
New Drimane Sesquiterpenes and Polyketides from Marine-Derived Fungus Penicillium sp. TW58-16 and Their Anti-Inflammatory and α-Glucosidase Inhibitory Effects


Abstract
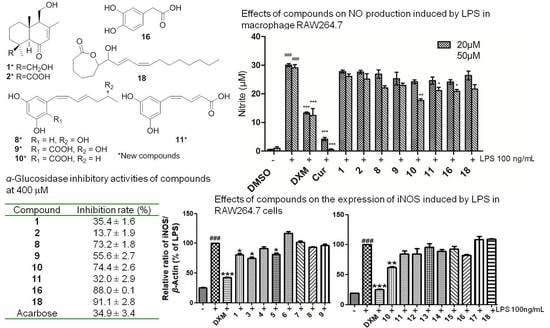
:
1. Introduction
2. Results
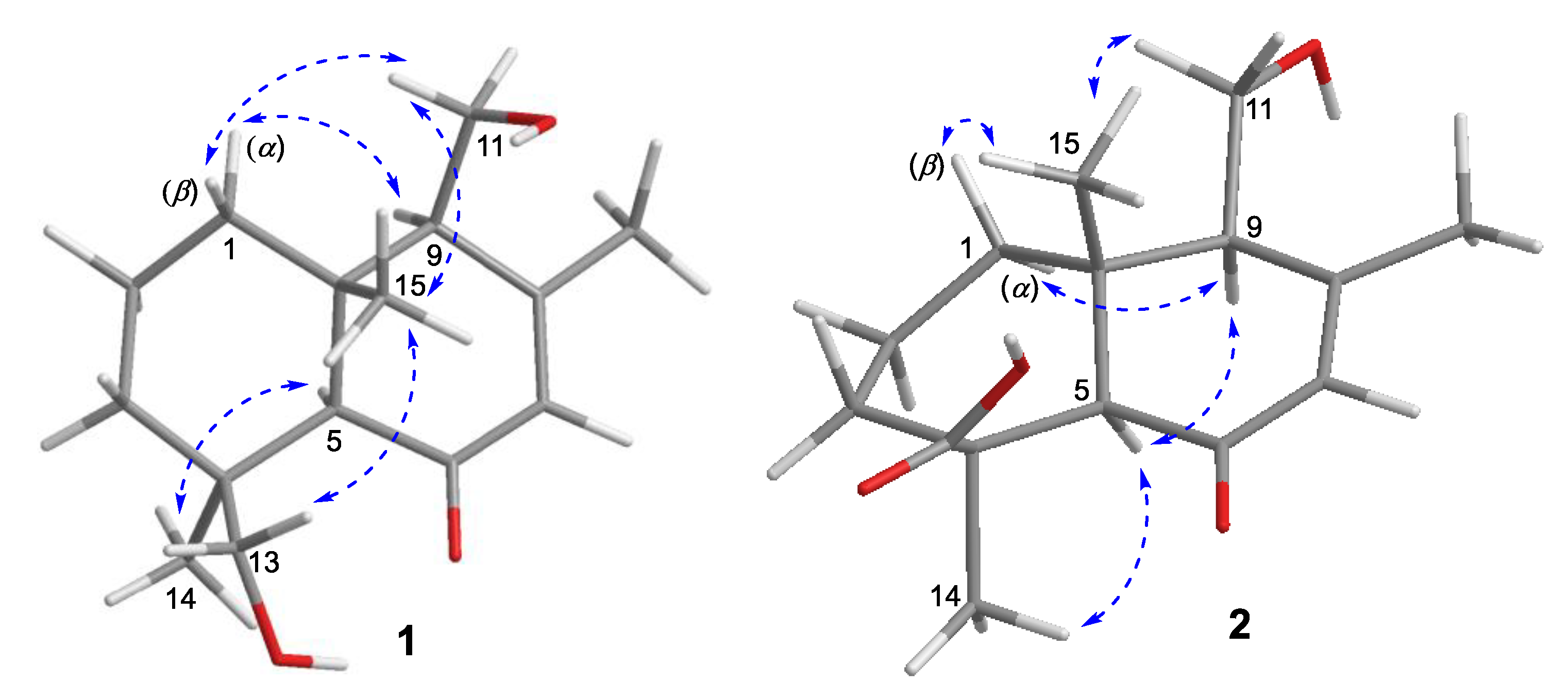
2.1. Structure Elucidation
2.2. Bioactivities
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedure
4.2. Fungal Material
4.3. Fermentation and Extraction
4.4. Compound Isolation
4.5. Spectroscopic Data of Compounds
4.6. Quantum Chemical ECD Calculations of Compounds 1, 2, 7, and 8
4.7. Cell Culture
4.8. Measurement of Cell Viability
4.9. NO Inhibition Assay
4.10. Western Blotting Assay
4.11. α-Glucosidase Inhibitory Assay
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shinde, P.; Banerjee, P.; Mandhare, A. Marine natural products as source of new drugs: A patent review (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 283–309. [Google Scholar] [CrossRef]
- Liu, Z.; Frank, M.; Yu, X.Q.; Yu, H.Q.; Tran-Cong, N.M.; Gao, Y.; Proksch, P. Secondary metabolites from marine-derived fungi from China. Prog. Chem. Org. Nat. Prod. 2020, 111, 81–153. [Google Scholar]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, S.S.; Gui, M.; Li, H.H.; Yu, C.B.; Li, H.J.; Zeng, Z.L.; Sun, P. Secondary metabolites from marine micromonospora: Chemistry and bioactivities. Chem. Biodivers. 2020, 17, e2000024. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, I.; Kim, S.K. Chemistry and Pharmacology of Naturally Occurring Bioactive Compounds; Brahmachari, G., Ed.; CRC Press: Boca Raton, FL, USA, 2013; Chapter 18; pp. 445–457. [Google Scholar]
- Pittayakhajonwut, P.; Dramae, A.; Intaraudom, C.; Boonyuen, N.; Nithithanasilp, S.; Rachtawee, P.; Laksanacharoen, P. Two new drimane sesquiterpenes, fudecadiones A and B, from the soil fungus Penicillium sp. BCC 17468. Planta Med. 2011, 77, 74–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, T.; Nagai, A.; Takagi, M.; Shin-ya, K. JBIR-137 and JBIR-138, new secondary metabolites from Aspergillus sp. fA75. J. Antibiot. 2012, 65, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Bian, X.Q.; Bai, J.; Hu, X.L.; Wu, X.; Xue, C.M.; Han, A.H.; Su, G.Y.; Hua, H.M.; Pei, Y.H. Penioxalicin, a novel 3-nor-2,3-seco-labdane type diterpene from the fungus Penicillium oxalicum TW01-1. Tetrahedron Lett. 2015, 56, 5013–5016. [Google Scholar] [CrossRef]
- Qi, B.; Jia, F.F.; Luo, Y.; Ding, N.; Li, S.; Shi, F.Y.; Hai, Y.; Wang, L.L.; Zhu, Z.X.; Liu, X.; et al. Two new diterpenoids from Penicillium chrysogenum MT-12, an endophytic fungus isolated from Huperzia serrata. Nat. Prod. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Liu, W.; Han, S.Y.; Zhang, J.; Xu, W.; Li, Q.; Cheng, Z.B. Penitholabene, a rare 19-nor labdane-type diterpenoid from the deep-sea-derived fungus Penicillium thomii YPGA3. Fitoterapia 2020, 146, 104691. [Google Scholar] [CrossRef]
- Jouda, J.B.; Kusari, S.; Lamshoeft, M.; Mouafo Talontsi, F.; Douala Meli, C.; Wandji, J.; Spiteller, M. Penialidins A-C with strong antibacterial activities from Penicillium sp. an endophytic fungus harboring leaves of Garcinia nobilis. Fitoterapia 2014, 98, 209–214. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Nonaka, G.; Nishioka, I. Studies on rhubarb (rhei rhizoma). V. Isolation and characterization of chromone and chromanone derivatives. Chem. Pharm. Bull. 1984, 32, 3493–3500. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Z.; Xiao, B.H.; Chen, Q.; Lian, X.Y. Synthesis and biological evaluation of caffeic acid 3,4-dihydroxyphenethyl ester. J. Nat. Prod. 2010, 73, 252–254. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.W.; Nagasawa, H.; Nagura, F.; Mohamad, S.B.; Uto, Y.; Ohkura, K.; Hori, H. Elucidation of strict structural requirements of brefeldin A as an inducer of differentiation and apoptosis. Bioorg. Med. Chem. 2000, 8, 455–463. [Google Scholar] [CrossRef]
- Zeng, F.R.; Chen, C.M.; Al Chnani, A.A.L.; Zhou, Q.; Tong, Q.Y.; Wang, W.J.; Zang, Y.; Gong, J.J.; Wu, Z.D.; Liu, J.J.; et al. Dibrefeldins A and B, A pair of epimers representing the first brefeldin A dimers with cytotoxic activities from Penicillium janthinellum. Bioorg. Chem. 2019, 86, 176–182. [Google Scholar] [CrossRef]
- Guzman-Gutierrez, S.L.; Nieto-Camacho, A.; Castillo-Arellano, J.I.; Huerta-Salazar, E.; HernaNdez-Pasteur, G.; Silva-Miranda, M.; ArguEllo-NaJera, O.; Sepulveda-Robles, O.; Espitia, C.I.; Reyes-Chilpa, R. Mexican propolis: A source of antioxidants and anti-inflammatory compounds, and isolation of a novel chalcone and ε-caprolactone derivative. Molecules 2018, 23, 334. [Google Scholar] [CrossRef] [Green Version]
- Shindo, M.; Makigawa, S.; Matsumoto, K.; Iwata, T.; Wasano, N.; Kano, A.; Morita, M.T.; Fujii, Y. Essential structural features of (2Z,4E)-5-phenylpenta-2,4-dienoic acid for inhibition of root gravitropism. Phytochemistry 2020, 172, 112287. [Google Scholar] [CrossRef]
- Okuma, K.; Tanaka, Y.; Ohta, H.; Matsuyama, H. Optical resolution of 2- and 3-hydroxyalkyltriphenylphosphonium salts. stereoselective synthesis of enantiomerically pure (E)- and (Z)-homoallylic alcohols. Bull. Chem. Soc. Jpn. 1993, 66, 2623–2632. [Google Scholar] [CrossRef]
- Lan, W.J.; Fu, S.J.; Xu, M.Y.; Liang, W.L.; Lam, C.K.; Zhong, G.H.; Xu, J.; Yang, D.P.; Li, H.J. Five new cytotoxic metabolites from the marine fungus neosartorya pseudofischeri. Mar. Drugs 2016, 14, 18. [Google Scholar] [CrossRef] [Green Version]
- Fan, A.L.; Mi, W.B.; Liu, Z.G.; Zeng, G.H.; Zhang, P.; Hu, Y.C.; Fang, W.G.; Yin, W.B. Deletion of a histone acetyltransferase leads to the pleiotropic activation of natural products in Metarhizium robertsii. Org. Lett. 2017, 19, 1686–1689. [Google Scholar] [CrossRef]
- Tian, J.F.; Li, P.J.; Li, X.X.; Sun, P.H.; Gao, H.; Liu, X.Z.; Huang, P.; Tang, J.S.; Yao, X.S. New antibacterial isocoumarin glycosides from a wetland soil derived fungal strain Metarhizium anisopliae. Bioorg. Med. Chem. Lett. 2016, 26, 1391–1396. [Google Scholar] [CrossRef]
- Lu, S.L.; Wang, J.M.; Sheng, R.L.; Fang, Y.W.; Guo, R.H. Novel bioactive polyketides isolated from marine actinomycetes: An update review from 2013 to 2019. Chem. Biodivers. 2020, 17, e2000562. [Google Scholar] [CrossRef]
- Yang, L.J.; Peng, X.Y.; Zhang, Y.H.; Liu, Z.Q.; Li, X.; Gu, Y.C.; Shao, C.L.; Han, Z.; Wang, C.Y. Antimicrobial and antioxidant polyketides from a deep-sea-derived fungus Aspergillus versicolor SH0105. Mar. Drugs 2020, 18, 636. [Google Scholar] [CrossRef]
- Risdian, C.; Mozef, T.; Wink, J. Biosynthesis of polyketides in streptomyces. Microorganisms 2019, 7, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.J.; Chen, S.H.; Liu, W.Y.; Liu, Y.Y.; Huang, X.S.; She, Z.G. Polyketides with immunosuppressive activities from mangrove endophytic fungus Penicillium sp. ZJ-SY2. Mar. Drugs 2016, 14, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.X.; Zheng, C.J.; Huang, G.L.; Mei, R.Q.; Nong, X.H.; Shao, T.M.; Chen, G.Y.; Wang, C.Y. Bioactive polyketide derivatives from the mangrove-derived fungus Daldinia eschscholtzii HJ004. J. Nat. Prod. 2019, 82, 2211–2219. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.M.; Chen, S.H.; Qiu, P.; Tan, C.B.; Long, Y.H.; Lu, Y.J.; She, Z.G. (+)- and (−)-Ascomlactone A: A pair of novel dimeric polyketides from a mangrove endophytic fungus Ascomycota sp. SK2YWS-L. Org. Biomol. Chem. 2017, 15, 10276–10280. [Google Scholar] [CrossRef]
- Jansen, B.J.M.; Groot, A.D. The occurrence and biological activity of drimane sesquiterpenoids. Nat. Prod. Rep. 1991, 8, 309–318. [Google Scholar] [CrossRef]
- Cui, H.; Liu, Y.Y.; Nie, Y.; Liu, Z.M.; Chen, S.H.; Zhang, Z.R.; Lu, Y.J.; He, L.; Huang, X.S.; She, Z.G. Polyketides from the mangrove-derived endophytic fungus Nectria sp. HN001 and their α-glucosidase inhibitory activity. Mar. Drugs 2016, 14, 86. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Y.; Yang, Q.; Xia, G.P.; Huang, H.B.; Li, H.X.; Ma, L.; Lu, Y.J.; He, L.; Xia, X.K.; She, Z.G. Polyketides with α-glucosidase inhibitory activity from a mangrove endophytic fungus, Penicillium sp. HN29-3B1. J. Nat. Prod. 2015, 78, 1816–1822. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Stephens, P.J.; Harada, N. ECD Cotton effect approximated by the Gaussian curve and other methods. Chirality 2010, 22, 229–233. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumloffel, A.; Hemberger, Y.; Bringmann, G. SpecDics: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Gao, E.; Zhou, Z.Q.; Zou, J.; Yu, Y.; Feng, X.L.; Chen, G.D.; He, R.R.; Yao, X.S.; Gao, H. Bioactive asarone-derived phenylpropanoids from the rhizome of Acorus tatarinowii Schott. J. Nat. Prod. 2017, 80, 2923–2929. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Inhibition Rate (%, 400 μM) a | Compound | Inhibition Rate (%, 400 μM) a |
|---|---|---|---|
| 1 | 35.4 ± 1.6 | 11 | 10.0 ± 3.2 |
| 2 | 13.7 ± 1.9 | 12 | 4.8 ± 3.3 |
| 3 | 73.2 ± 1.8 | 13 | 17.9 ± 4.1 |
| 4 | 55.6 ± 2.7 | 14 | 36.9 ± 3.1 |
| 5 | 74.4 ± 2.6 | 15 | 5.3 ± 2.2 |
| 6 | 32.0 ± 2.9 | 16 | 88.0 ± 0.1 |
| 7 | 5.1 ± 2.3 | 17 | 0.4 ± 3.6 |
| 8 | 2.5 ± 1.1 | 18 | 91.1 ± 2.8 |
| 9 | 5.7 ± 0.2 | Acarbose | 34.9 ± 3.4 |
| 10 | 10.3 ± 2.0 |
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 40.1 | 1.48 (1H, m, Ha); 2.06 (1H, brd, 13.1, Hb) | 40.0 | 1.49 (1H, dd, 13.3, 3.6, Ha); 2.05 (1H, brd, 12.8, Hb) |
| 2 | 18.9 | 1.48 (1H, m, Ha); 1.60 (1H, m, Hb) | 20.0 | 1.54 (1H, m, Ha); 1.72 (1H, m, Hb) |
| 3 | 37.2 | 1.94 (1H, brd, 13.5, Ha); 0.96 (1H, m, Hb) | 40.1 | 1.22 (1H, td, 13.8, 3.5, Ha); 2.16 (1H, brd, 13.8, Hb) |
| 4 | 39.1 | 44.6 | ||
| 5 | 65.2 | 2.44 (1H, s) | 64.7 | 2.63 (1H, s) |
| 6 | 203.2 | 203.7 | ||
| 7 | 129.0 | 5.80 (1H, s) | 128.4 | 5.95 (1H, s) |
| 8 | 162.2 | 164.4 | ||
| 9 | 59.7 | 2.39 (1H, brs) | 59.2 | 2.44 (1H, brs) |
| 10 | 43.5 | 44.0 | ||
| 11 | 59.9 | 3.91 (1H, dd, 11.5, 2.7, Ha); 3.76 (1H, dd, 11.5, 6.0, Hb) | 59.7 | 3.93 (1H, dd, 11.6, 2.9, Ha); 3.78 (1H, dd, 11.6, 6.0, Hb) |
| 12 | 22.3 | 2.08 (3H, s) | 22.5 | 2.13 (3H, s) |
| 13 | 63.8 | 4.13 (1H, d, 11.0, Ha); 3.65 (1H, d, 11.0, Hb) | 179.7 | |
| 14 | 27.4 | 1.15 (3H, s) | 29.6 | 1.42 (3H, s) |
| 15 | 17.0 | 0.90 (3H, s) | 16.0 | 0.95 (3H, s) |
| No. | 3 b | 4 a | 5 a | 6 b | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 158.3 | 162.3 | 162.0 | 158.4 | ||||
| 2 | 101.4 | 6.11 (1H, d, 2.8) | 102.5 | 6.21 (1H, brs) | 102.5 | 6.21 (1H, brs) | 102.4 | 6.20 (1H, brs) |
| 3 | 158.3 | 159.3 | 158.6 | 158.4 | ||||
| 4 | 106.8 | 6.18 (1H, d, 2.1) | 108.5 | 108.5 | 107.1 | 6.20 (1H, brs) | ||
| 5 | 138.7 | 143.6 | 143.7 | 137.8 | ||||
| 6 | 106.8 | 6.18 (1H, d, 2.1) | 111.9 | 6.22 (1H, brs) | 111.7 | 6.21 (1H, brs) | 107.1 | 6.20 (1H, brs) |
| 7 | 127.9 | 6.11 (1H, d, 11.0) | 132.1 | 6.82 (1H, d, 11.0) | 131.7 | 6.82 (1H, d, 10.4) | 136.5 | 6.60 (1H, d, 11.4) |
| 8 | 129.8 | 6.12 (1H, m) | 128.9 | 6.13 (1H, t, 11.0) | 128.9 | 6.10 (1H, t, 10.4) | 127.4 | 6.32 (1H, t, 11.4) |
| 9 | 128.0 | 6.57 (1H, dd, 15.0, 10.2) | 130.6 | 6.38 (1H, dd, 14.9, 11.3) | 128.6 | 6.34 (1H, dd, 14.8, 11.3) | 138.1 | 7.53 (1H, dd, 14.8, 12.2) |
| 10 | 134.7 | 5.87 (1H, dt, 15.0, 7.4) | 132.7 | 5.79 (1H, dt, 14.9, 7.3) | 136.5 | 5.76 (1H, dt, 14.8, 7.1) | 126.8 | 6.02 (1H, d, 14.8) |
| 11 | 42.6 | 2.17 (2H, m) | 43.6 | 2.21 (2H, m) | 36.0 | 2.05 (2H, m) | 168.7 | |
| 12 | 65.9 | 3.66 (1H, m) | 68.6 | 3.77 (1H, m) | 23.6 | 1.41 (2H, m) | ||
| 13 | 23.2 | 1.04 (3H, d, 6.2) | 23.0 | 1.14 (3H, d, 4.2) | 14.0 | 0.91 (3H, t, 7.4) | ||
| 14 | 166.1 | 165.9 | ||||||
| No. | 7 | 8 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δC | δH (J in Hz) | |
| 1 | 168.6 | 169.3 | ||
| 2 | - | - | - | - |
| 3 | 76.7 | 5.30 (1H, m) | 78.1 | 4.56 (1H, m) |
| 4 | 31.7 | 3.10 (1H, dd, 16.5, 3.7); 2.95 (1H, dd, 16.5, 10.0) | 32.0 | 2.92 (1H, dd, 16.4, 2.9); 2.83 (1H, dd, 16.4, 11.4) |
| 4a | 141.2 | 142.1 | ||
| 5 | 107.2 | 6.26 (1H, brs) | 107.0 | 6.23 (1H, brs) |
| 6 | 165.3 | 165.0 | ||
| 7 | 101.1 | 6.19 (1H, d, 1.6) | 100.9 | 6.17 (1H, brs) |
| 8 | 163.4 | 163.4 | ||
| 8a | 99.8 | 99.9 | ||
| 9 | 140.1 | 6.68 (1H, dd, 15.6, 4.1) | 29.6 | 1.92 (2H, m) |
| 10 | 126.6 | 6.00 (1H, d, 15.6) | 29.6 | 2.35 (2H, m) |
| 11 | 167.6 | 174.3 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gou, X.; Tian, D.; Wei, J.; Ma, Y.; Zhang, Y.; Chen, M.; Ding, W.; Wu, B.; Tang, J. New Drimane Sesquiterpenes and Polyketides from Marine-Derived Fungus Penicillium sp. TW58-16 and Their Anti-Inflammatory and α-Glucosidase Inhibitory Effects. Mar. Drugs 2021, 19, 416. https://doi.org/10.3390/md19080416
Gou X, Tian D, Wei J, Ma Y, Zhang Y, Chen M, Ding W, Wu B, Tang J. New Drimane Sesquiterpenes and Polyketides from Marine-Derived Fungus Penicillium sp. TW58-16 and Their Anti-Inflammatory and α-Glucosidase Inhibitory Effects. Marine Drugs. 2021; 19(8):416. https://doi.org/10.3390/md19080416
Chicago/Turabian StyleGou, Xiaoshuang, Danmei Tian, Jihua Wei, Yihan Ma, Yixue Zhang, Mei Chen, Wenjuan Ding, Bin Wu, and Jinshan Tang. 2021. "New Drimane Sesquiterpenes and Polyketides from Marine-Derived Fungus Penicillium sp. TW58-16 and Their Anti-Inflammatory and α-Glucosidase Inhibitory Effects" Marine Drugs 19, no. 8: 416. https://doi.org/10.3390/md19080416
APA StyleGou, X., Tian, D., Wei, J., Ma, Y., Zhang, Y., Chen, M., Ding, W., Wu, B., & Tang, J. (2021). New Drimane Sesquiterpenes and Polyketides from Marine-Derived Fungus Penicillium sp. TW58-16 and Their Anti-Inflammatory and α-Glucosidase Inhibitory Effects. Marine Drugs, 19(8), 416. https://doi.org/10.3390/md19080416