Density Functional Theory (DFT)-Aided Structure Elucidation of Linear Diterpenes from the Irish Brown Seaweed Bifurcaria bifurcata

 , , and
, , and 
Abstract
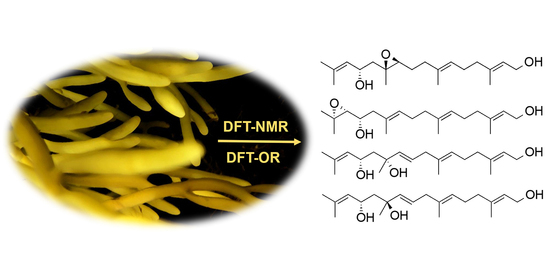
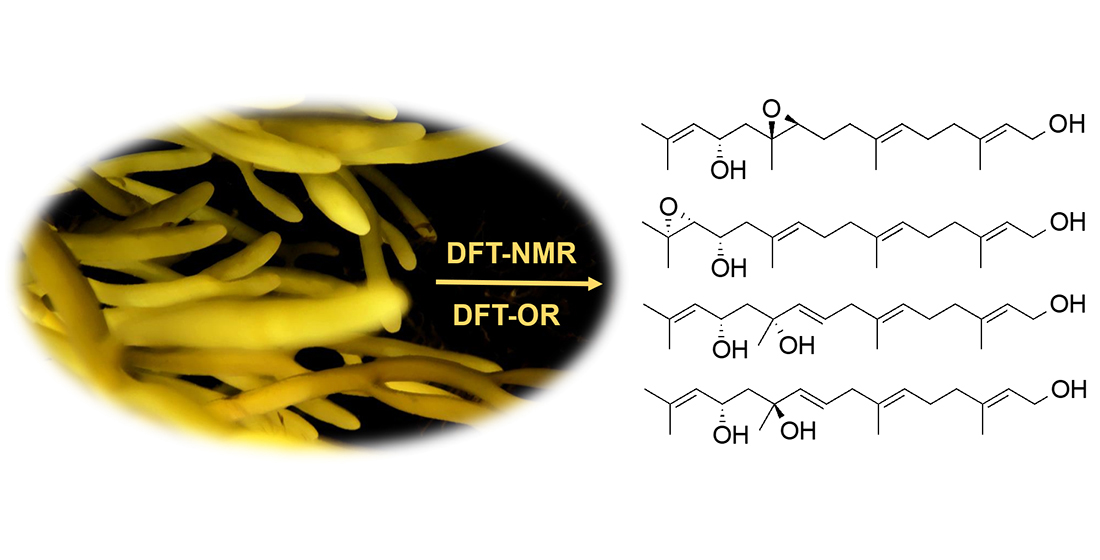
1. Introduction
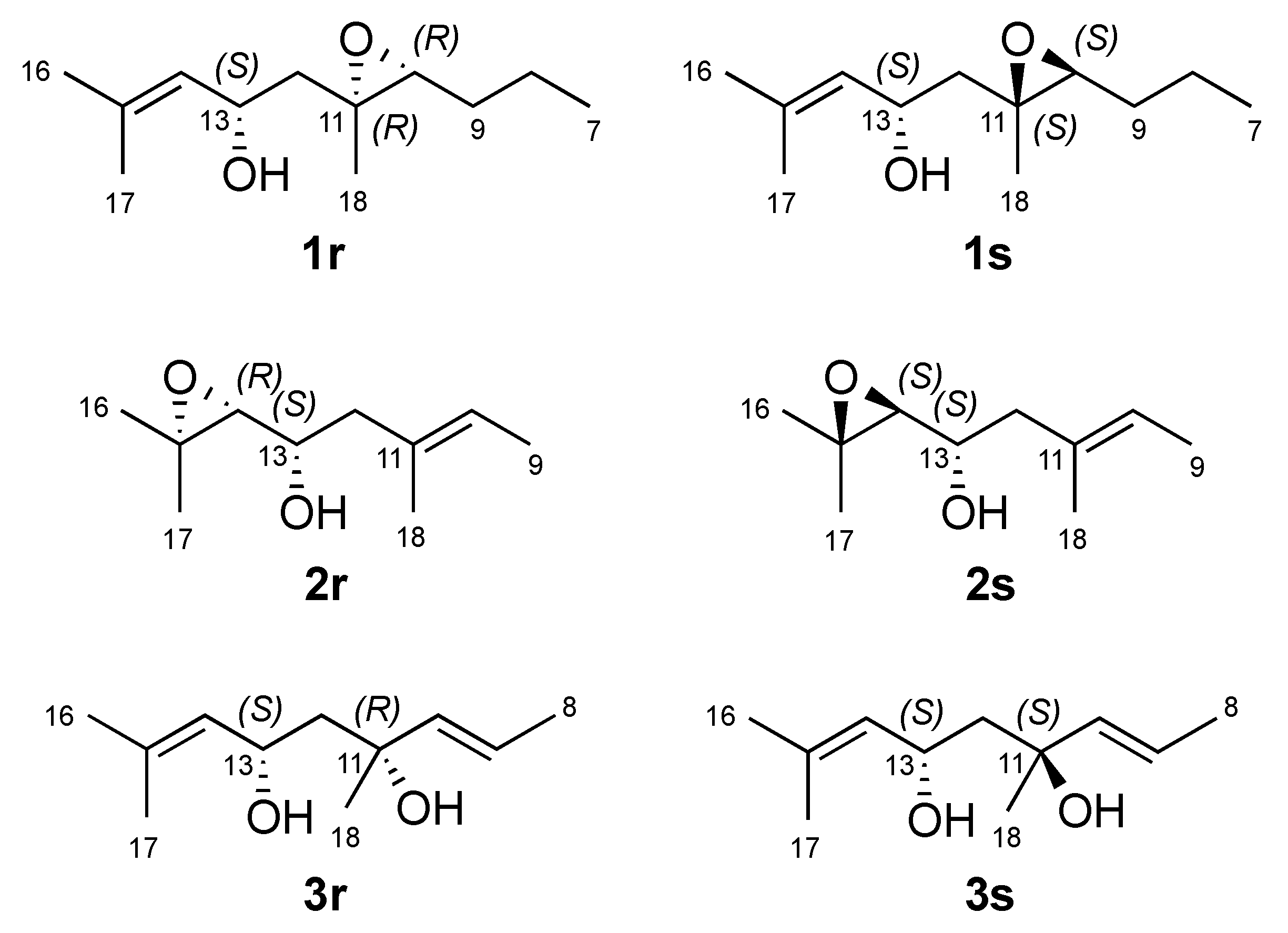
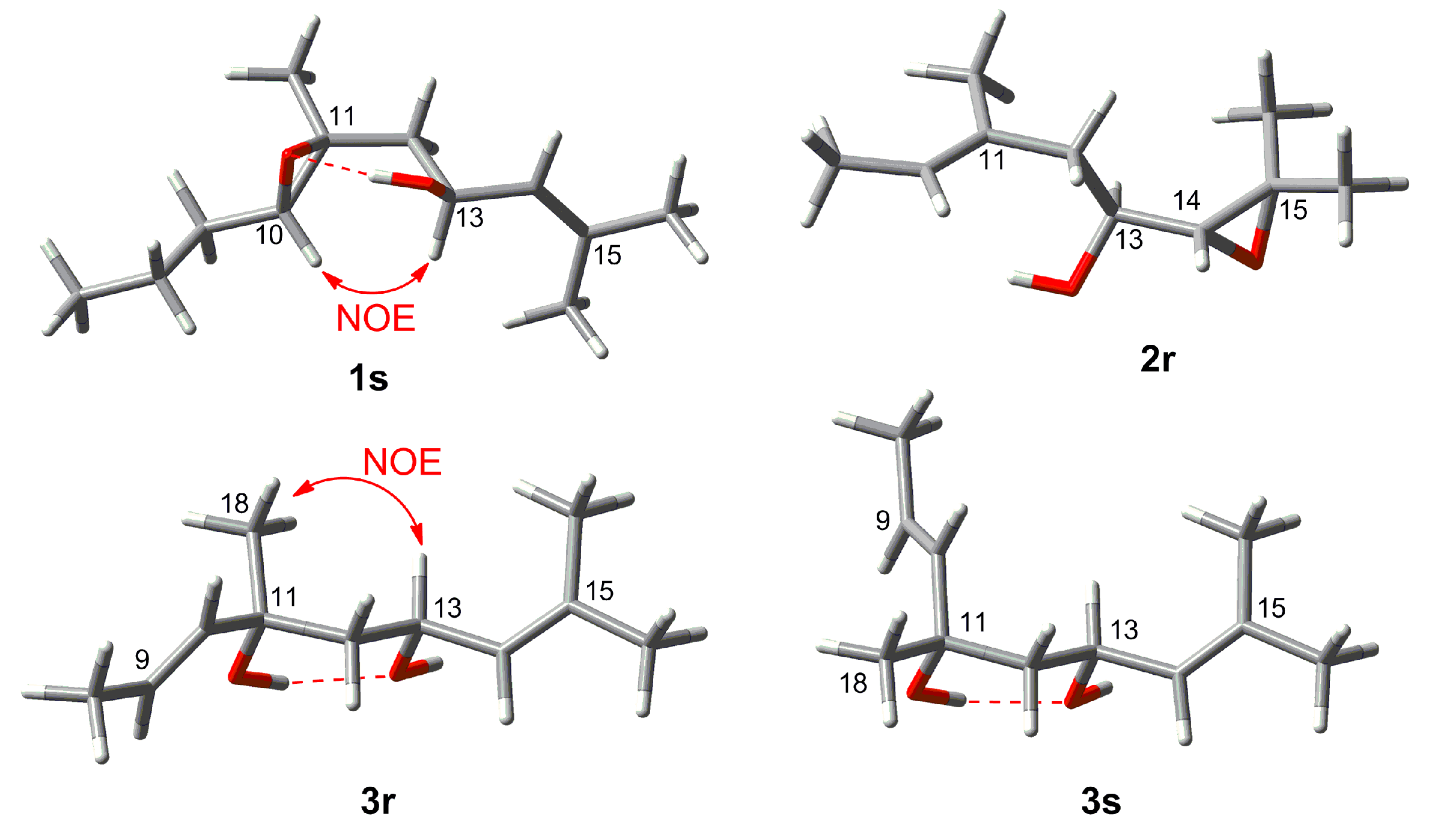
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Algal Material
3.3. Extraction and Isolation
3.4. Computational Studies
3.5. Anticancer Activity Assessments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Seaweed Site: Information on Marine Algae. Available online: http://www.seaweed.ie/descriptions/Bifurcaria_bifurcata.php (accessed on 30 October 2020).
- Pais, A.C.S.; Saraiva, J.A.; Rocha, S.M.; Silvestre, A.J.D.; Santos, S.A.O. Current research on the bioprospection of linear diterpenes from Bifurcaria bifurcata: From extraction methodologies to possible applications. Mar. Drugs 2019, 17, 556. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.; Culioli, G.; Köck, M. Linear diterpenes from the marine brown alga Bifurcaria bifurcata: A chemical perspective. Phytochem. Rev. 2013, 12, 407–424. [Google Scholar] [CrossRef]
- Valls, R.; Piovetti, L.; Banaigs, B.; Archavlis, A.; Pellegrini, M. (S)-13-hydroxygeranylgeraniol-derived furanoditerpenes from Bifurcaria bifurcata. Phytochemistry 1995, 39, 145–149. [Google Scholar] [CrossRef]
- Culioli, G.; Mesguiche, V.; Piovetti, L.; Valls, R. Geranylgeraniol and geranylgeraniol-derived diterpenes from the brown alga Bifurcaria bifurcata (Cystoseiraceae). Biochem. Syst. Ecol. 1999, 27, 665–668. [Google Scholar] [CrossRef]
- Culioli, G.; Daoudi, M.; Mesguiche, V.; Valls, R.; Piovetti, L. Geranylgeraniol-derived diterpenoids from the brown alga Bifurcaria bifurcata. Phytochemistry 1999, 52, 1447–1454. [Google Scholar] [CrossRef]
- Santos, S.A.O.; Trindade, S.S.; Oliveira, C.S.D.; Parreira, P.; Rosa, D.; Duarte, M.F.; Ferreira, I.; Cruz, M.T.; Rego, A.M.; Abreu, M.H.; et al. Lipophilic fraction of cultivated Bifurcaria bifurcata R. Ross: Detailed composition and in vitro prospection of current challenging bioactive properties. Mar. Drugs 2017, 15, 340. [Google Scholar] [CrossRef]
- Silva, J.; Alves, C.; Freitas, R.; Martins, A.; Pinteus, S.; Ribeiro, J.; Gaspar, H.; Alfonso, A.; Pedrosa, R. Antioxidant and neuroprotective potential of the brown seaweed Bifurcaria bifurcata in an in vitro Parkinson’s disease model. Mar. Drugs 2019, 17, 85. [Google Scholar] [CrossRef]
- Valls, R.; Banaigs, B.; Piovetti, L.; Archavlis, A.; Artaud, J. Linear diterpene with antimitotic activity from the brown alga Bifurcaria bifurcata. Phytochemistry 1993, 34, 1585–1588. [Google Scholar] [CrossRef]
- Merten, C.; Smyrniotopoulos, V.; Tasdemir, D. Assignment of absolute configurations of highly flexible linear diterpenes from the brown alga Bifurcaria bifurcata by VCD spectroscopy. Chem. Commun. 2015, 51, 16217–16220. [Google Scholar] [CrossRef]
- Smyrniotopoulos, V.; Merten, C.; Kaiser, M.; Tasdemir, D. Bifurcatriol, a new antiprotozoal acyclic diterpene from the brown alga Bifurcaria bifurcata. Mar. Drugs 2017, 15, 245. [Google Scholar] [CrossRef]
- Smyrniotopoulos, V.; Merten, C.; Firsova, D.; Fearnhead, H.; Tasdemir, D. Oxygenated acyclic diterpenes with anticancer activity from the Irish brown seaweed Bifurcaria bifurcata. Mar. Drugs 2020, 18, 581. [Google Scholar] [CrossRef]
- Grauso, L.; Li, Y.; Scarpato, S.; Shulha, O.; Rárová, L.; Strnad, M.; Teta, R.; Mangoni, A.; Zidorn, C. Structure and conformation of zosteraphenols, tetracyclic diarylheptanoids from the seagrass Zostera marina: An NMR and DFT study. Org. Lett. 2020, 22, 78–82. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
- Vinale, F.; Nicoletti, R.; Borrelli, F.; Mangoni, A.; Parisi, O.A.; Marra, R.; Lombardi, N.; Lacatena, F.; Grauso, L.; Finizio, S.; et al. Co-culture of plant beneficial microbes as source of bioactive metabolites. Sci. Rep. 2017, 7, 14330. [Google Scholar] [CrossRef] [PubMed]
- Costantino, V.; Della Sala, G.; Saurav, K.; Teta, R.; Bar-Shalom, R.; Mangoni, A.; Steindler, L. Plakofuranolactone as a quorum quenching agent from the Indonesian sponge Plakortis cf. lita. Mar. Drugs 2017, 15, 59. [Google Scholar] [CrossRef]
- Fan, B.; Dewapriya, P.; Li, F.; Grauso, L.; Blümel, M.; Mangoni, A.; Tasdemir, D. Pyrenosetin D, a new pentacyclic decalinoyltetramic acid derivative from the algicolous fungus Pyrenochaetopsis sp. FVE-087. Mar. Drugs 2020, 18, 281. [Google Scholar] [CrossRef]
- Grauso, L.; Teta, R.; Esposito, G.; Menna, M.; Mangoni, A. Computational prediction of chiroptical properties in structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 1005–1030. [Google Scholar] [CrossRef]
- Göthel, Q.; Lichte, E.; Köck, M. Further eleganolone-derived diterpenes from the brown alga Bifurcaria bifurcata. Tetrahedron Lett. 2012, 53, 1873–1877. [Google Scholar] [CrossRef]
- Handley, J.T.; Blackman, A.J. Secondary metabolites from the marine alga Caulerpa brownii (Chlorophyta). Austr. J. Chem. 2005, 58, 39–46. [Google Scholar] [CrossRef]
- Bohlmann, F.; Abraham, W.R.; Robinson, H.; King, R.M. Heliangolides and other constituents from Bejaranoa semistriata. Phytochemistry 1981, 20, 1639–1642. [Google Scholar] [CrossRef]
- Kamo, T.; Sato, K.; Sen, K.; Shibata, H.; Hirota, M. Geranylgeraniol-type diterpenoids, boletinins A-J, from Boletinus cavipes as inhibitors of superoxide anion generation in macrophage cells. J. Nat. Prod. 2004, 67, 958–963. [Google Scholar] [CrossRef]
- Murata, M.; Nakai, Y.; Kawazu, K.; Ishizaka, M.; Kajiwara, H.; Abe, H.; Takeuchi, K.; Ichinose, Y.; Mitsuhara, I.; Mochizuki, A.; et al. Loliolide, a carotenoid metabolite, is a potential endogenous inducer of herbivore resistance. Plant Physiol. 2019, 179, 1822–1833. [Google Scholar] [CrossRef]
- Mori, K.; Ooi, T.; Hiraoka, M.; Oka, N.; Hamada, H.; Tamura, M.; Kusumi, T. Fucoxanthin and its metabolites in edible brown algae cultivated in deep seawater. Mar. Drugs 2004, 2, 63–72. [Google Scholar] [CrossRef]
- Englert, G.; Bjoernland, T.; Liaaen-Jensen, S. 1D and 2D NMR study of some allenic carotenoids of the fucoxanthin series. Magn. Res. Chem. 1990, 28, 519–528. [Google Scholar] [CrossRef]
- Zhang, R.; He, H.P.; Di, Y.T.; Li, S.L.; Zuo, G.Y.; Zhang, Y.; Hao, X.J. Chemical constituents from Aphanamixis grandifolia. Fitoterapia 2014, 92, 100–104. [Google Scholar] [CrossRef]
- Bhowmick, S.; Mazumdar, A.; Moulick, A.; Vojtech, A. Algal metabolites: An inevitable substitute for antibiotics. Biotech. Adv. 2020, 43, 107571. [Google Scholar] [CrossRef]
- Lefranc, F.; Koutsaviti, A.; Ioannou, E.; Kornienko, A.; Roussis, V.; Kiss, R.; Newman, D. Algae metabolites: From in vitro growth inhibitory effects to promising anticancer activity. Nat. Prod. Rep. 2019, 36, 810–841. [Google Scholar] [CrossRef]
- Lever, J.; Brkljava, R.; Kraft, G.; Urban, S. Natural products of marine macroalgae from South Eastern Australia, with emphasis on the Port Phillip Bay and heads regions of Victoria. Mar. Drugs 2020, 18, 142. [Google Scholar] [CrossRef]
- Culioli, G.; Ortalo-Magne, A.; Daoudi, M.; Thomas-Guyon, H.; Valls, R.; Piovetti, L. Trihydroxylated linear diterpenes from the brown alga Bifurcaria bifurcata. Phytochemistry 2004, 65, 2063–2069. [Google Scholar] [CrossRef]




{kind=link}
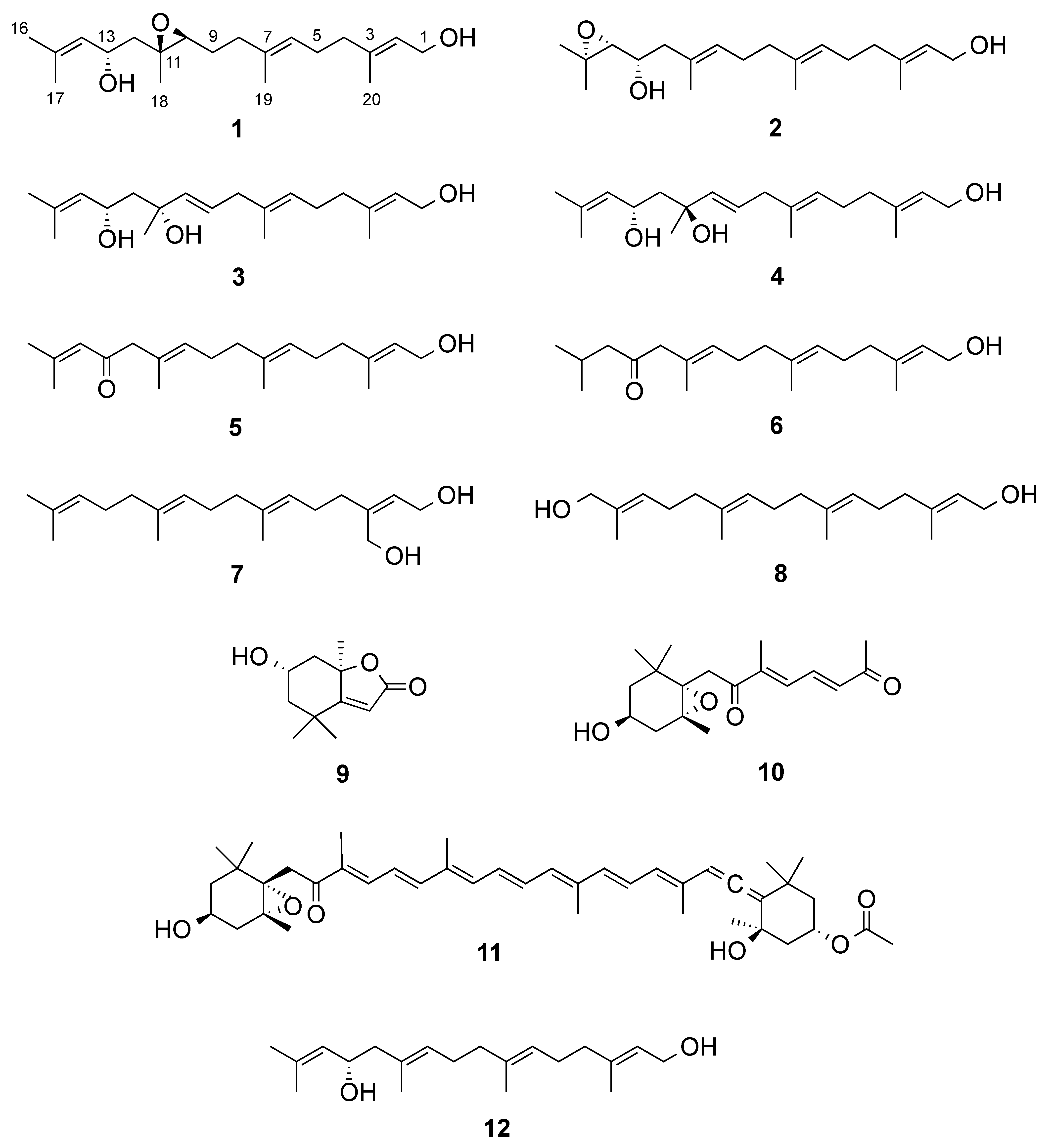{kind=link}
{kind=link}
{kind=link}
| C | 1 | 2 a | 3 b | 4 a | 4 b |
|---|---|---|---|---|---|
| δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | |
| 1 | 4.13, br. d (6.9) | 4.00, d (6.7) | 4.13, br. d (6.8) | 3.97, d (6.6) | 4.13, br. d (6.8) |
| 2 | 5.38, br. t (6.9) | 5.43, t (6.7) | 5.37, br. t (6.8) | 5.39, br. t (6.6) | 5.37, br. t (6.8) |
| 3 | - | - | - | - | - |
| 4 | 2.03, br. t (7.4) | 2.02, m | 2.03, m | 2.00, m | 2.03, m |
| 5 | 2.12, m | 2.12, m | 2.10, m | 2.13, m | 2.12, m |
| 6 | 5.16, m | 5.22, t (6.9) | 5.10, br. t (6.9) | 5.28, br. t (7.0) | 5.15, br. t (7.1) |
| 7 | - | - | - | - | - |
| 8 | 2.13, m | 2.03, m | 2.65, br. d (6.4) | 2.75, br. d (7.0) | 2.71 br. d (6.9) |
| 9 | 1.64, m | 2.13, m | 5.60, dt (15.6, 6.4) | 5.95, dt (15.3, 7.0) | 5.7, dt (15.4, 6.9) |
| 10 | 2.98, t (6.2) | 5.23, t (5.8) | 5.54, br. d (15.6) | 5.47, br. d (15.3) | 5.52, br. d (15.4) |
| 11 | - | - | - | - | - |
| 12 | 1.81, dd (14.6, 3.4) 1.74, dd (14.6, 9.1) | 2.23, dd (13.3, 7.7) 2.10 m | 1.78, dd (14.6, 10.0) 1.52, dd (14.6, 2.9) | 1.83, dd (14.4, 10.8) 1.39, dd (14.4, 1.9) | 1.79, dd (14.5, 10.5) 1.48, dd (14.5, 1.8) |
| 13 | 4.44 td (9.1, 3.4) | 3.51, ddd (7.7, 7.7, 5.6) | 4.74, ddd (10.0, 8.6, 2.9) | 4.70, ddd (10.8, 8.5, 1.9) | 4.62 ddd (10.5, 8.6, 1.8) |
| 14 | 5.15, m | 2.66, d (7.7) | 5.19, dhept (8.6, 1.2) | 5.25, dhept (8.5, 1.2) | 5.18, dhept (8.6, 1.2) |
| 15 | - | - | - | - | - |
| 16 | 1.70, br. s | 1.09, s | 1.69, d (1.2) | 1.56, d (1.2) | 1.68, d (1.2) |
| 17 | 1.66, br. s | 1.07, s | 1.67, d (1.2) | 1.52, d (1.2) | 1.61, d (1.2) |
| 18 | 1.30, s | 1.52, br. s | 1.37, s | 1.26, s | 1.25, s |
| 19 | 1.61, br. s | 1.56, br. s | 1.56, br. s | 1.60, br. s | 1.59, br. s |
| 20 | 1.65, br. s | 1.48, br. s | 1.65, br. s | 1.45, br. s | 1.55, br. s |
| C | 1 | 2 a | 3 b | 4 a |
|---|---|---|---|---|
| δC, type | δC, type | δC, type | δC, type | |
| 1 | 59.4, CH2 | 59.3, CH2 | 59.4, CH2 | 59.5, CH2 |
| 2 | 123.6, CH | 125.1, CH | 123.9, CH | 125.4, CH |
| 3 | 139.3, C | 137.8, C | 139.0, C | 137.3, C |
| 4 | 39.3, CH2 | 39.7, CH2 | 39.2, CH2 | 39.5, CH2 |
| 5 | 26.1, CH2 | 26.5, CH2 | 25.9, CH2 | 26.2, CH2 |
| 6 | 124.7, CH | 124.8, CH | 124.8, CH | 124.8, CH |
| 7 | 134.3, C | 134.9, C | 134.0, C | 134.6, C |
| 8 | 36.2, CH2 | 39.8, CH2 | 42.2, CH2 | 42.8, CH2 |
| 9 | 26.9, CH2 | 26.7, CH2 | 125.7, CH | 126.9, CH |
| 10 | 62.1, CH | 128.1, CH | 138.9, CH | 138.2, CH |
| 11 | 60.4, C | 131.1, C | 73.0, C | 73.6, C |
| 12 | 44.1, CH2 | 44.8, CH2 | 47.6, CH2 | 47.8, CH2 |
| 13 | 65.5, CH | 69.2, CH | 66.3, CH | 67.5, CH |
| 14 | 127.4, CH | 67.5, CH | 127.8, CH | 129.5, CH |
| 15 | 134.7, C | 58.3, C | 134.6, C | 132.7, C |
| 16 | 25.7, CH3 | 24.9, CH3 | 25.7, CH3 | 25.6, CH3 |
| 17 | 18.2, CH3 | 19.4, CH3 | 18.2, CH3 | 18.1, CH3 |
| 18 | 18.3, CH3 | 16.6, CH3 | 27.0, CH3 | 31.0, CH3 |
| 19 | 16.0, CH3 | 16.0, CH3 | 16.2, CH3 | 16.7, CH3 |
| 20 | 16.2, CH3 | 16.1, CH3 | 16.1, CH3 | 16.2, CH3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smyrniotopoulos, V.; Firsova, D.; Fearnhead, H.; Grauso, L.; Mangoni, A.; Tasdemir, D. Density Functional Theory (DFT)-Aided Structure Elucidation of Linear Diterpenes from the Irish Brown Seaweed Bifurcaria bifurcata. Mar. Drugs 2021, 19, 42. https://doi.org/10.3390/md19010042
Smyrniotopoulos V, Firsova D, Fearnhead H, Grauso L, Mangoni A, Tasdemir D. Density Functional Theory (DFT)-Aided Structure Elucidation of Linear Diterpenes from the Irish Brown Seaweed Bifurcaria bifurcata. Marine Drugs. 2021; 19(1):42. https://doi.org/10.3390/md19010042
Chicago/Turabian StyleSmyrniotopoulos, Vangelis, Daria Firsova, Howard Fearnhead, Laura Grauso, Alfonso Mangoni, and Deniz Tasdemir. 2021. "Density Functional Theory (DFT)-Aided Structure Elucidation of Linear Diterpenes from the Irish Brown Seaweed Bifurcaria bifurcata" Marine Drugs 19, no. 1: 42. https://doi.org/10.3390/md19010042
APA StyleSmyrniotopoulos, V., Firsova, D., Fearnhead, H., Grauso, L., Mangoni, A., & Tasdemir, D. (2021). Density Functional Theory (DFT)-Aided Structure Elucidation of Linear Diterpenes from the Irish Brown Seaweed Bifurcaria bifurcata. Marine Drugs, 19(1), 42. https://doi.org/10.3390/md19010042