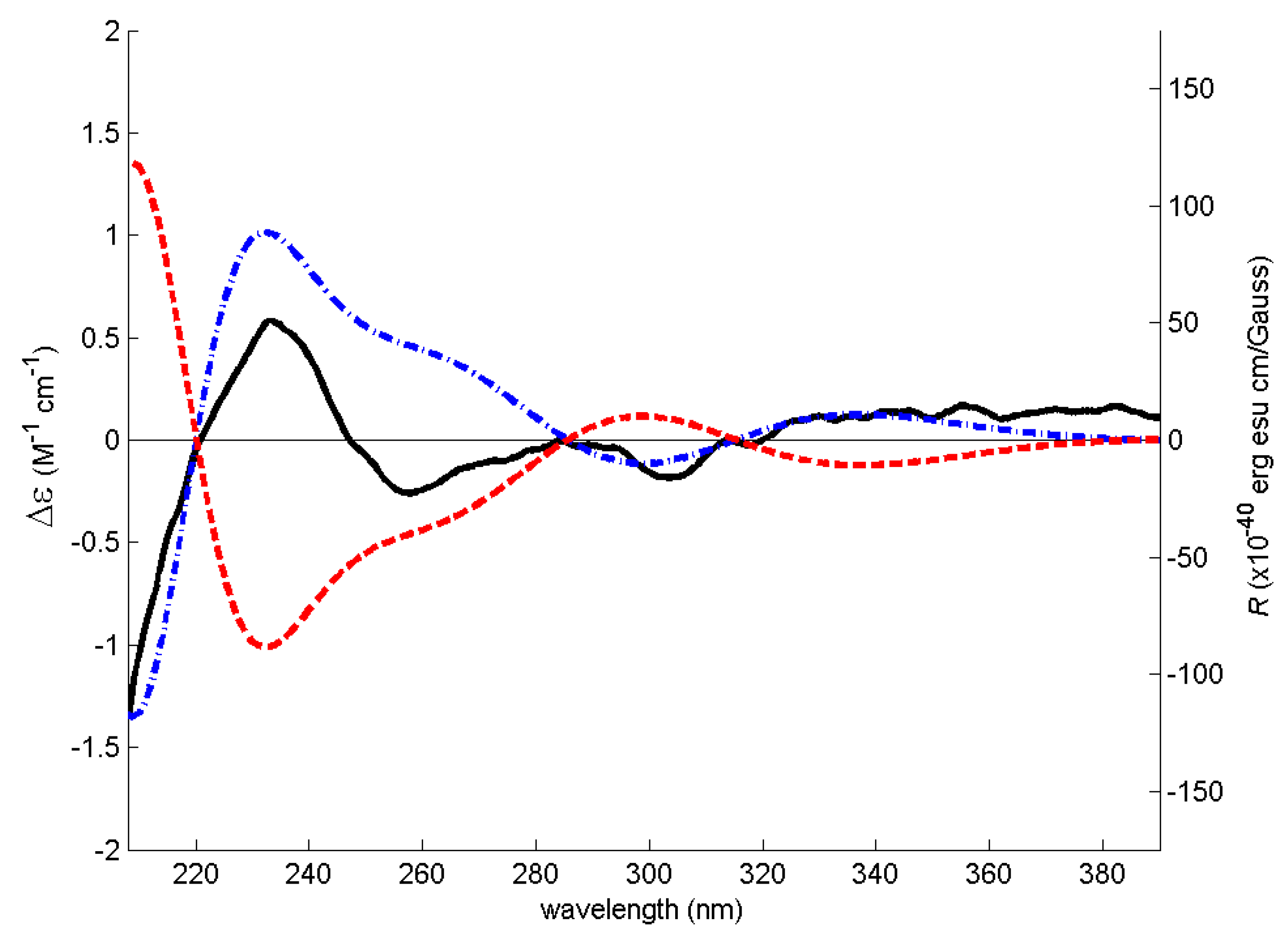
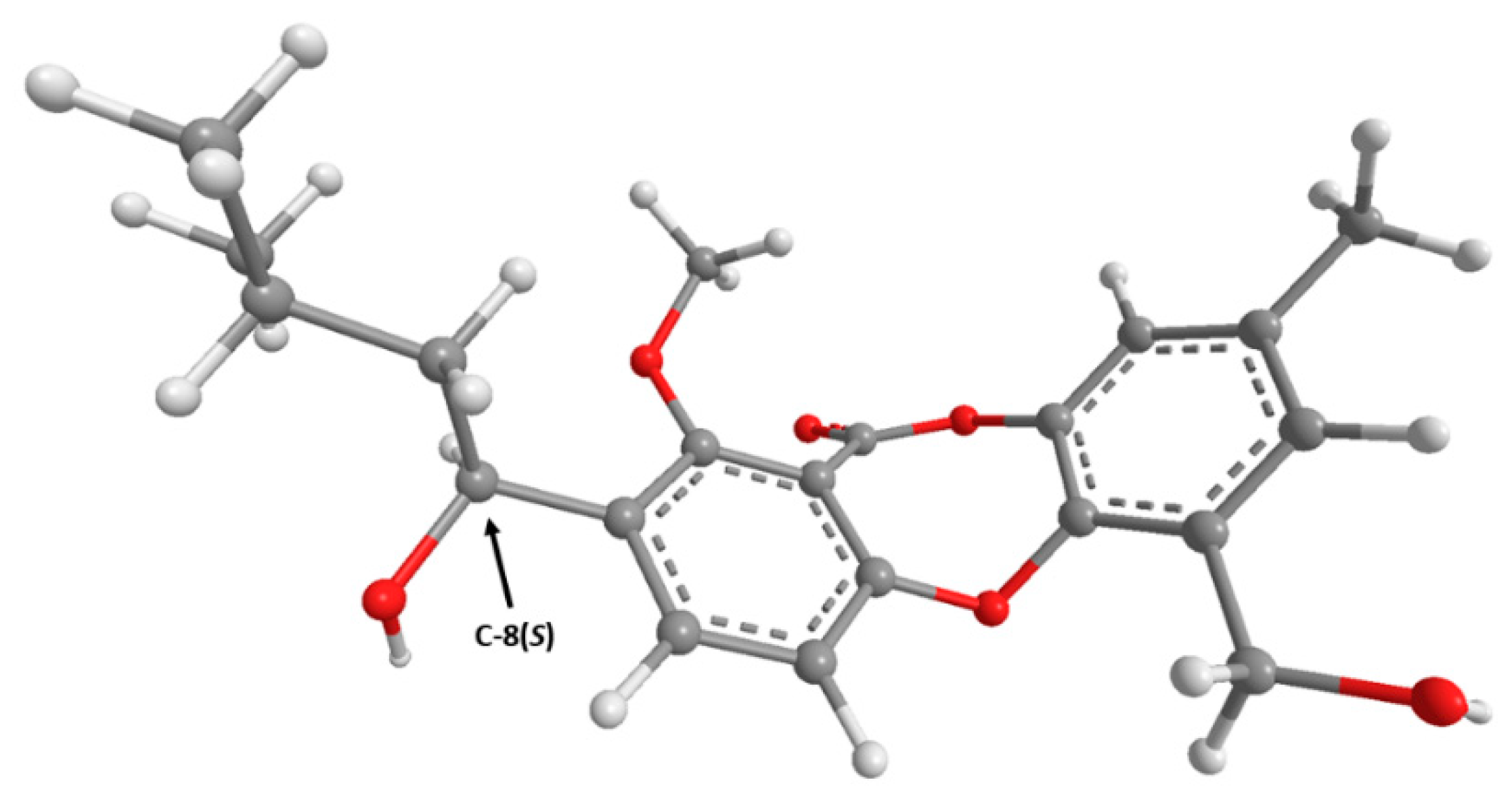
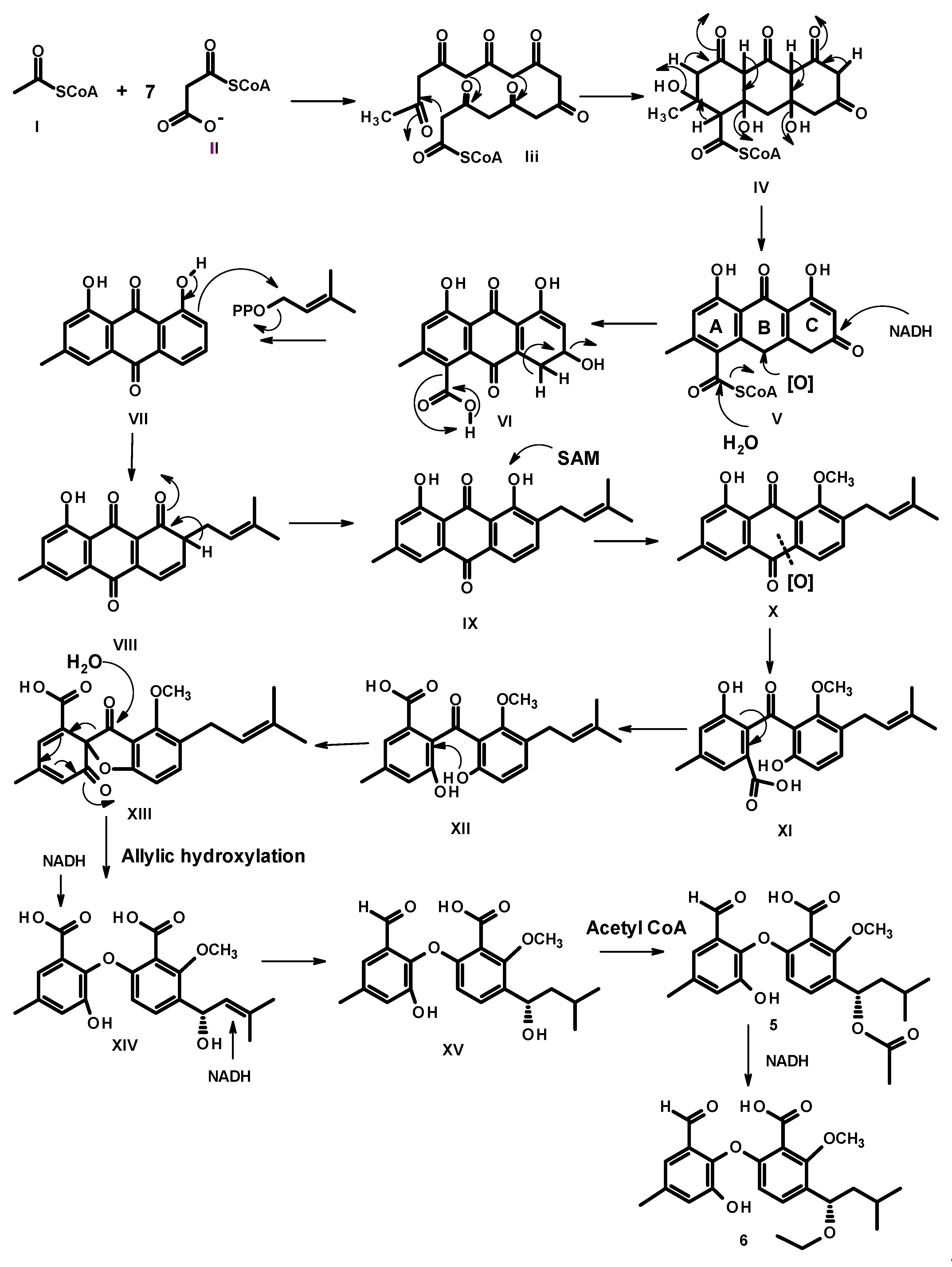
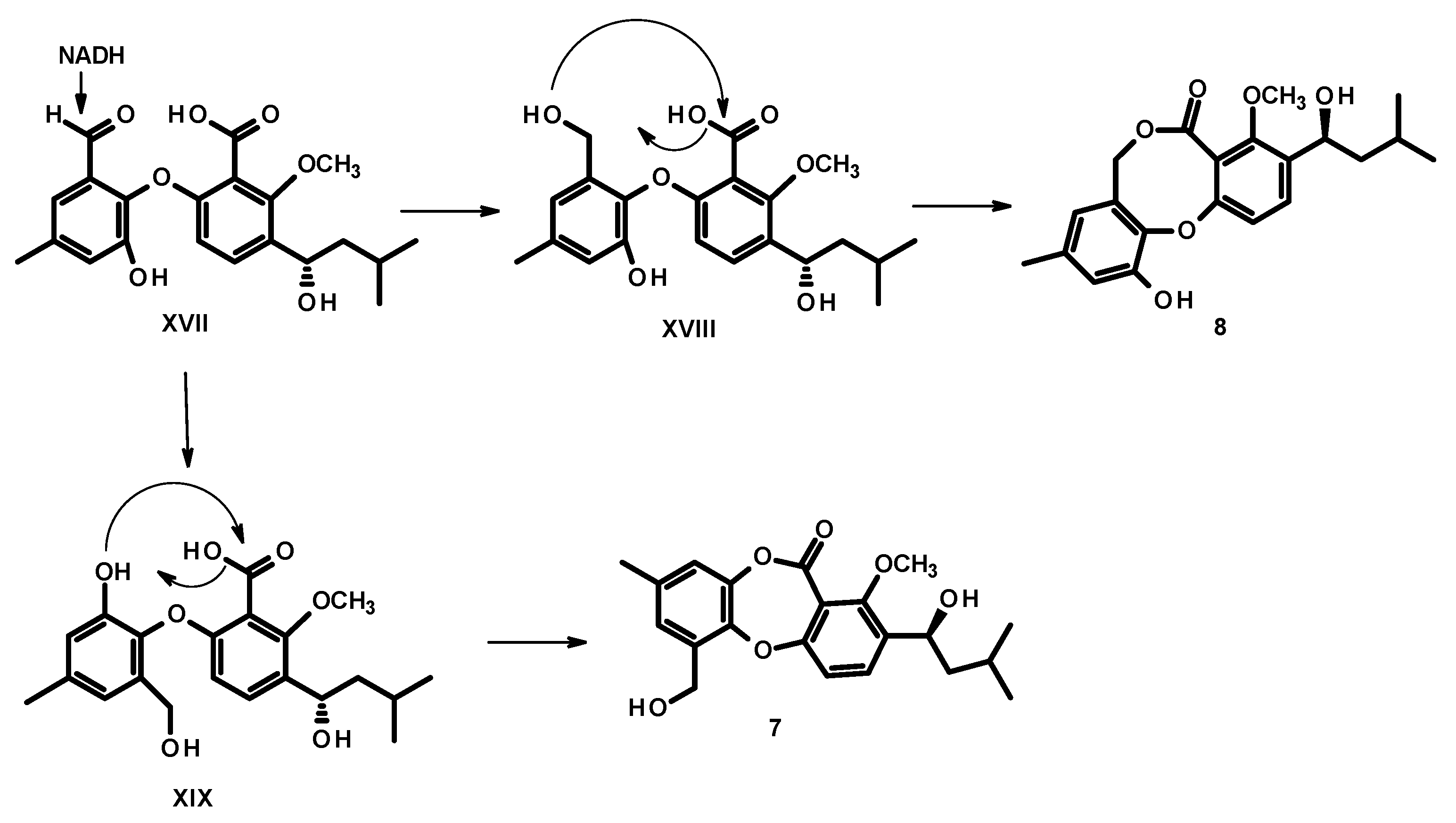
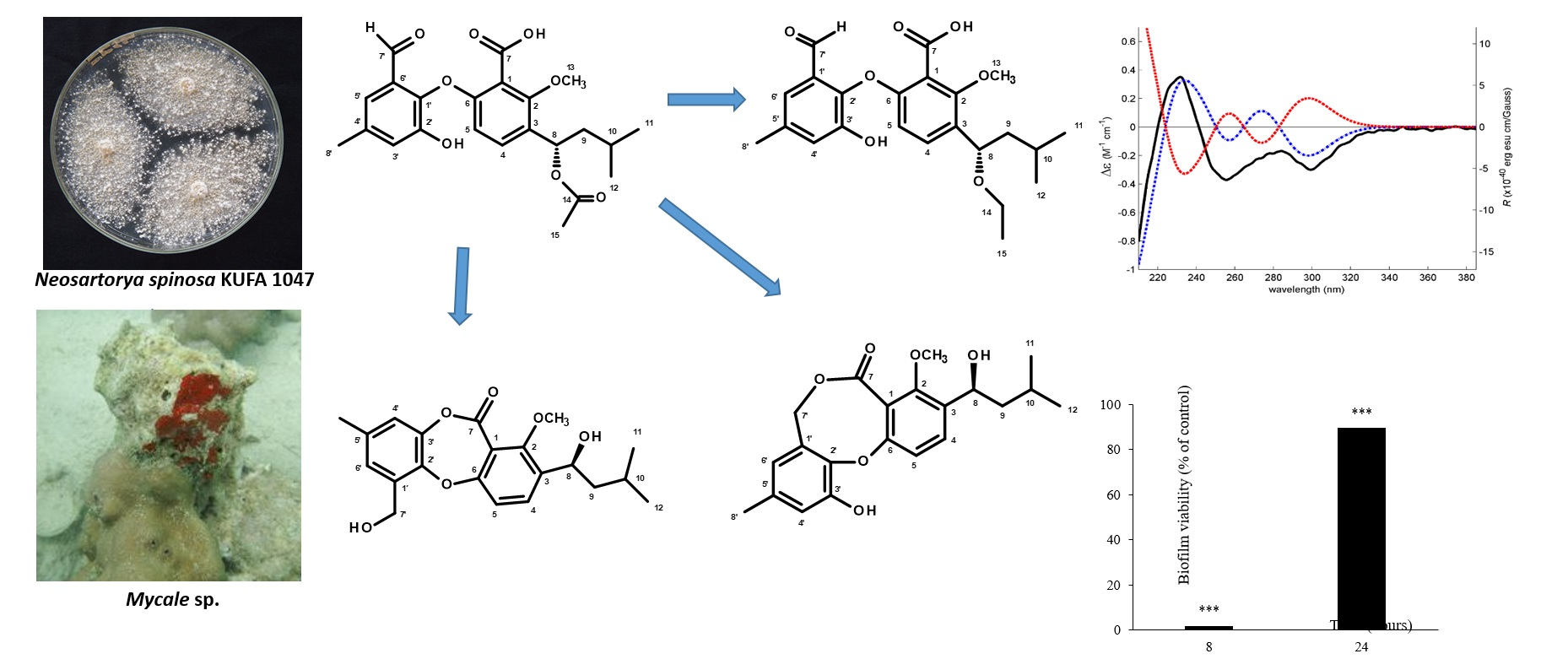
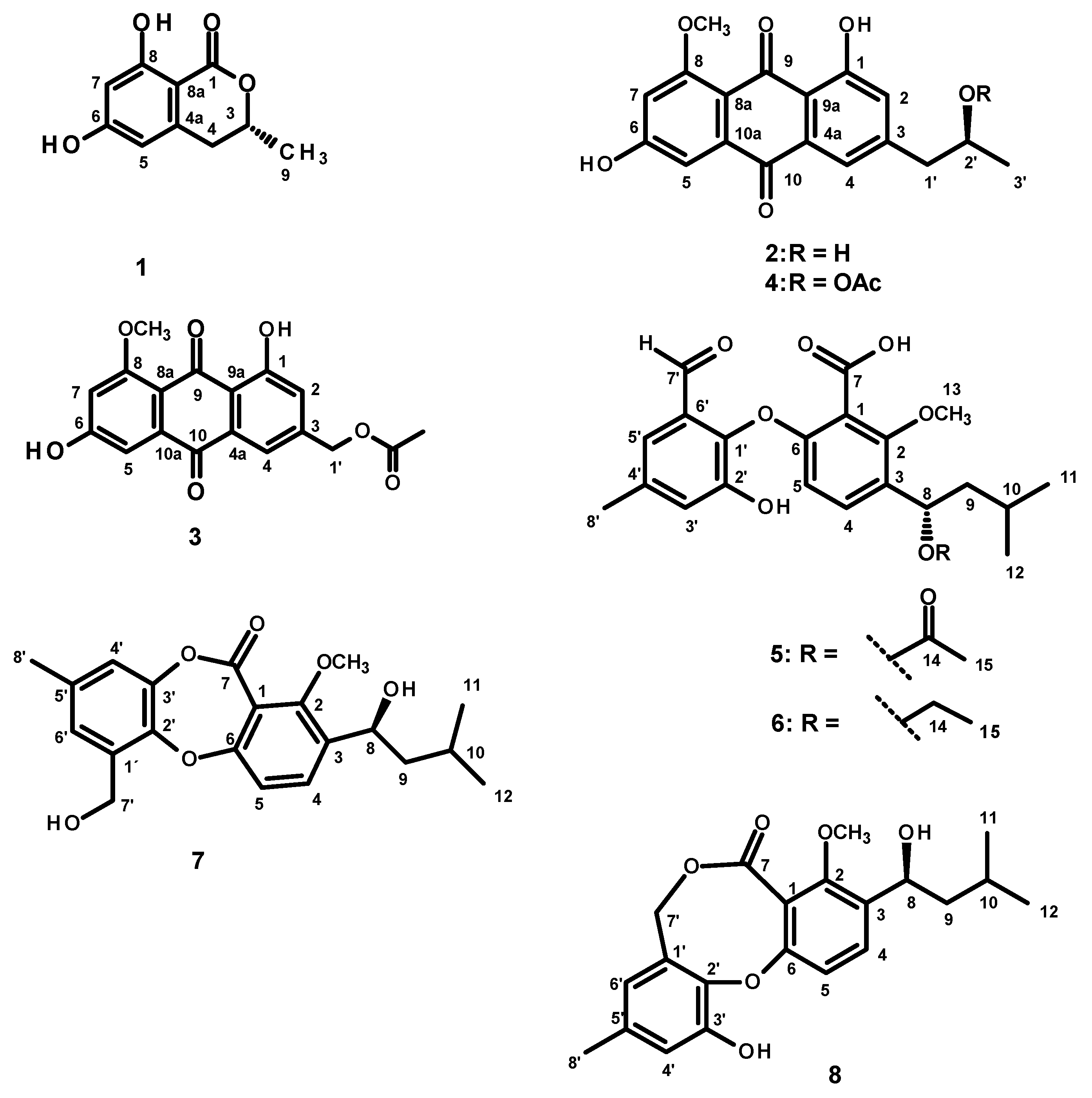
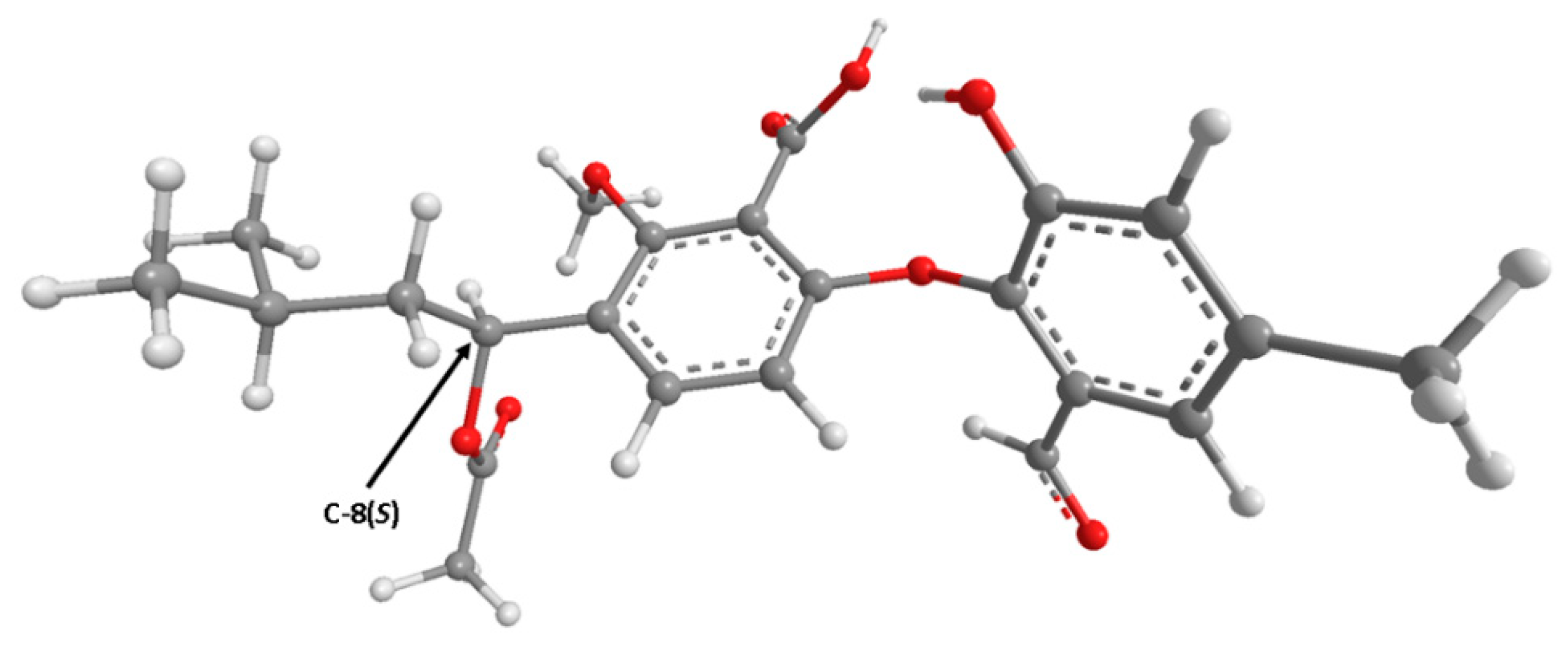
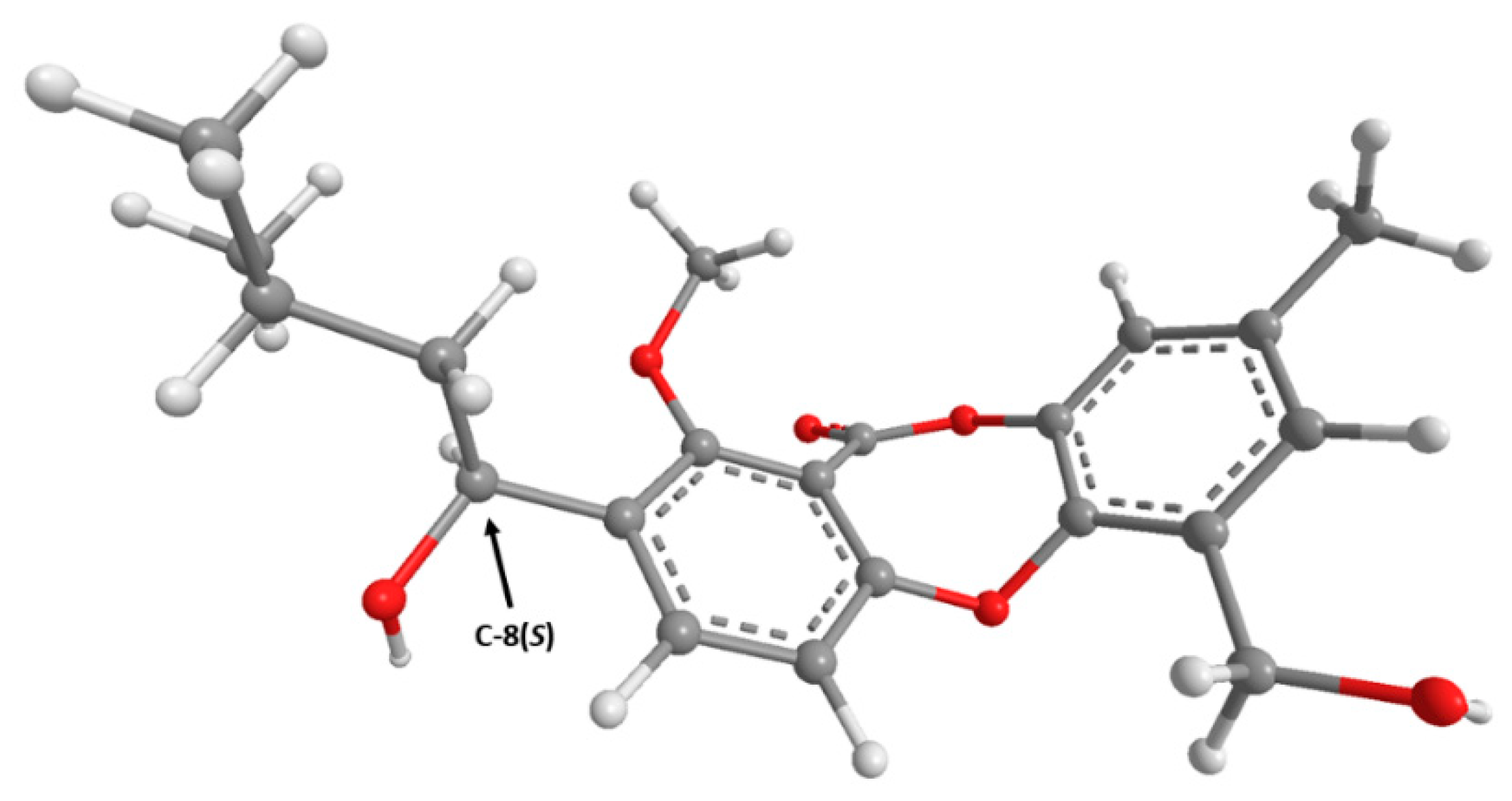
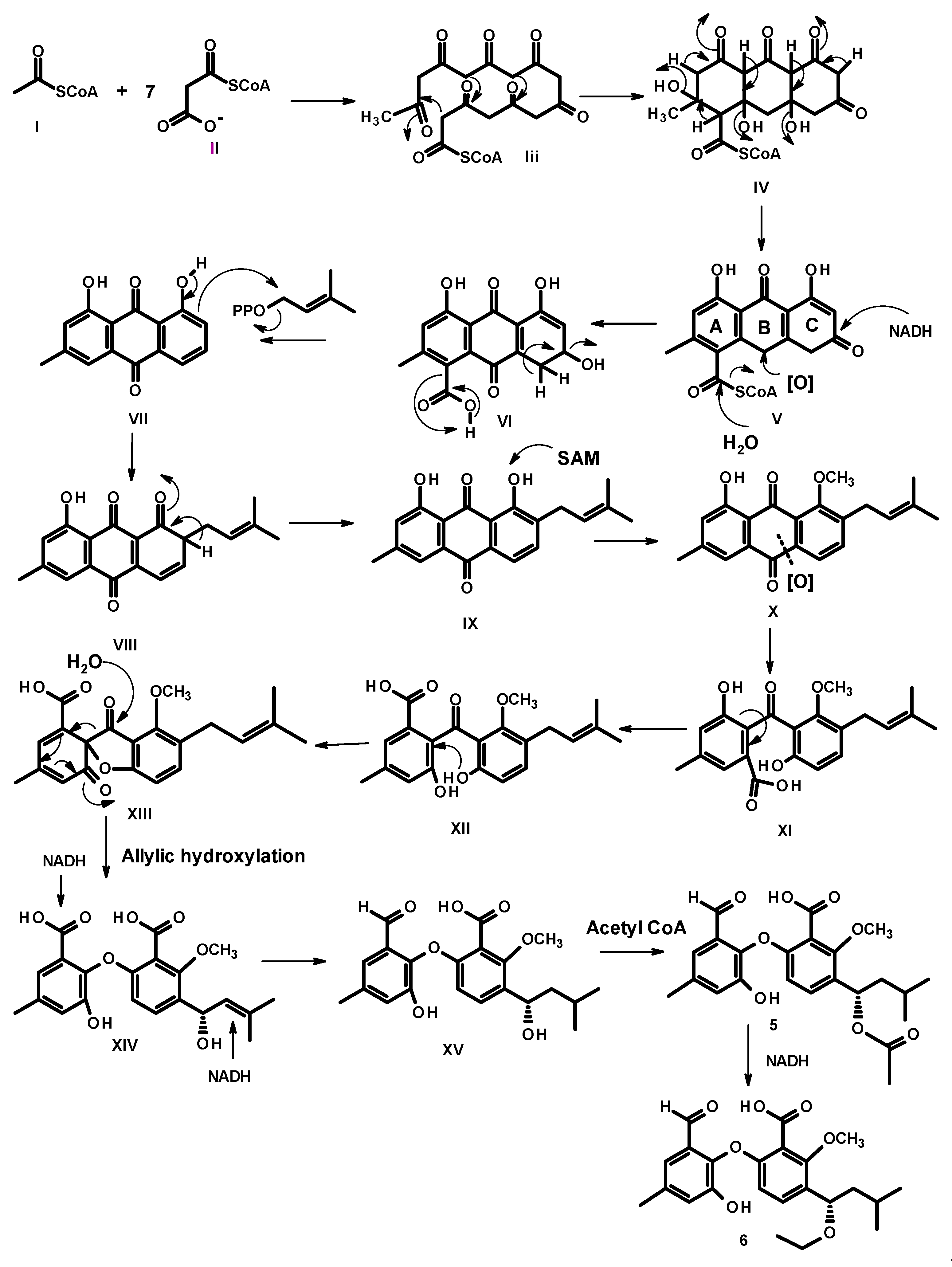
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
3.3.1. Acetylpenipurdin A (4)
3.3.2. Neospinosic Acid (6)
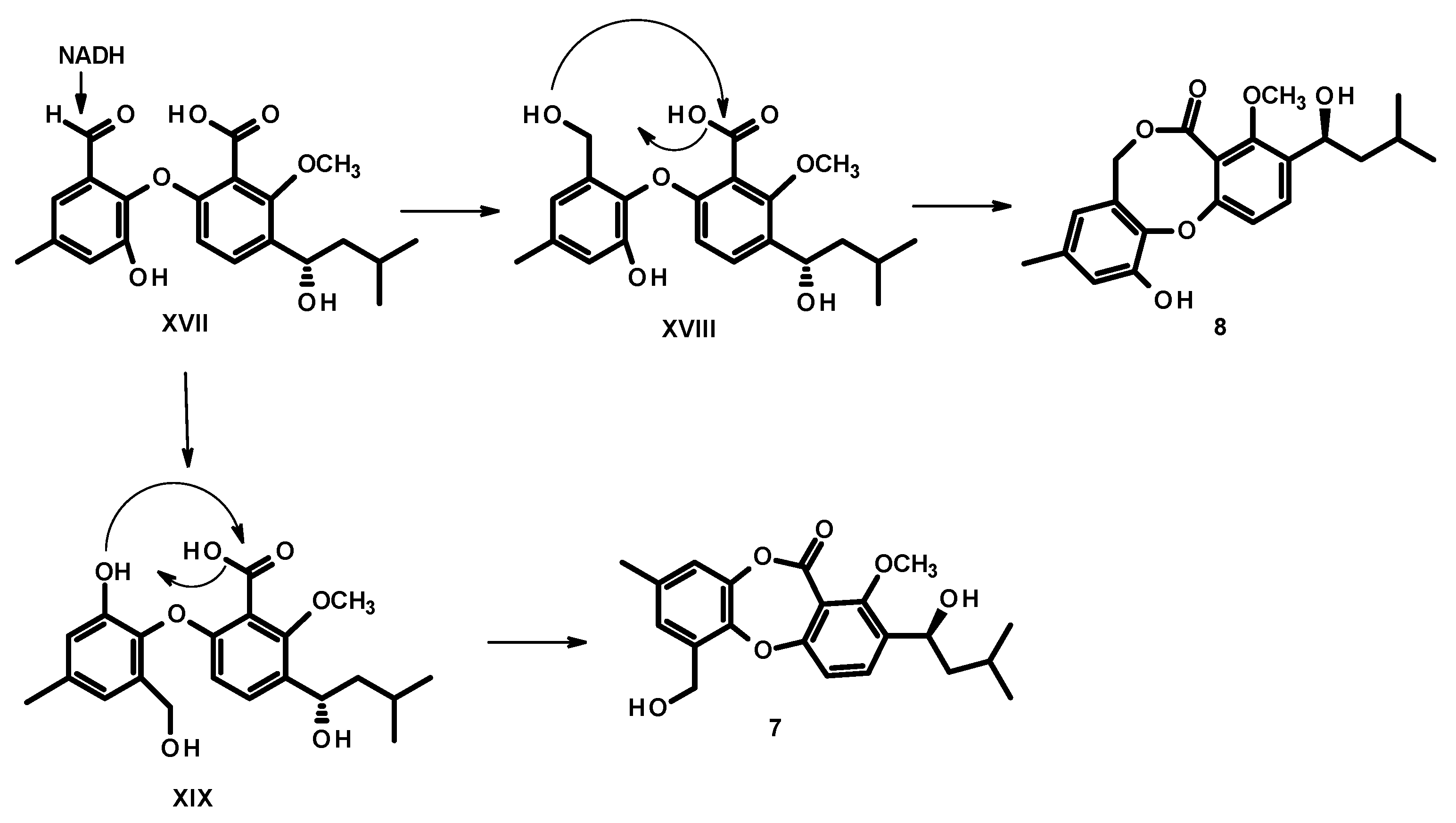
3.3.3. Spinolactone (7)
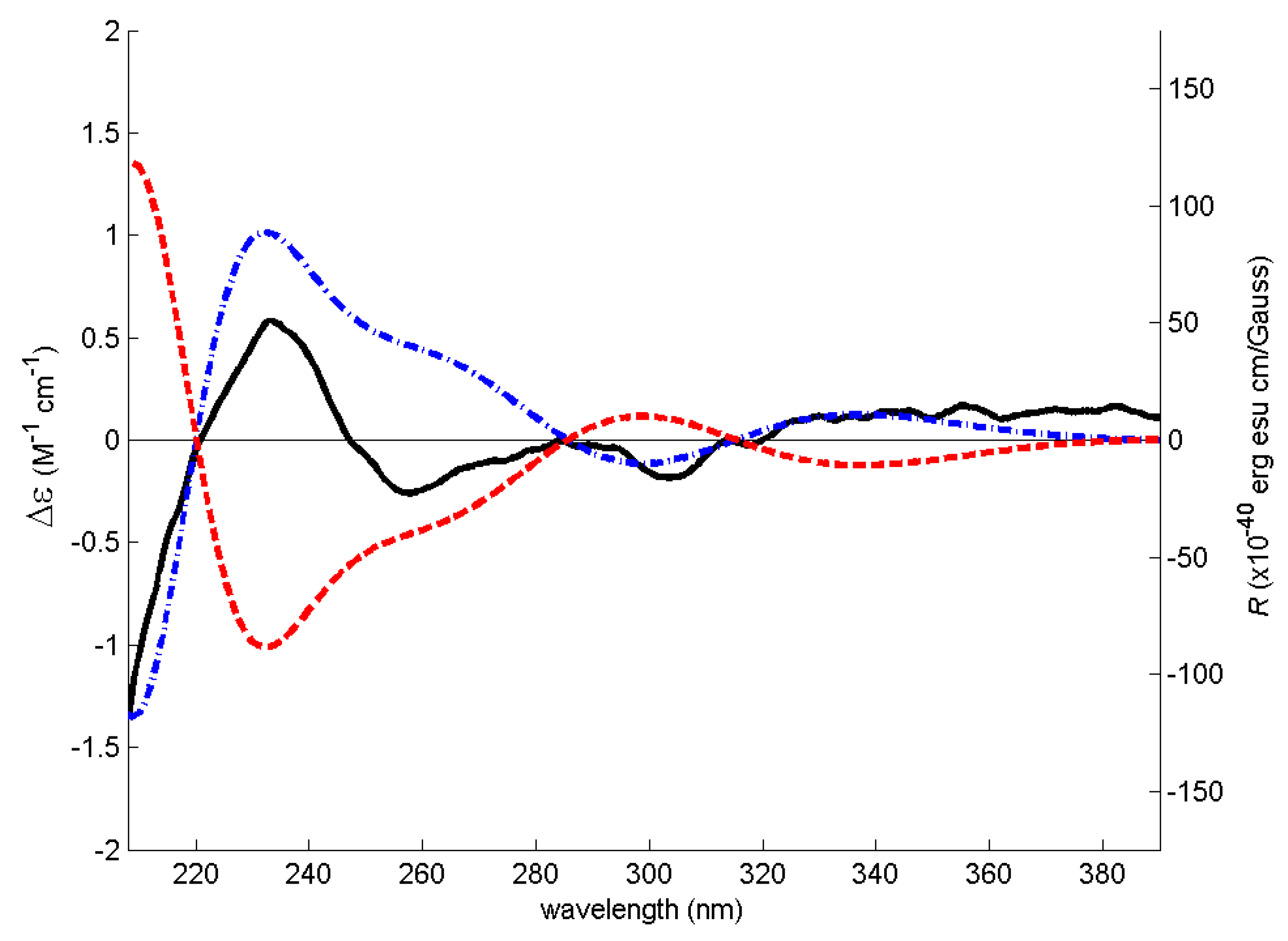
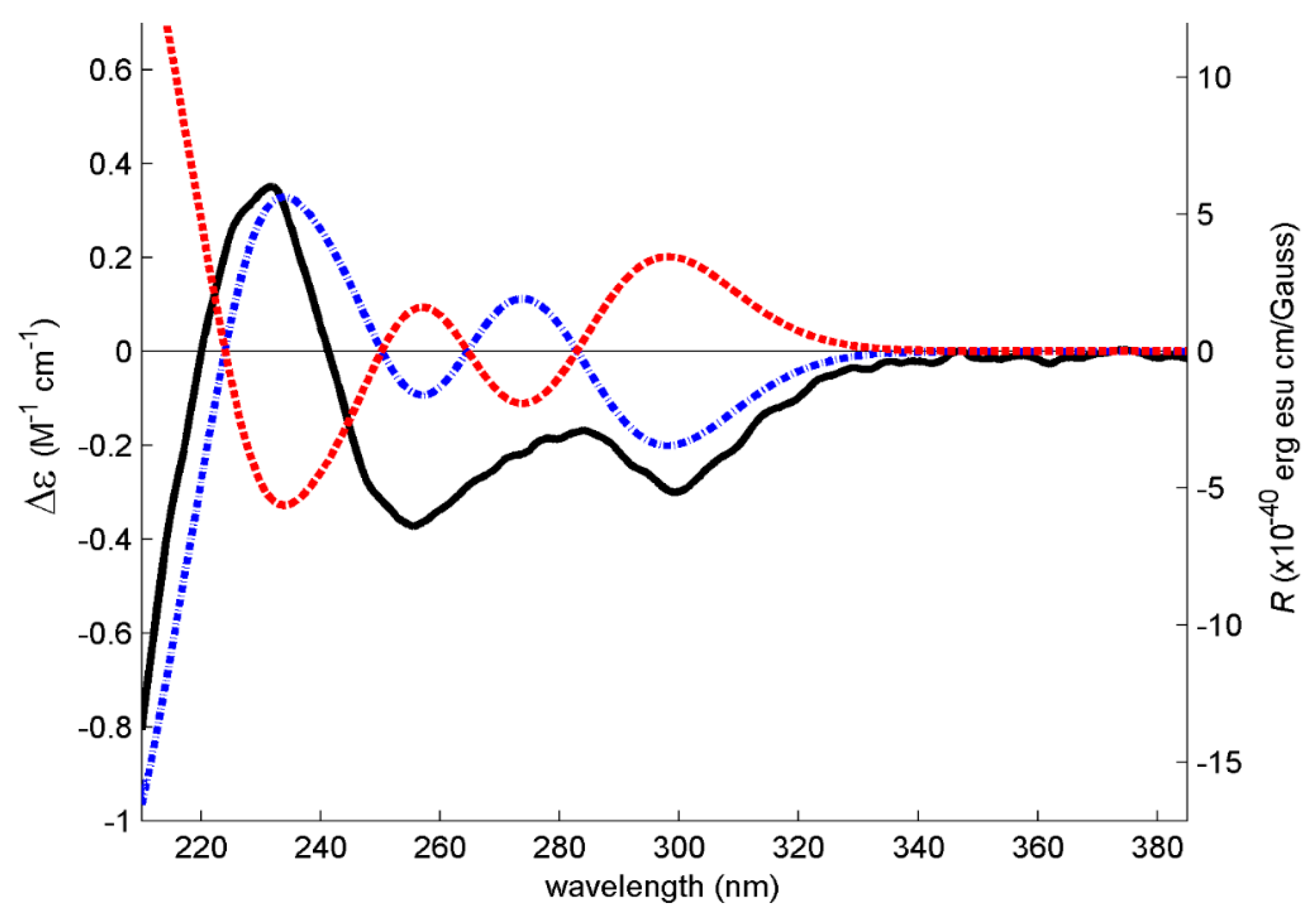
3.4. Electronic Circular Dichroism (ECD)
3.5. Antibacterial Activity Bioassays
3.5.1. Bacterial Strains and Growth Conditions
3.5.2. Antimicrobial Susceptibility Testing
3.5.3. Antibiotic Synergy Testing
3.5.4. Biofilm Formation Inhibition Assay
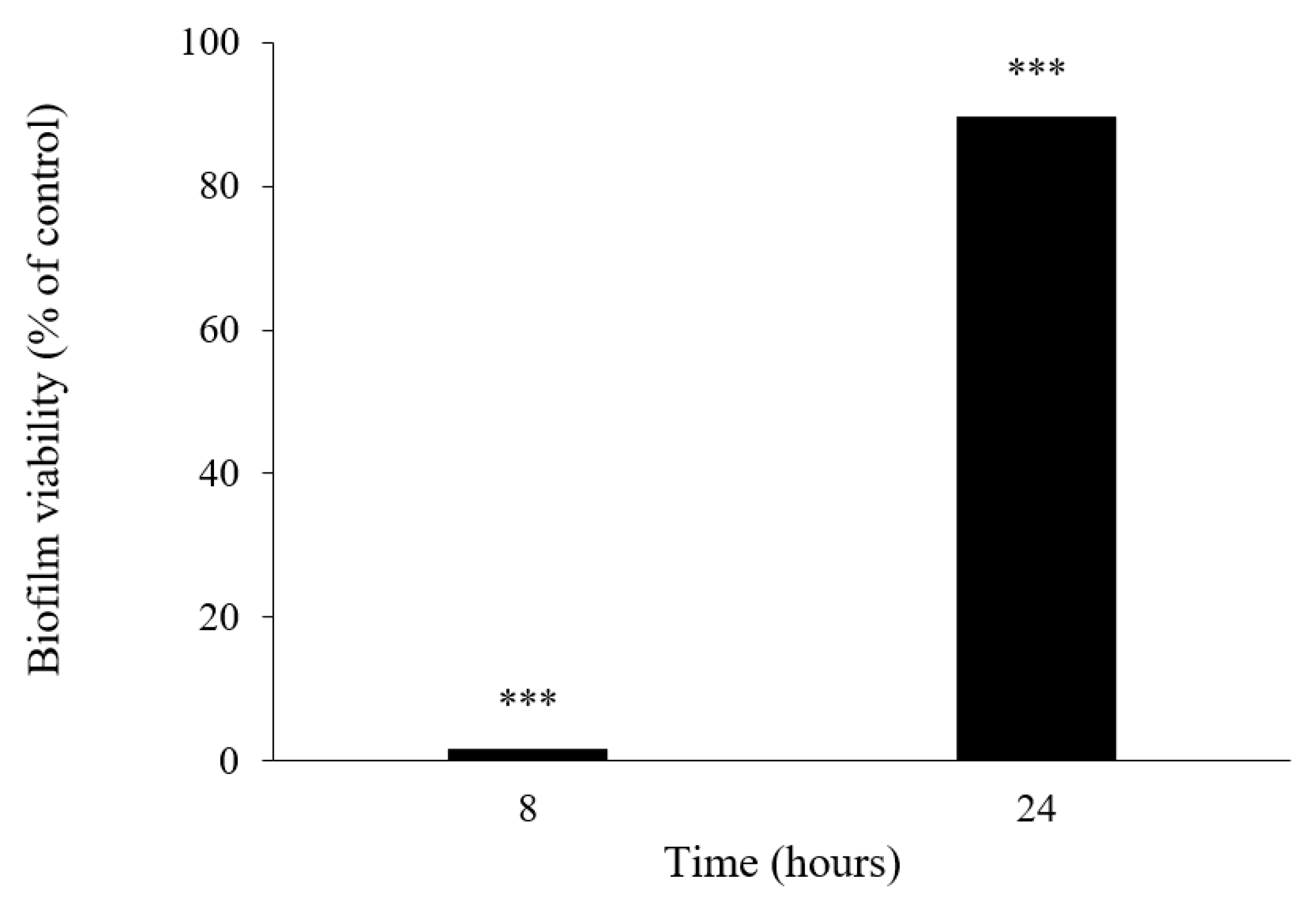
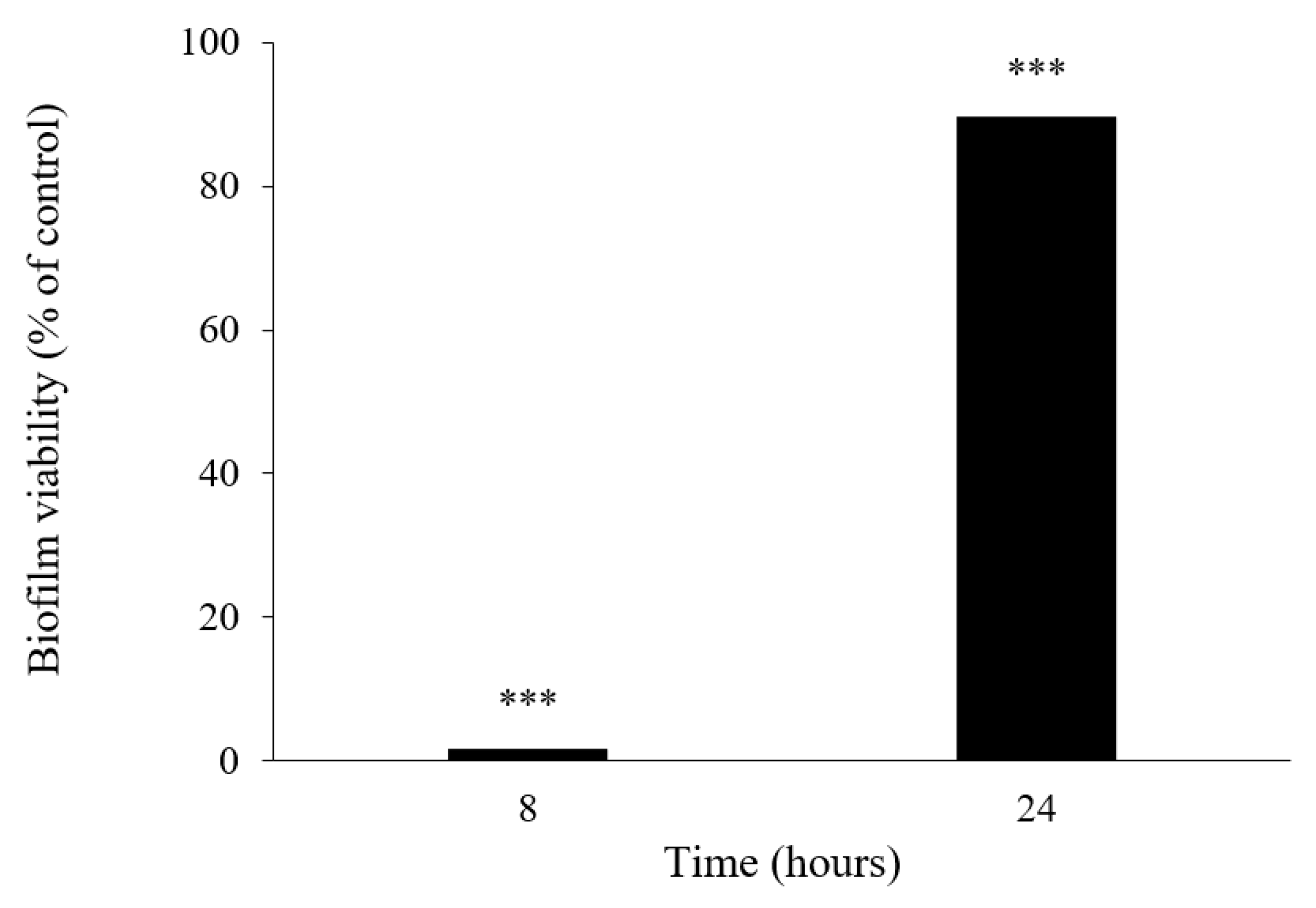
3.5.5. Biofilm Viability Assay
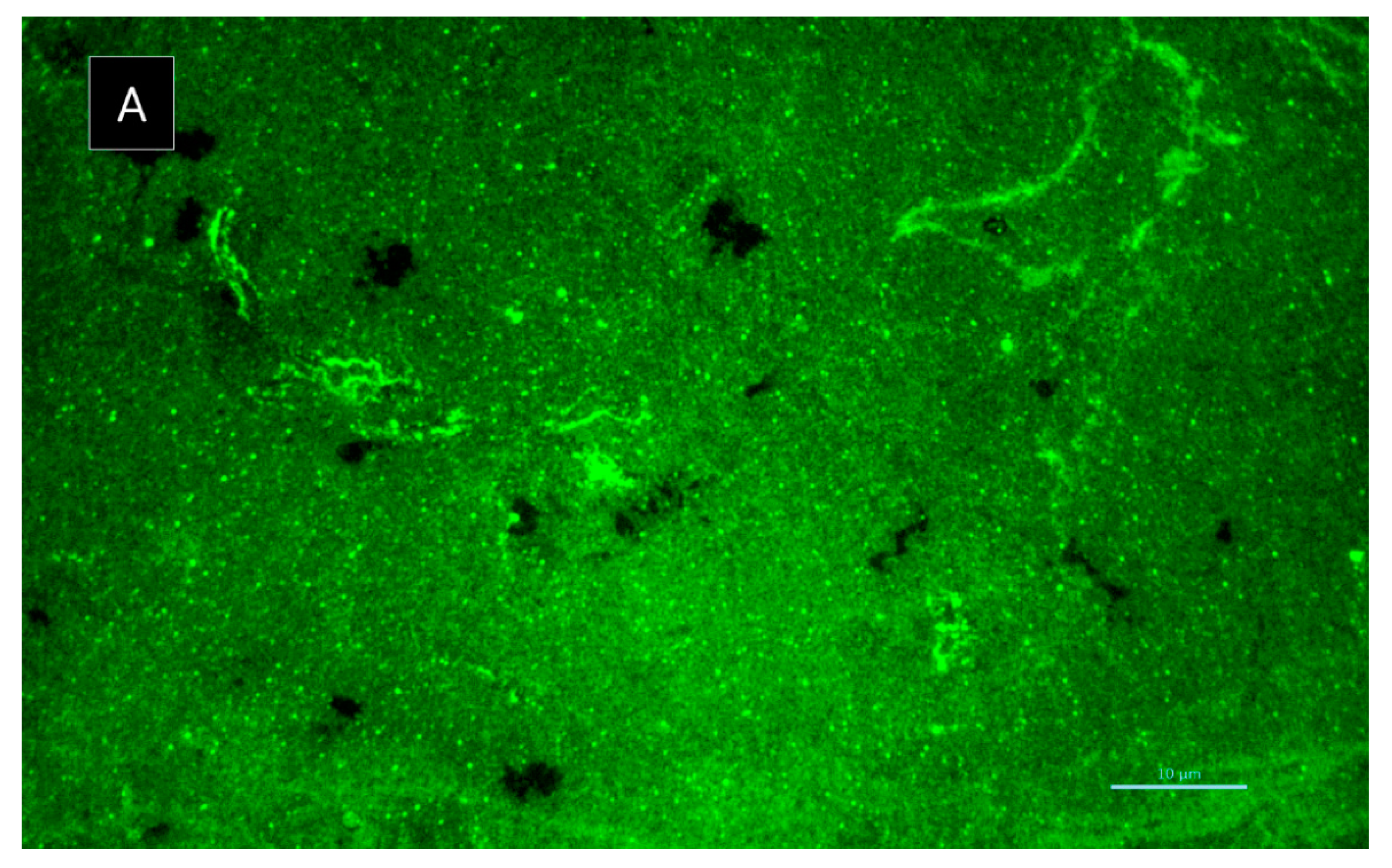
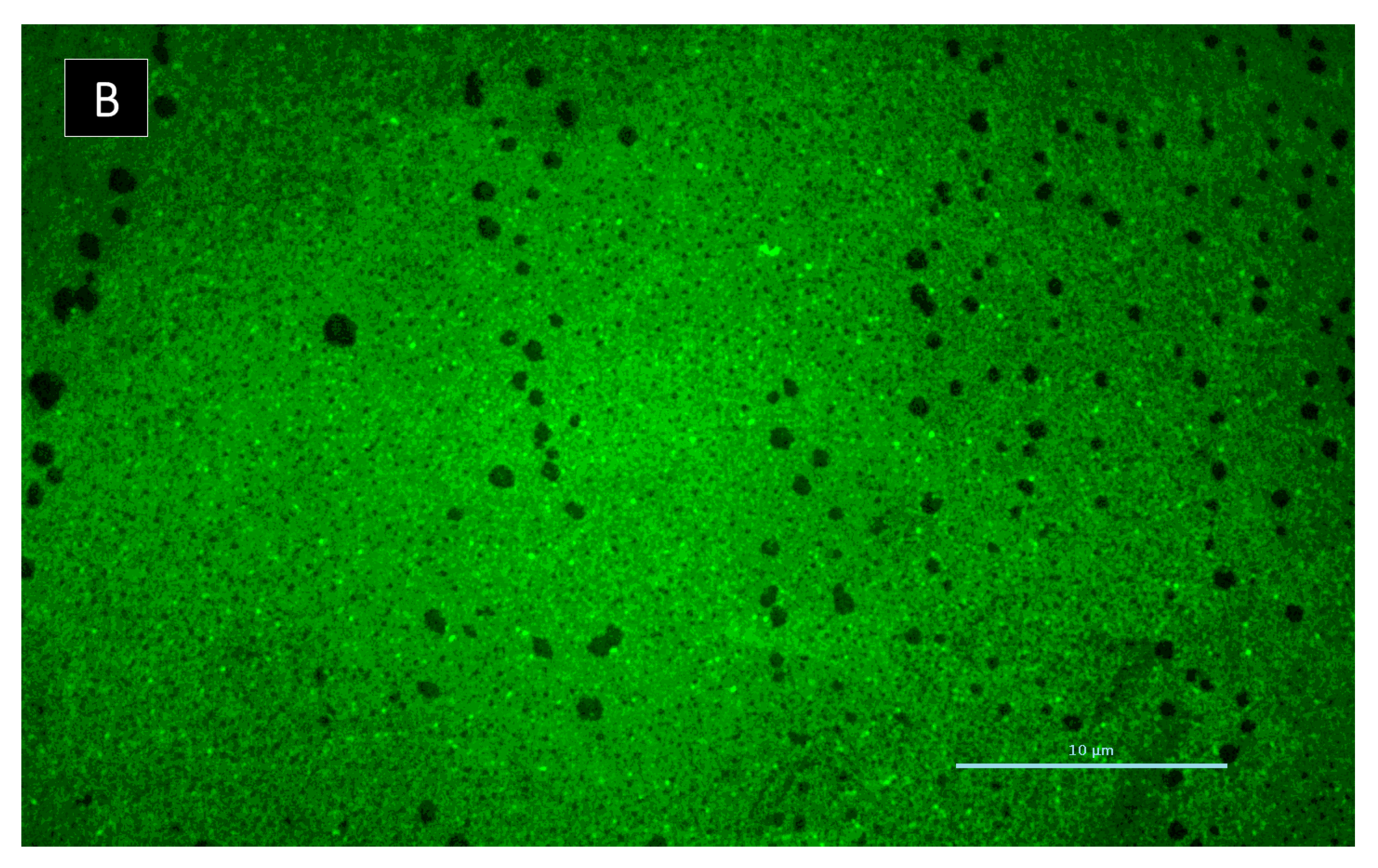
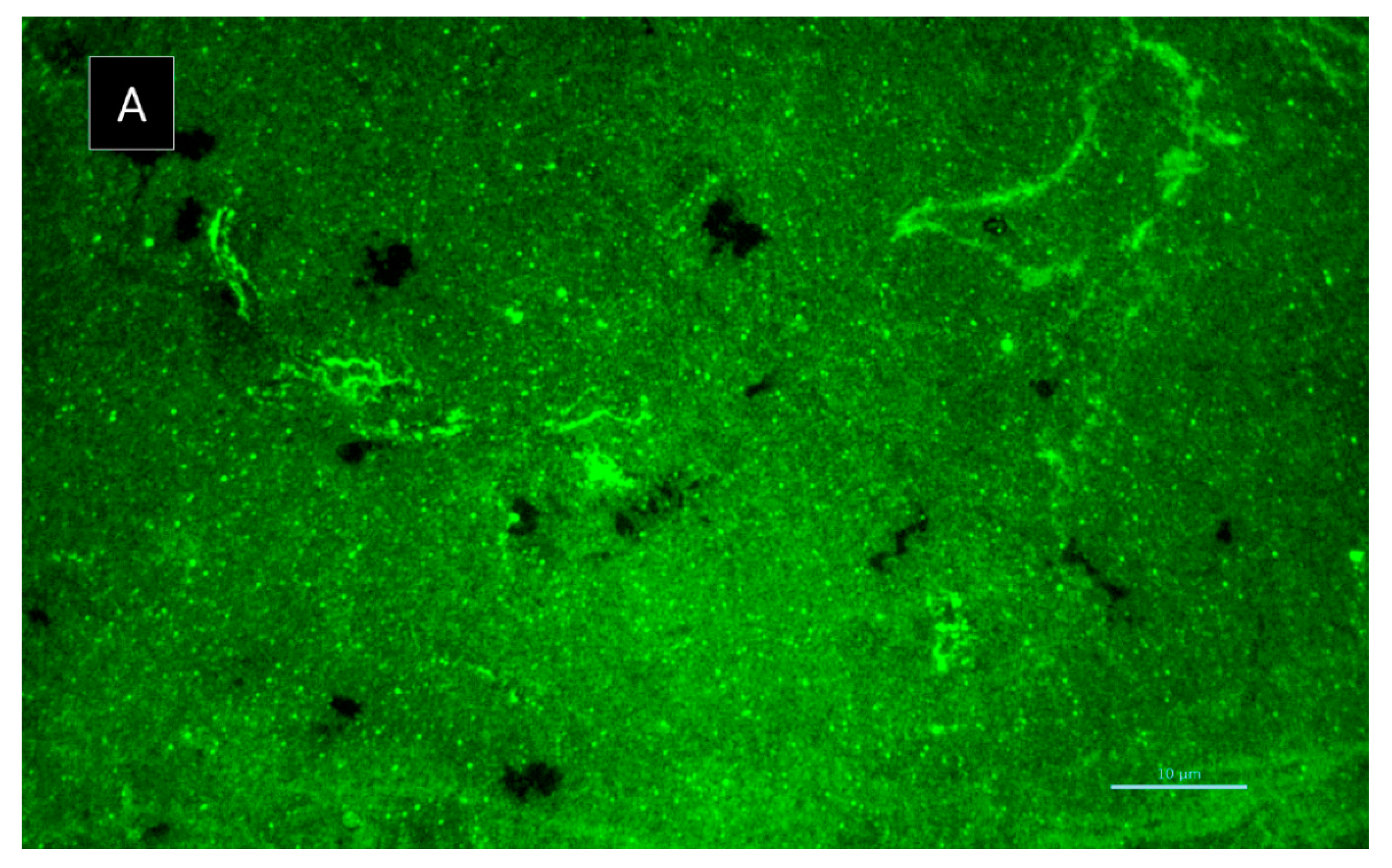
3.5.6. Biofilm Matrix Visualization
3.6. Acetylcholinesterase Inhibitory Activity Assay
3.7. Tyrosinase Inhibitory Activity Assay
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Brakhage, A.A. Regulation of fungal secondary metabolism. Nat. Ver. Microbiol. 2013, 11, 21–32. [Google Scholar] [CrossRef]
- Beekman, A.M.; Barrow, R.A. Fungal Metabolites as Pharmaceuticals. Aust. J. Chem. 2014, 67, 827–843. [Google Scholar] [CrossRef]
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef] [PubMed]
- Prompanya, C.; Fernandes, C.; Cravo, S.; Pinto, M.M.M.; Dethoup, T.; Silva, A.M.S.; Kijjoa, A. A new cyclic hexapeptide and a new isocoumarin derivative from the marine sponge-associated fungus Aspergillus similanensis KUFA 0013. Mar. Drugs. 2015, 13, 1432–1450. [Google Scholar] [CrossRef] [Green Version]
- May Zin, W.W.; Prompanya, C.; Buttachon, S.; Kijjoa, A. Bioactive secondary metabolites from a Thai collection of soil and marine-derived fungi of the genera Neosartorya and Aspergillus. Curr. Drug Deliv. 2016, 13, 378–388. [Google Scholar] [CrossRef]
- Kumla, D.; Aung, T.S.; Buttachon, S.; Dethoup, T.; Gales, L.; Pereira, J.A.; Inácio, Â.; Costa, P.M.; Lee, M.; Sekeroglu, N.; et al. A New Dihydrochromone Dimer and Other Secondary Metabolites from Cultures of the Marine Sponge-Associated Fungi Neosartorya fennelliae KUFA 0811 and Neosartorya tsunodae KUFC 9213. Mar. Drugs 2017, 15, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucinda, J.; Bessa, L.J.; Buttachon, S.; Dethoup, T.; Martins, R.; Vasconcelos, V.; Kijjoa, A.; Costa, P.M. Neofiscalin A and fiscalin C are potential novel indole alkaloid alternatives for the treatment of multidrug resistant Gram-positive bacterial infections. FEMS Microbiol. Lett. 2016, 363, fnw150. [Google Scholar] [CrossRef] [Green Version]
- Rajachan, O.-T.; Kanokmedhakul, K.; Sanmanoch, W.; Boonlue, S.; Hannongbua, S.; Saparpakorn, P.; Kanokmedhakul, S. Chevalone C analogues and globoscinic acid derivatives from the fungus Neosartorya spinosa KKU-1NK1. Phytochemistry 2016, 132, 68–75. [Google Scholar] [CrossRef] [PubMed]
- May Zin, W.W.; Buttachon, S.; Buaruang, J.; Gale, L.; Pereira, J.A.; Pinto, M.M.M.; Silva, A.M.S.; Kijjoa, A. A new meroditerpene and a new tryptoquivaline analog from the algicolous fungus Neosartorya takakii KUFC 7898. Mar. Drugs 2015, 13, 3776–3790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.S.; Ishigami, K.; Watanabe, H. Synthesis of (−)-mellein, (+)-ramulosin, and related natural products. Tetrahedron 2007, 63, 1074–1079. [Google Scholar] [CrossRef]
- Xue, J.; Fu, Y.; Wu, P.; Xu, L.; Huang, R.; Wei, X.; Li, H. Two new anthraquinones from the soil fungus Penicillium purpurigenum SC0070. J. Antibiot. 2015, 68, 598–599. [Google Scholar] [CrossRef]
- May Zin, W.W.; Buttachon, S.; Dethoup, T.; Pereira, J.A.; Gales, L.; Inácio, Â.; Paulo, M.; Costa, P.M.; Lee, M.; Sekeroglu, N.; et al. Antibacterial and antibiofilm activities of the metabolites isolated from the culture of the mangrove-derived endophytic fungus Eurotium chevalieri KUFA0006. Phytochemistry 2017, 141, 86–97. [Google Scholar] [CrossRef]
- Oh, H.; Kwon, T.O.; Gloer, J.B.; Marvanová, L.; Shearer, S.A. Tenellic Acids A-D: New Bioactive Diphenyl Ether Derivatives from the Aquatic Fungus Dendrospora tenella. J. Nat. Prod. 1999, 62, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Proksa, B.; Uhrín, D.; Adamcová, J.; Fuska, J. Vermixocins A and B, two novel metabolites from Penicillium vermiculatum. J. Antibiot. 1992, 45, 1268–1272. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Nozawa, K.; Udagawa, S.; Nakajima, S.; Kawai, K. Penicillide and dehydroisopenicillide from Talaromyces derxii. Phytochemistry 1991, 30, 2096–2098. [Google Scholar] [CrossRef]
- Daengrot, C.; Rukachaisirikul, V.; Tadpetch, K.; Phongpaichit, S.; Bowornwiriyapan, K.; Sakayarojc, J.; Shend, X. Penicillanthone and penicillidic acids A-C from the soil-derived fungus Penicillium aculeatum PSU-RSPG105. RSC Adv. 2016, 6, 39700. [Google Scholar] [CrossRef]
- Singh, A.J.; Gorka, A.P.; Bokesch, H.R.; Wamiru, A.; O’Keefe, B.R.; Schnermann, M.J.; Gustafson, K.R. Harnessing natural product diversity for fluorophore discovery: Naturally occurring fFluorescent hydroxyanthraquinones from the marine crinoid Pterometra venusta. J. Nat. Prod. 2018, 81, 2750–2755. [Google Scholar] [CrossRef]
- Nishida, H.; Tomoda, H.; Cao, J.; Okuda, S.; Omura, S. Purpactins, new inhibitors of acyl-CoA: Cholesterol acyltransterase produced by Penicillium purpurogenum II. Structure elucidation of purpactins A, B, C. J. Antibiot. 1991, 44, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Han, L.; Shao, C.-L.; She, Z.-G.; Wang, C.-Y. Bioactive diphenyl ether derivatives from a gorgonian-derived fungus Talaromyces sp. Chem. Biodivers. 2015, 12, 443–450. [Google Scholar] [CrossRef]
- Davey, M.E.; Caiazza, N.C.; O’Toole, G.A. Rhamnolipid surfactant production affects biofilm architecture in Pseudomonas aeruginosa PAO1. J. Bacteriol. 2003, 185, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Periasamy, S.; Joo, H.-S.; Duong, A.C.; Bach, T.-H.L.; Tan, V.Y.; Chatterjee, S.S.; Cheung, G.Y.C.; Otto, M. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1281–1286. [Google Scholar] [CrossRef] [Green Version]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–90. [Google Scholar] [CrossRef]
- Likhitwitayawuid, K.; Sritularak, B. A new dimeric stilbene with tyrosinase inhibitiory activity from Artocarpus gomezianus. J. Nat. Prod. 2001, 64, 1457–1459. [Google Scholar] [CrossRef]
- Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 1980, 8, 4321–4325. [Google Scholar] [CrossRef] [Green Version]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 72, 5463–5467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Harada, N. ECD Cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef]
- Simões, R.R.; Aires-de-Sousa, M.; Conceição, T.; Antunes, F.; da Costa, P.M.; de Lencastre, H. High prevalence of EMRSA-15 in Portuguese public buses: A worrisome finding. PLoS ONE 2011, 6, e17630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessa, L.J.; Barbosa-Vasconcelos, A.; Mendes, Â.; Vaz-Pires, P.; Da Costa, P.M. High prevalence of multidrug-resistant Escherichia coli and Enterococcus spp. in river water, upstream and downstream of a wastewater treatment plant. J. Water Health 2014, 12, 426–435. [Google Scholar] [CrossRef] [PubMed]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 27th ed.; CLSI supplement M100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 11th ed.; CLSI document M02-A11; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically, 10th ed.; CLSI document M07-A10; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015. [Google Scholar]
- CLSI. Methods for Determining Bactericidal Activity of Antimicrobial Agents; Approved Guideline, CLSI Document M26-A; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 1999. [Google Scholar]
- Kumla, D.; Dethoup, T.; Gales, L.; Pereira, J.A.; Freitas-Silva, J.; Costa, P.M.; Silva, A.; Pinto, M.M.; Kijjoa, A. Erubescensoic Acid, a new polyketide and a xanthonopyrone SPF-3059-26 from the culture of the marine sponge-associated fungus Penicillium erubescens KUFA 0220 and antibacterial activity evaluation of some of its constituents. Molecules 2019, 24, 208. [Google Scholar] [CrossRef] [Green Version]
- Buttachon, S.; Ramos, A.A.; Inácio, Â.; Dethoup, T.; Gales, L.; Lee, M.; Costa, P.M.; Silva, A.M.S.; Sekeroglu, N.; Rocha, E.; et al. Bis-indolyl benzenoids, hydroxypyrrolidine derivatives and other constituents from cultures of the marine sponge-associated fungus Aspergillus candidus KUFA0062. Mar. Drugs 2018, 16, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odds, F.C. Synergy, antagonism, and what the chequer board puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef]
- Stepanović, S.; Vuković, D.; Hola, V.; Di Bonaventura, G.; Djuki’c, S.; Cirković, I.; Ruzicka, F. Quantification of biofilm in microtiter plates: Overview of testing conditions and practical recommendations for assessment of biofilm production by staphylococci. Apmis 2007, 115, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Grela, E.; Kozłowska, J.; Grabowiecka, A. Current methodology of MTT assay in bacteria—A review. Acta Histochem. 2018, 120, 303–331. [Google Scholar] [CrossRef] [PubMed]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell viability assays. In The Assay Guidance Manual [Internet]; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004; 1 May 2013; Last Update: 1 July 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK144065/ (accessed on 17 June 2021).
- Bessa, L.J.; Grande, R.; Di Iorio, D.; Di Giulio, M.; Di Campli, E.; Cellini, L. Helicobacter pylori free-living and biofilm modes of growth: Behavior in response to different culture media. Apmis 2013, 121, 149–160. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC, Type | δH, J in Hz | COSY | HMBC |
|---|---|---|---|---|
| 1 | 162.1, C | |||
| 2 | 125.1, CH | 7.15, d (1.5) | H-4 | C-1, 1′, 4, 9a |
| 3 | 146.6, C | |||
| 4 | 119.6, CH | 7.47, d (1.5) | H-2 | C-1′, 2, 9a, 10 |
| 4a | 132.6, C | |||
| 5 | 108.3, CH | 7.16, d (2.2) | H-7 | C-7, 8a, 10 |
| 6 | 166.6, C | |||
| 7 | 105.5, CH | 6.78, d (2.2) | H-5 | C-5, 6, 8a |
| 8 | 164.1, C | |||
| 8a | 112.2, C | |||
| 9 | 186.4, CO | |||
| 9a | 115.6, C | |||
| 10 | 183.0, CO | |||
| 10a | 137.2, C | |||
| OMe-8 | 56.7, CH3 | 3.89, s | C-8 | |
| 1′ | 41.5, CH2 | 2.92, m | H-2′ | C-2. 3, 4 |
| 2′ | 70.7, CH | 5.06, m | H2-1′, H3-3′ | C-3, CO (Ac) |
| 3′ | 20.0, CH3 | 1.20, d (6.3) | H-2′ | C-1′, 2′ |
| CO (Ac) | 170.2, CO | |||
| Me (Ac) | 21.4, CH3 | 1.94, s | ||
| OH-1′ | - | 13.40, s | C-1, 2, 9a |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 119.3, C | - | ||
| 2 | 155.1, C | - | ||
| 3 | 130.5, C | |||
| 4 | 128.5, CH | 7.25, d (8.7) | H-5 | C-2, 6, 8 |
| 5 | 110.3, CH | 6.32, d (8.7) | H-4 | C-1, 3, 6, 7 (w) |
| 6 | 154.8, C | - | ||
| 7 | 167.2, CO | - | ||
| 8 | 73.1, CH | 4.58, dd (9.2, 3.8) | H-9a, 9b | C-3, 9, 14 |
| 9a, b | 47.1, CH2 | 1.57, ddd (13.8, 9.2, 5.0) 1.27, ddd (13.8, 8.9, 3.9) | C-8, 9, 11, 12 | |
| 10 | 24.9, CH | 1.71, m | H-9a, 9b, 11, 12 | |
| 11 | 23.7, CH3 | 0.88, d (6.6) | H-10 | C-9, 10, 12 |
| 12 | 22.2, CH3 | 0.92, d (6.6) | H-10 | C.9, 10, 11 |
| 13 | 62.8, OMe | 3.83, s | ||
| 14 | 63.9, CH2 | 3.25, q (7.0) | H-15 | C-8, 15 |
| 15 | 15.7, CH3 | 1.06, t (7.0) | H-14 | C-14 |
| 1′ | 129.8, C | - | ||
| 2′ | 142.3, C | - | ||
| 3′ | 150.9, C | - | ||
| 4′ | 124.2, CH | 7.14, d (2.0) | H-6′, 8′ | C-2′, 3′, 6′, 8′ |
| 5′ | 136.7, C | - | ||
| 6′ | 118.4, CH | 7.11, d (2.0) | H-4′, 6′ | C-2′, 3′ (w), 7′, 8′ |
| 7′ | 189.8, CHO | 10.15, s | C-1′, 5′ (w), 6′ | |
| 8′ | 21.0, CH3 | 2.31, s | H-4′, 6′ |
| Position | δC, Type | δH, (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 114.3, C | - | ||
| 2 | 157.9, C | - | ||
| 3 | 138.3, C | - | ||
| 4 | 132.8, CH | 7.67, d (8.6) | H-5 | C-1, 3, 6 |
| 5 | 115.6, CH | 7.21, d (8.6) | H-4 | C-2, 6, 8 |
| 6 | 160.2, C | - | ||
| 7 | 161.6, CO | - | ||
| 8 | 64.4, CH | 4.87, ddd (9.2, 4.9, 4.2) | H-9a, 9b, OH-8 | |
| 9a, b | 48.3, CH2 | 1.24, ddd (13.7, 9.2, 4.2) 1.44, ddd (13.7, 9.2, 4.9) | H-8, 9b, 10 H-8, 9a, 10 | |
| 10 | 24.8, CH | 1.72, m | H-9a, b, Me -11, 12 | |
| 11 | 23.9, CH3 | 0.86, d (6.7) | H-10 | C-9, 12 |
| 12 | 22.1, CH3 | 0.90, d (6.7) | H-10 | C-9, 11 |
| 13 | 63.1, OMe | 3.76, s | C-2 | |
| 1′ | 135.6, C | - | ||
| 2′ | 145.9, C | - | ||
| 3′ | 143.4, C | - | ||
| 4′ | 119.9, CH | 7.11, brs | C-2′, 3′, 6′ | |
| 5′ | 136.3, C | - | ||
| 6′ | 125.9, CH | 7.12, d (0.5) | C-2′, 4′, 7′,8′ | |
| 7′ | 57.9, CH2 | 4.72, d (5.8) | OH-7′ | C-1′, 2′, 6′ |
| 8′ | 20.9, CH3 | 2.28, s | C-4′, 5′, 6′ | |
| OH-7′ | 5.34, t (5.8) | H-7′ | C-7′ | |
| OH-8 | 5.13, d (4.9) | H-8 | C-9 |
| Compound | E. faecalis ATCC 29212 | E. faecalis B3/101 (VRE) | S. aureus ATCC 29213 | S. aureus 66/1 (MRSA) |
|---|---|---|---|---|
| 2 | >64 | >64 | >64 | >64 |
| 5 | >64 | >64 | >64 | >64 |
| 6 | >64 | >64 | >64 | >64 |
| 7 | >64 | 64 | >64 | >64 |
| 8 | >64 | >64 | >64 | >64 |
| CAZ | - | - | 8 | - |
| KAN | 32 | - | - | - |
| Compound | Concentration (µg/mL) | Biofilm Biomass (% of Control) | ||
|---|---|---|---|---|
| E. coli ATCC 25922 | E. faecalis ATCC 29212 | S. aureus ATCC 29213 | ||
| 5 | 64 | 88.39 ± 0.09 *** | 75.89 ± 0.10 *** | 84.46 ± 0.10 *** |
| 6 | 64 | 83.89 ± 0.19 *** | - | 56.00 ± 0.06 *** |
| Compounds | % Inhibition at 200 µM | IC50 (µM) |
|---|---|---|
| 2 | 11.56 ± 2.05 * | n.d. |
| 5 | 4.58 ± 0.07 *** | n.d. |
| 6 | n.a. | - |
| 7 | 5.33 ± 0.18 *** | n.d. |
| 8 | 53.1 ± 1.17 *** | 177.03 ± 8.17 ** |
| Kojic acid (positive control) | 95.04 ± 0.018 **** | 14.00 ± 0.12 **** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Sá, J.D.M.; Pereira, J.A.; Dethoup, T.; Cidade, H.; Sousa, M.E.; Rodrigues, I.C.; Costa, P.M.; Mistry, S.; Silva, A.M.S.; Kijjoa, A. Anthraquinones, Diphenyl Ethers, and Their Derivatives from the Culture of the Marine Sponge-Associated Fungus Neosartorya spinosa KUFA 1047. Mar. Drugs 2021, 19, 457. https://doi.org/10.3390/md19080457
de Sá JDM, Pereira JA, Dethoup T, Cidade H, Sousa ME, Rodrigues IC, Costa PM, Mistry S, Silva AMS, Kijjoa A. Anthraquinones, Diphenyl Ethers, and Their Derivatives from the Culture of the Marine Sponge-Associated Fungus Neosartorya spinosa KUFA 1047. Marine Drugs. 2021; 19(8):457. https://doi.org/10.3390/md19080457
Chicago/Turabian Stylede Sá, Joana D. M., José A. Pereira, Tida Dethoup, Honorina Cidade, Maria Emília Sousa, Inês C. Rodrigues, Paulo M. Costa, Sharad Mistry, Artur M. S. Silva, and Anake Kijjoa. 2021. "Anthraquinones, Diphenyl Ethers, and Their Derivatives from the Culture of the Marine Sponge-Associated Fungus Neosartorya spinosa KUFA 1047" Marine Drugs 19, no. 8: 457. https://doi.org/10.3390/md19080457
APA Stylede Sá, J. D. M., Pereira, J. A., Dethoup, T., Cidade, H., Sousa, M. E., Rodrigues, I. C., Costa, P. M., Mistry, S., Silva, A. M. S., & Kijjoa, A. (2021). Anthraquinones, Diphenyl Ethers, and Their Derivatives from the Culture of the Marine Sponge-Associated Fungus Neosartorya spinosa KUFA 1047. Marine Drugs, 19(8), 457. https://doi.org/10.3390/md19080457