Protective Effect of Astaxanthin on Blue Light Light-Emitting Diode-Induced Retinal Cell Damage via Free Radical Scavenging and Activation of PI3K/Akt/Nrf2 Pathway in 661W Cell Model

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Astaxanthin Prevented Blue Light LED-Induced 661W Cell Death
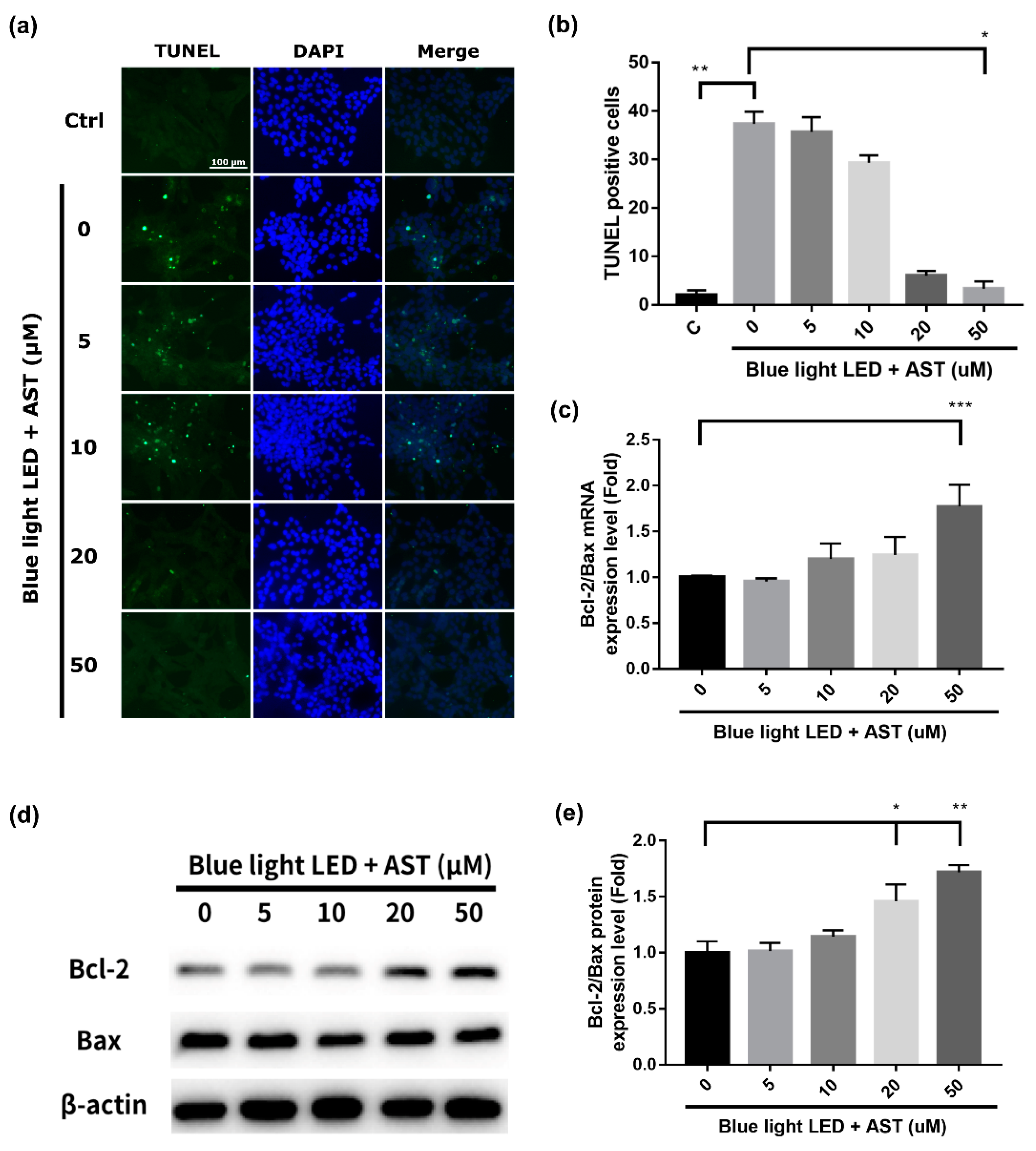
2.2. Astaxanthin Inhibited Blue Light LED-Induced 661W Cell Apoptosis
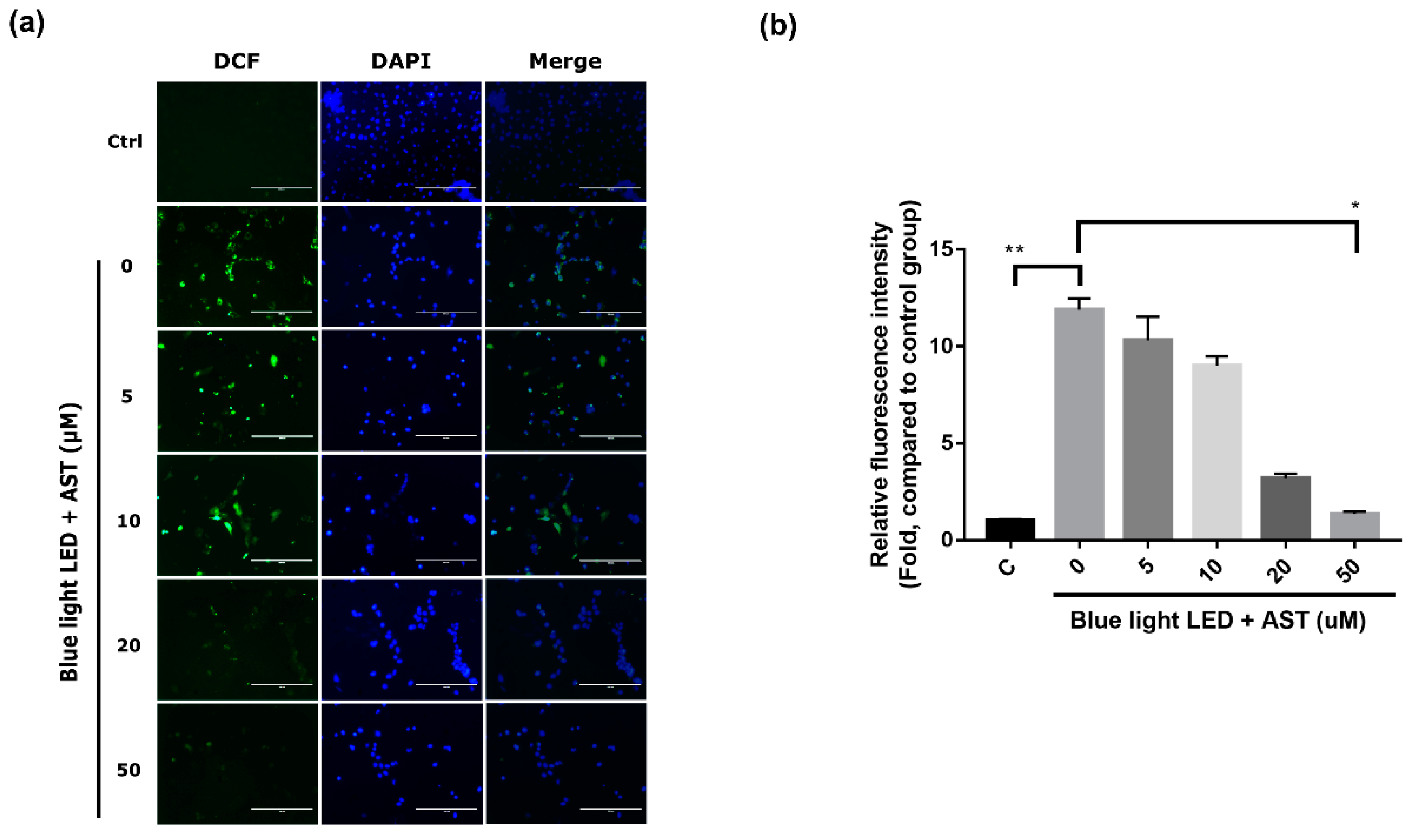
2.3. Astaxanthin Inhibited ROS Production in 661W Cells
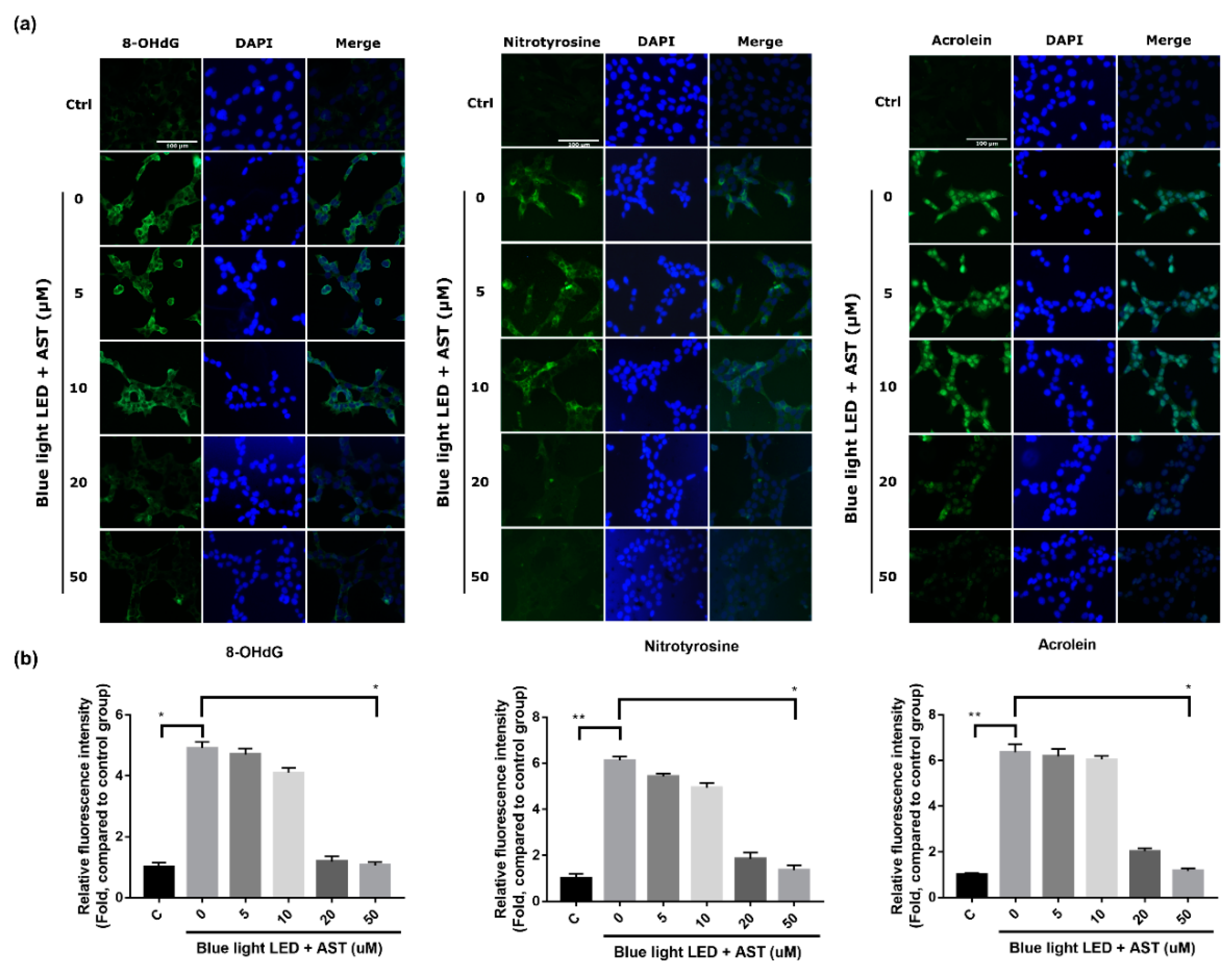
2.4. Astaxanthin Inhibited Oxidative Stress Biomarkers in 661W Cells
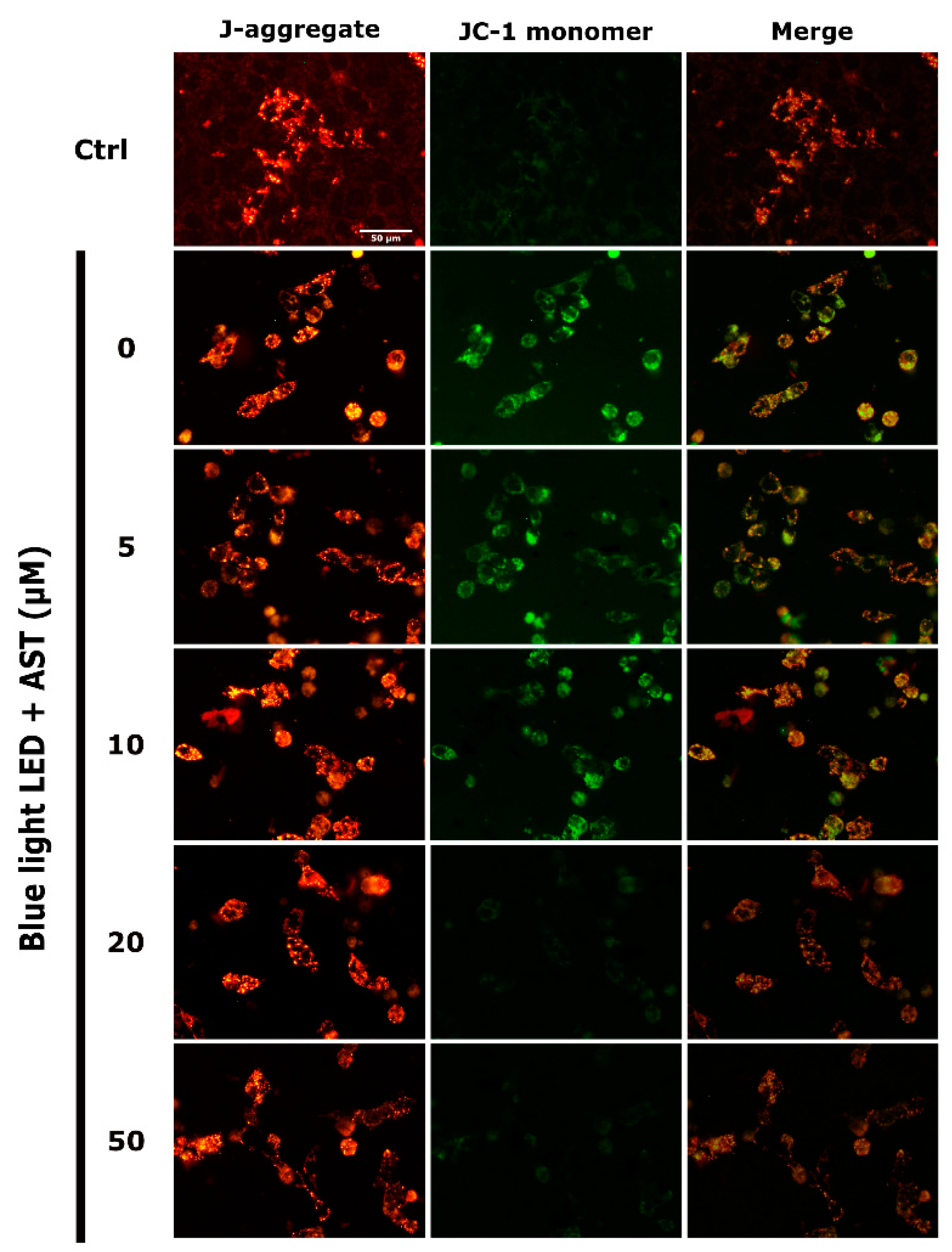
2.5. Astaxanthin Suppressed Blue Light LED-Induced Mitochondrial Damage in 661W Cells
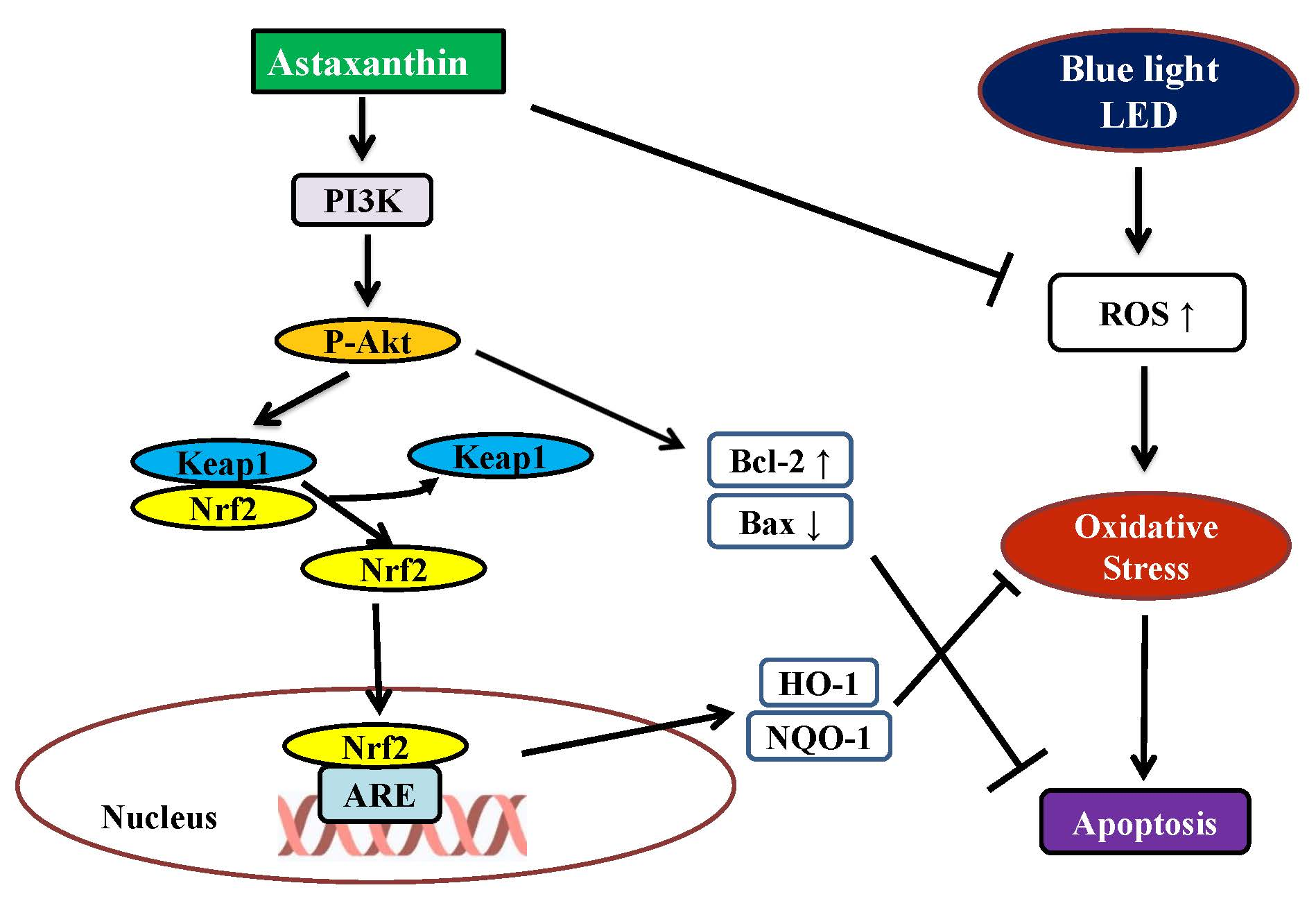
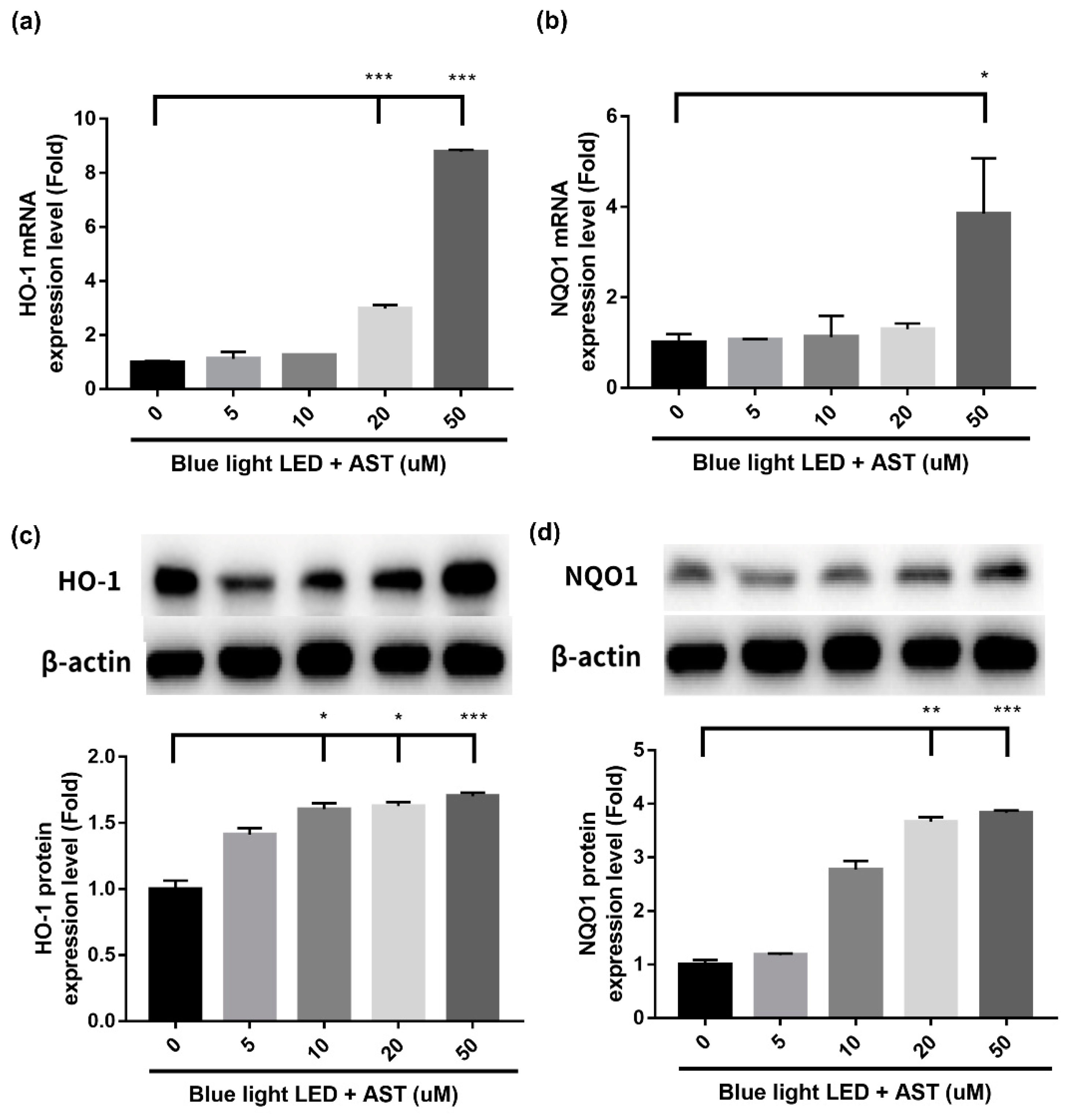
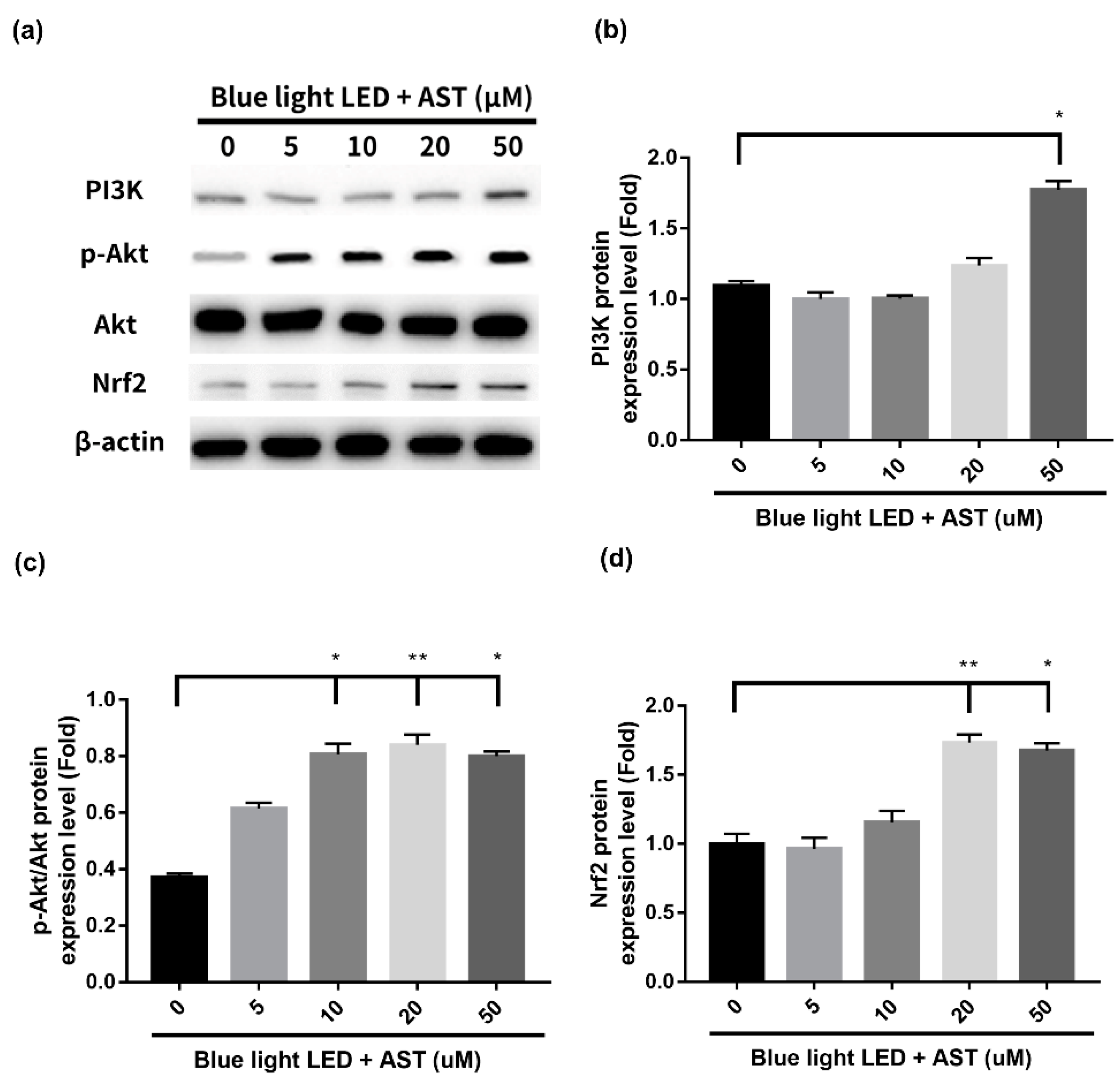
2.6. Astaxanthin Induced Phase II Antioxidant Enzyme Expression and Activated PI3K/Akt/Nrf2 Pathway in 661W Cells
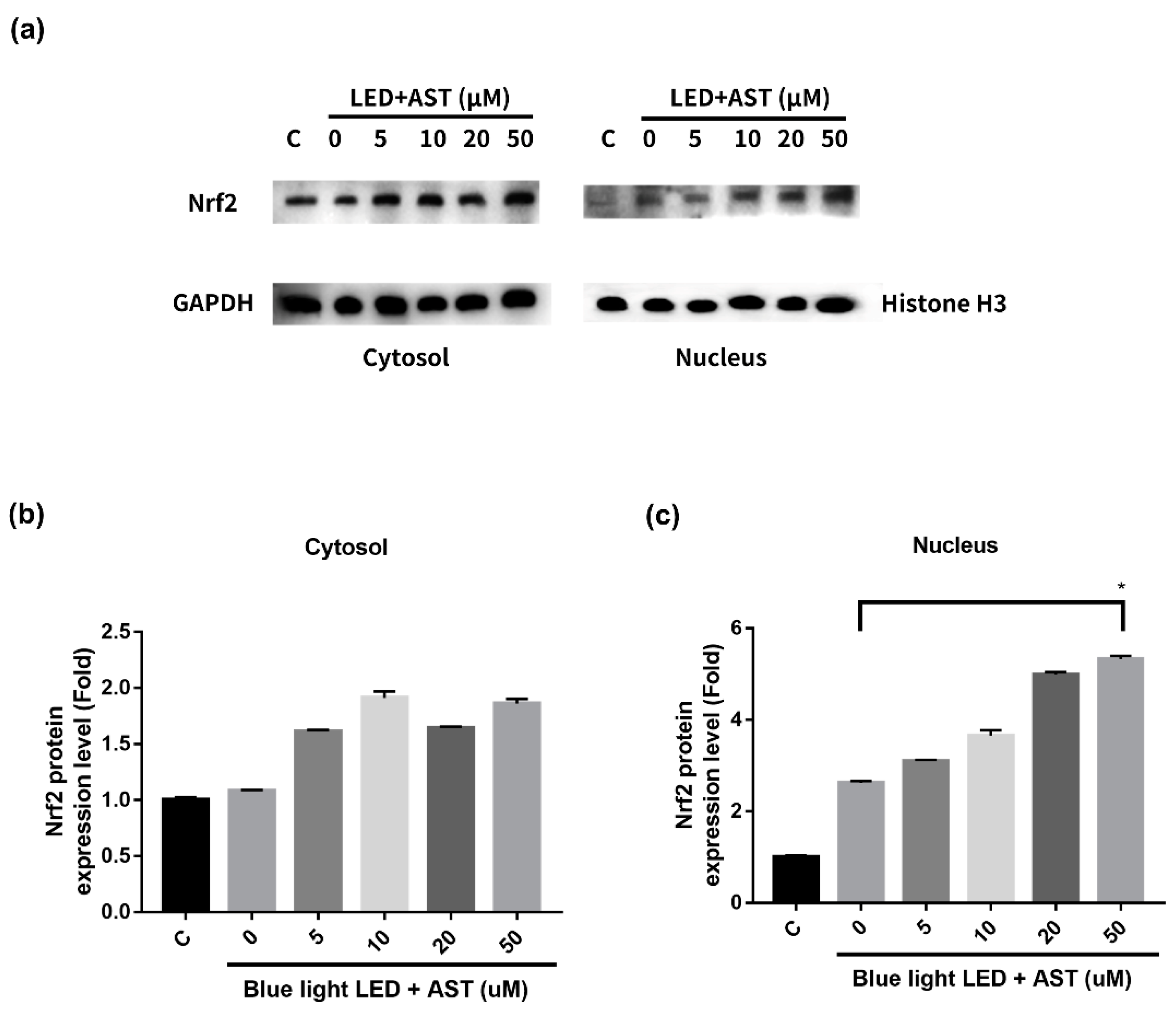
2.7. Astaxanthin-Induced Nuclear Translocation of Nrf2 in 661W Cells
3. Discussion
4. Materials and Methods
4.1. 661W Cell Culture
4.2. Blue Light LED and Astaxanthin Treatment of 661W Cells
4.3. CCK-8 Assay for Cell Viability
4.4. TUNEL Assay
4.5. Detection of Intracellular ROS
4.6. Immunofluorescence (IF) Detection of Oxidative Stress Biomarkers
4.7. Determination of Mitochondrial Dysfunction
4.8. RNA Extraction and Quantitative Reverse Transcription Polymerase Chain Reaction
4.9. Western Blot Analysis
4.10. Nuclear and Cytosolic Protein Extraction and Analysis of Nrf2 Protein Expression
4.11. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chamorro, E.; Bonnin-Arias, C.; Pérez-Carrasco, M.J.; Luna, J.M.; Vázquez, D.; Sánchez-Ramose, C. Effects of light-emitting diode radiations on human retinal pigment epithelial cells in vitro. Photochem. Photobiol. 2013, 89, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Parver, L.M.; Auker, C.R.; Fine, B.S. Observations on monkey eyes exposed to light from an operating microscope. Ophthalmology 1983, 90, 964–972. [Google Scholar] [CrossRef]
- Jaadane, I.; Boulenguez, P.; Chahory, S.; Carre, S.; Savoldelli, M.; Jonet, L.; Behar-Cohen, F.; Martinsons, C.; Torriglia, A. Retinal damage induced by commercial light emitting diodes (LEDs). Free Radic. Biol. Med. 2015, 84, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.M.; Wang, G.S.; Sliney, D.; Yang, C.H.; Lee, L.L. White light-emitting diodes (LEDs) at domestic lighting levels and retinal injury in a rat model. Environ. Health Perspect. 2014, 122, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Seregard, S.; Algvere, P.V. Photochemical damage of the retina. Surv. Ophthalmol. 2006, 51, 461–481. [Google Scholar] [CrossRef]
- Organisciak, D.T.; Vaughan, D.K. Retinal light damage: Mechanisms and protection. Prog. Retin. Eye Res. 2010, 29, 113–134. [Google Scholar] [CrossRef]
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med. 2008, 14, 194–198. [Google Scholar] [CrossRef]
- Nakamura, M.; Kuse, Y.; Tsuruma, K.; Shimazawa, M.; Hara, H. The involvement of the oxidative stress in murine blue LED light-induced retinal damage model. Biol. Pharm. Bull. 2017, 40, 1219–1225. [Google Scholar] [CrossRef]
- Friedman, D.S.; O’Colmain, B.J.; Muñoz, B.; Tomany, S.C.; McCarty, C.; de Jong, P.T.; Nemesure, B.; Mitchell, P.; Kempen, J. Prevalence of age-related macular degeneration in the United States. Arch. Ophthalmol. 2004, 122, 564–572. [Google Scholar] [CrossRef]
- Bloch, S.B.; Larsen, M.; Munch, I.C. Incidence of legal blindness from age-related macular degeneration in Denmark: Year 2000 to 2010. Am. J. Ophthalmol. 2012, 153, 209–213.e2. [Google Scholar] [CrossRef]
- Gao, X.; Talalay, P. Induction of phase 2 genes by sulforaphane protects retinal pigment epithelial cells against photooxidative damage. Proc. Natl. Acad. Sci. USA 2004, 102, 10446–10451. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Huang, H.H.; Yang, C.M.; Yang, C.H. Protective effect of chitosan oligosaccharides on blue light light-emitting diode induced retinal pigment epithelial cell damage. J. Funct. Foods 2018, 49, 12–19. [Google Scholar] [CrossRef]
- Ogawa, K.; Kuse, Y.; Tsuruma, K.; Kobayashi, S.; Shimazawa, M.; Hara, H. Protective effects of bilberry and lingonberry extracts against blue light-emitting diode light-induced retinal photoreceptor cell damage in vitro. BMC Complement. Altern. Med. 2014, 2, 120. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Koo, S. Efficient Syntheses of the Keto-carotenoids Canthaxanthin, Astaxanthin, and Astacene. J. Org. Chem. 2005, 70, 3328–3331. [Google Scholar] [CrossRef]
- Jackson, H.; Braun, C.L.; Ernst, H. The chemistry of novel xanthophyll carotenoids. Am. J. Cardiol. 2008, 101, 50D–57D. [Google Scholar] [CrossRef]
- Kurashige, M.; Okimasu, E.; Inoue, M.; Utsumi, K. Inhibition of oxidative injury of biological membranes by astaxanthin. Physiol. Chem. Phys. Med. NMR 1990, 22, 27–38. [Google Scholar]
- Ohgami, K.; Shiratori, K.; Kotake, S.; Nishida, T.; Mizuki, N.; Yazawa, K.; Ohno, S. Effects of astaxanthin on lipopolysaccharide-induced inflammation in vitro and in vivo. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2694–2701. [Google Scholar] [CrossRef]
- Spiller, G.A.; Dewell, A. Safety of an astaxanthin-rich Haematococcus pluvialis algal extract: A randomized clinical trial. J. Med. Food. 2003, 6, 51–56. [Google Scholar] [CrossRef]
- Stewart, J.S.; Lignell, A.; Pettersson, A.; Elfving, E.; Soni, M.G. Safety assessment of astaxanthin-rich microalgae biomass: Acute and subchronic toxicity studies in rats. Food Chem. Toxicol. 2008, 46, 3030–3036. [Google Scholar] [CrossRef]
- Inoue, Y.; Shimazawa, M.; Nagano, R.; Kuse, Y.; Takahashi, K.; Tsuruma, K.; Hayashi, M.; Ishibashi, T.; Maoka, T.; Hara, H. Astaxanthin analogs, adonixanthin and lycopene, activate Nrf2 to prevent light-induced photoreceptor degeneration. J. Pharmacol. Sci. 2017, 134, 147–157. [Google Scholar] [CrossRef]
- Opatrilova, R.; Kubatka, P.; Caprnda, M.; Büsselberg, D.; Krasnik, V.; Vesely, P.; Saxena, S.; Ruia, S.; Mozos, I.; Rodrigo, L.; et al. Nitric oxide in the pathophysiology of retinopathy: Evidences from preclinical and clinical researches. Acta Ophthalmol. 2018, 96, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, T.; Shimazawa, M.; Inoue, Y.; Nakano, Y.; Ojino, K.; Izawa, H.; Tsuruma, K.; Ishibashi, T.; Hara, H. Astaxanthin protects against retinal damage: Evidence from in vivo and in vitro retinal ischemia and reperfusion models. Curr. Eye Res. 2016, 41, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Baccouche, B.; Benlarbi, M.; Barber, A.J.; Ben Chaouacha-Chekir, R. Short-Term Administration of astaxanthin attenuates retinal changes in diet-induced diabetic Psammomys obesus. Curr. Eye Res. 2018, 43, 1177–1189. [Google Scholar] [CrossRef]
- Benlarbi-Ben Khedher, M.; Hajri, K.; Dellaa, A.; Baccouche, B.; Hammoum, I.; Boudhrioua-Mihoubi, N.; Dhifi, W.; Ben Chaouacha-Chekir, R. Astaxanthin inhibits aldose reductase activity in Psammomys obesus, a model of type 2 diabetes and diabetic retinopathy. Food Sci. Nutr. 2019, 7, 3979–3985. [Google Scholar] [CrossRef] [PubMed]
- Giannaccare, G.; Pellegrini, M.; Senni, C.; Bernabei, F.; Scorcia, V.; Cicero, A.F.G. Clinical applications of astaxanthin in the treatment of ocular diseases: Emerging insights. Mar. Drugs 2020, 18, 239. [Google Scholar] [CrossRef]
- Landon, R.; Gueguen, V.; Petite, H.; Letourneur, D.; Pavon-Djavid, G.; Anagnostou, F. Impact of astaxanthin on diabetes pathogenesis and chronic complications. Mar. Drugs 2020, 18, 357. [Google Scholar] [CrossRef]
- Shang, Y.M.; Wang, G.S.; Sliney, D.; Yang, C.H.; Lee, L.L. Light-emitting-diode induced retinal damage and its wavelength dependency in vivo. Int. J. Ophthalmol. 2017, 10, 191–202. [Google Scholar] [CrossRef]
- Cutler, R.G. Oxidative stress profiling: Part I. Its potential importance in the optimization of human health. Ann. N. Y. Acad. Sci. 2005, 1055, 93–135. [Google Scholar] [CrossRef]
- Godley, B.F.; Shamsi, F.A.; Liang, F.Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J. Biol. Chem. 2005, 280, 21061–21066. [Google Scholar] [CrossRef]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef]
- Park, S.H.; Jang, J.H.; Chen, C.Y.; Na, H.K.; Surh, Y.J. A formulated red ginseng extract rescues PC12 cells from PCB-induced oxidative cell death through Nrf2-mediated upregulation of heme oxygenase-1 and glutamate cysteine ligase. Toxicology 2010, 278, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.N.; Chen, Y.; Cai, J.; Sternberg, P., Jr. Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: Implication for protection against oxidative stress. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2709–2715. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, X.; Liu, H.; Chen, X.; Shi, H.; Fan, Y.; Hou, D.; Zhang, X. Astaxanthin protects ARPE-19 cells from oxidative stress via upregulation of Nrf2-regulated phase II enzymes through activation of PI3K/Akt. Mol. Vis. 2013, 19, 1656–1666. [Google Scholar]
- Zhao, C.R.; Gao, Z.H.; Qu, X.J. NRF2-ARE signaling pathway and natural products for cancer chemoprevention. Cancer Epidemiol. 2010, 34, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, D.N.; Jena, G.B. Astaxanthin intervention ameliorates cyclophosphamide-induced oxidative stress, DNA damage and early hepatocarcinogenesis in rat: Role of Nrf2, p53, p38 and phase-II enzymes. Mutat. Res. 2010, 696, 69–80. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, X.S.; Wang, H.D.; Zhang, X.; Yu, Q.; Li, W.; Zhou, M.L.; Wang, X.L. Astaxanthin activates nuclear factor erythroid-related factor 2 and the antioxidant responsive element (Nrf2-ARE) pathway in the brain after subarachnoid hemorrhage in rats and attenuates early brain injury. Mar. Drugs 2014, 12, 6125–6141. [Google Scholar] [CrossRef]
- Wang, H.Q.; Sun, X.B.; Xu, Y.X.; Zhao, H.; Zhu, Q.Y.; Zhu, C.Q. Astaxanthin upregulates heme oxygenase-1 expression through ERK1/2 pathway and its protective effect against beta-amyloid-induced cytotoxicity in SH-SY5Y cells. Brain Res. 2010, 1360, 159–167. [Google Scholar] [CrossRef]
- Wu, H.; Niu, H.; Shao, A.; Wu, C.; Dixon, B.J.; Zhang, J.; Yang, S.; Wang, Y. Astaxanthin as a Potential Neuroprotective Agent for Neurological Diseases. Mar. Drugs 2015, 13, 5750–5766. [Google Scholar] [CrossRef]
- Kim, J.H.; Nam, S.W.; Kim, B.W.; Choi, W.; Lee, J.H.; Kim, W.J.; Choi, Y.H. Astaxanthin Improves Stem Cell Potency via an Increase in the Proliferation of Neural Progenitor Cells. Int. J. Mol. Sci. 2010, 11, 5109–5119. [Google Scholar] [CrossRef]
- Kim, Y.H.; Koh, H.K.; Kim, D.S. Down-regulation of IL-6 production by astaxanthin via ERK-, MSK-, and NF-κB-mediated signals in activated microglia. Int. Immunopharmacol. 2010, 10, 1560–1572. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Sternberg, P.; Cai, J. Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Higuera-Ciapara, I.; Felix-Valenzuela, L.; Goycoolea, F.M. Astaxanthin: A review of its chemistry and applications. Crit. Rev. Food Sci. Nutr. 2006, 46, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health promoting effects of astaxanthin: A high-value carotenoid mostly from microalgae. Mol. Nutr. Food Res. 2011, 55, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Guerin, M.; Huntley, M.E.; Olaizola, M. Haematococcus astaxanthin: Applications for human health and nutrition. Trends Biotechnol. 2003, 21, 210–216. [Google Scholar] [CrossRef]
- Fassett, R.G.; Coombes, J.S. Astaxanthin: A potential therapeutic agent in cardiovascular disease. Mar. Drugs 2011, 9, 447–465. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Inokuchi, Y.; Shimazawa, M.; Otsubo, K.; Ishibashi, T.; Hara, H. Astaxanthin, a dietary carotenoid, protects retinal cells against oxidative stress in-vitro and in mice in-vivo. J. Pharm. Pharmacol. 2008, 60, 1365–1374. [Google Scholar] [CrossRef]
- Tan, E.; Ding, X.Q.; Saadi, A.; Agarwal, N.; Naash, M.I.; Al-Ubaidi, M.R. Expression of cone-photoreceptor-specific antigens in a cell line derived from retinal tumors in transgenic mice. Investig. Ophthalmol. Vis. Sci. 2004, 45, 764–768. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-W.; Yang, C.-M.; Yang, C.-H. Protective Effect of Astaxanthin on Blue Light Light-Emitting Diode-Induced Retinal Cell Damage via Free Radical Scavenging and Activation of PI3K/Akt/Nrf2 Pathway in 661W Cell Model. Mar. Drugs 2020, 18, 387. https://doi.org/10.3390/md18080387
Lin C-W, Yang C-M, Yang C-H. Protective Effect of Astaxanthin on Blue Light Light-Emitting Diode-Induced Retinal Cell Damage via Free Radical Scavenging and Activation of PI3K/Akt/Nrf2 Pathway in 661W Cell Model. Marine Drugs. 2020; 18(8):387. https://doi.org/10.3390/md18080387
Chicago/Turabian StyleLin, Chao-Wen, Chung-May Yang, and Chang-Hao Yang. 2020. "Protective Effect of Astaxanthin on Blue Light Light-Emitting Diode-Induced Retinal Cell Damage via Free Radical Scavenging and Activation of PI3K/Akt/Nrf2 Pathway in 661W Cell Model" Marine Drugs 18, no. 8: 387. https://doi.org/10.3390/md18080387
APA StyleLin, C.-W., Yang, C.-M., & Yang, C.-H. (2020). Protective Effect of Astaxanthin on Blue Light Light-Emitting Diode-Induced Retinal Cell Damage via Free Radical Scavenging and Activation of PI3K/Akt/Nrf2 Pathway in 661W Cell Model. Marine Drugs, 18(8), 387. https://doi.org/10.3390/md18080387