Efficient Preparation of Bafilomycin A1 from Marine Streptomyces lohii Fermentation Using Three-Phase Extraction and High-Speed Counter-Current Chromatography

 ,
,
Abstract
1. Introduction
2. Results
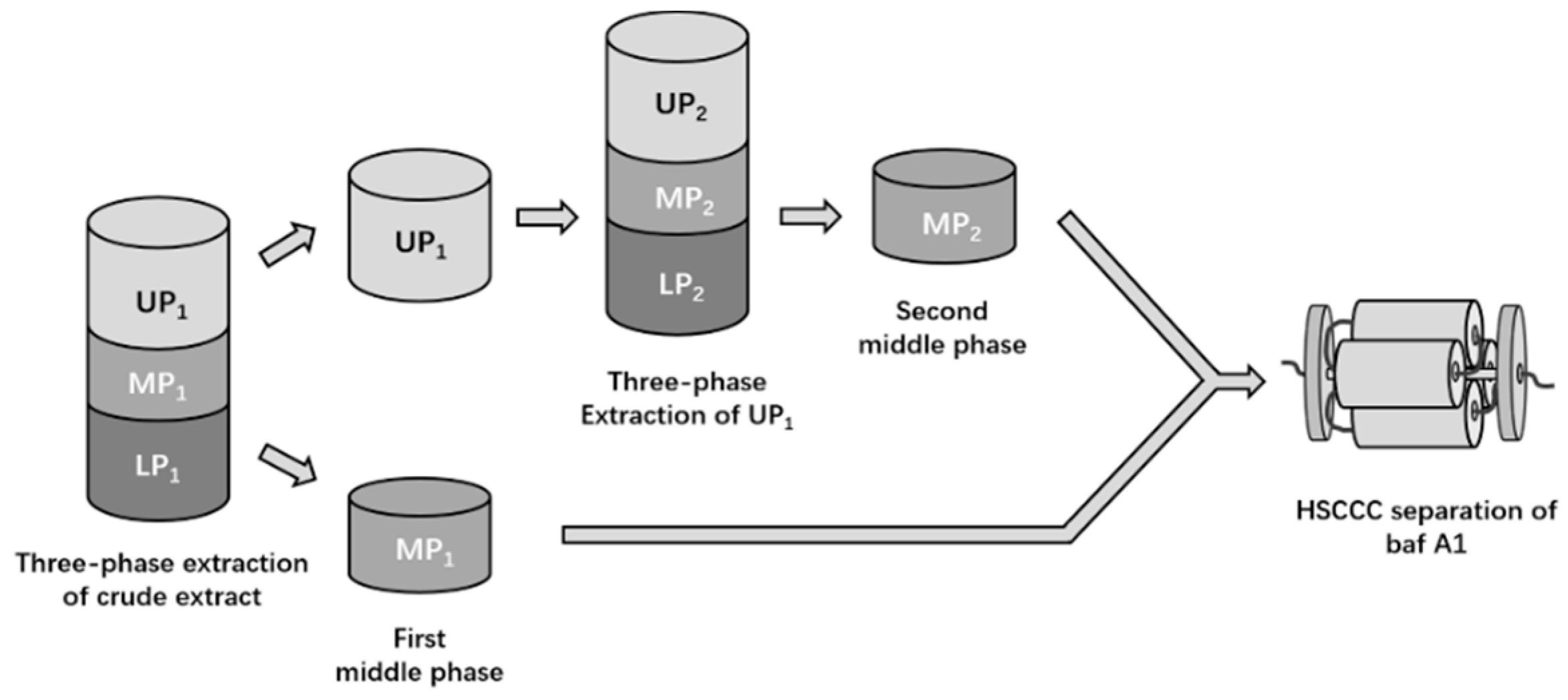
2.1. Selection of a Three-Phase Solvent System for Extraction
2.2. Determination of Partition Coefficient Values (KD) and HSCCC Separation
2.3. Structural Identification of baf A1 after HSCCC
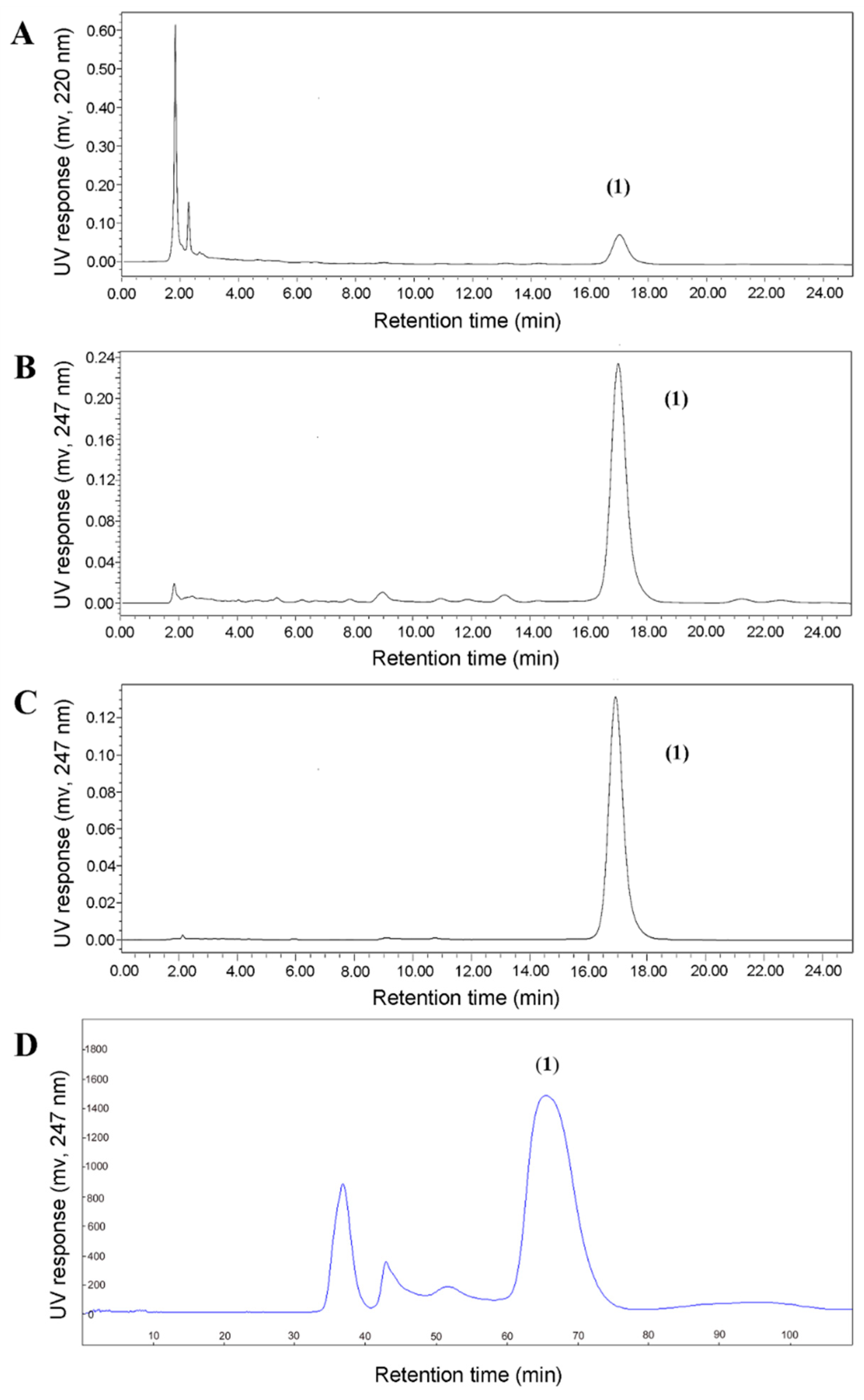
2.4. Comparison of HSCCC and Semi-Preparative HPLC for the Preparative Separation of baf A1
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Crude Extract
4.3. Apparatus
4.4. Selection of Three-Phase Solvent System for Extraction
4.5. Preparation of Enriched Example
4.6. Selection of Partition Coefficient Values (KD)
4.7. Preparation of the Two-Phase Solvent System and Sample Solution
4.8. HSCCC Separation
4.9. HPLC Analysis, Semi-Preparative HPLC Purification of baf A1 in Fractions from HSCCC Separation, and Compound Identification by MS and NMR
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Werner, G.; Hagenmaier, H.; Albert, K.; Kohlshorn, H. The structure of the bafilomycins, a new group of macrolide antibiotics. Tetrahedron Lett. 1983, 24, 5193–5196. [Google Scholar] [CrossRef]
- Werner, G.; Hagenmaier, H.; Drautz, H.; Baumgartner, A.; Zähner, H. Metabolic products of microorganisms. 224. Bafilomycins, a new group of macrolide antibiotics. Production, isolation, chemical structure and biological activity. J. Antibiot. 1984, 37, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Bowman, E.J.; Siebers, A.; Altendorf, K. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 7972–7976. [Google Scholar] [CrossRef] [PubMed]
- Dröse, S.; Altendorf, K. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J. Exp. Biol. 1997, 200, 1–8. [Google Scholar] [PubMed]
- Pérez-Sayáns, M.; Somoza-Martín, J.M.; Barros-Angueira, F.; Rey, J.M.; García-García, A. V-ATPase inhibitors and implication in cancer treatment. Cancer Treat. Rev. 2009, 35, 707–713. [Google Scholar] [CrossRef]
- Mauvezin, C.; Nagy, G.; Juhász, P.; Neufeld, T.P. Autophagosome–lysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun. 2015, 6, 7007. [Google Scholar] [CrossRef]
- Scheidt, K.A.; Bannister, T.D.; Tasaka, A.; Wendt, M.D.; Savall, B.M.; Fegley, G.J.; Roush, W.R. Total Synthesis of (-)-Bafilomycin A 1. J. Am. Chem. Soc. 2002, 124, 6981–6990. [Google Scholar] [CrossRef]
- Toshima, K.; Jyojima, T.; Yamaguchi, H.; Noguchi, Y.; Yoshida, T.; Murase, H.; Nakata, M.; Matsumura, S. Total synthesis of Bafilomycin A(1). J. Org. Chem. 1997, 62, 3271–3284. [Google Scholar] [CrossRef]
- Gagliardi, S.; Gatti, P.A.; Belfiore, P.; Zocchetti, A.; Clarke, G.D.; Farina, C. Synthesis and structure-activity relationships of bafilomycin A1 derivatives as inhibitors of vacuolar H+-ATPase. J. Med. Chem. 1998, 41, 1883–1893. [Google Scholar] [CrossRef]
- Li, Z.; Du, L.; Zhang, W.; Zhang, X.W.; Jiang, Y.Y.; Liu, K.; Men, P.; Xu, H.F.; Fortman, J.L.; Sherman, D.H.; et al. Complete elucidation of the late steps of bafilomycin biosynthesis in Streptomyces lohii. J. Biol. Chem. 2017, 292, 7095–7104. [Google Scholar] [CrossRef]
- Castro, L.D.; Priego-Capote, F. Ultrasound assistance to liquid-liquid extraction: A debatable analytical tool. Anal. Chim. Acta 2007, 583, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Greer, D.; Pfahl, L.; Rieck, J.; Daniels, T.; Garza, O. Comparison of a novel distillation method versus a traditional distillation method in a model gin system using liquid/liquid extraction. J. Agric. Food Chem. 2008, 56, 9030–9036. [Google Scholar] [CrossRef] [PubMed]
- Shibusawa, Y.; Yamakawa, Y.; Noji, R.; Yanagida, A.; Shindo, H.; Ito, Y. Three-phase solvent systems for comprehensive separation of a wide variety of compounds by high-speed counter-current chromatography. J. Chromatogr. A 2006, 1133, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhang, S.; Long, L.J.; Yin, H.; Tian, X.P.; Luo, X.M.; Nan, H.H.; He, S. The separation of flavonoids from Pongamia pinnata using combination columns in high-speed counter-current chromatography with a three-phase solvent system. J. Chromatogr. A 2013, 1315, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Chao, Z.M.; Wang, C.; Yu, L. Separation of chemical constituents from three plant medicines by counter-current chromatography using a three-phase solvent system at a novel ratio. J. Chromatogr. A 2015, 1384, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.L.; Zhu, X.L.; Zhang, F.; Du, Y.R.; Ma, J.B.; Jiang, R.W. An efficient strategy based on liquid-liquid extraction with three-phase solvent system and high- speed counter-current chromatography for rapid enrichment and separation of epimers of minor bufadienolide from toad meat. J. Agric. Food Chem. 2018, 66, 1008–1014. [Google Scholar] [CrossRef]
- Liu, Y.J.; Chen, X.F.; Liu, J.X.; Di, D.L. Three-phase solvent systems for the comprehensive separation of a wide variety of compounds from Dicranostigma leptopodum by high-speed counter-current chromatography. J. Sep. Sci. 2015, 38, 2038–2045. [Google Scholar] [CrossRef]
- Hamzaoui, M.; Renault, J.H.; Nuzillard, J.M.; Reynaud, R.; Hubert, J. Stepwise elution of a three-phase solvent system in centrifugal partition extraction: A new strategy for the fractionation and phytochemical screening of a crude bark extract. Phytochem. Anal. 2013, 24, 367–373. [Google Scholar] [CrossRef]
- Wang, F.Z.; Li, R.; Long, L.J.; Tian, X.P.; Xiao, Z.H.; Zhang, S.; Yin, H. A three-phase solvent system in high-speed counter-current chromatographic for the separation and purification of bioactive constituents from Acanthus ilicifolius. Chromatographia 2015, 78, 1401–1407. [Google Scholar] [CrossRef]
- Ito, Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography. J. Chromatogr. A 2005, 1065, 145–168. [Google Scholar] [CrossRef]
- OuYang, X.K.; Jin, M.C.; He, C.H. Preparative separation of four major alkaloids from medicinal plant of Tripterygium Wilfordii Hook F using high-speed counter-current chromatography. Sep. Purif. Technol. 2007, 56, 319–324. [Google Scholar] [CrossRef]
- He, S.; Wang, H.Q.; Yan, X.J.; Zhu, P.; Chen, J.J.; Yang, R. Preparative isolation and purification of macrolactin antibiotics from marine bacterium Bacillus amyloliquefaciens using high-speed counter-current chromatography in stepwise elution mode. J. Chromatogr. A 2013, 1272, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.J.; He, S.; Yan, X.J. Efficient preparation of pseudoalteromone A from marine Pseudoalteromonas rubra QD1-2 by combination of response surface methodology and high-speed counter-current chromatography: A comparison with high-performance liquid chromatography. Appl. Microbiol. Biot. 2014, 98, 4369–4377. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.B.; Zhang, Y.Y.; Ding, L.J.; He, S.; Wu, B.; Dong, J.D.; Zhu, P.; Chen, J.J.; Zhang, J.R.; Yan, X.J. Preparative separation of sulfur-containing diketopiperazines from marine fungus Cladosporium sp. using high-speed counter-current chromatography in stepwise elution mode. Mar. Drugs 2015, 13, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.J.; Luo, Q.J.; Ding, L.J.; Fang, F.; Yuan, Y.; Chen, J.J.; Zhang, J.R.; Jin, H.X.; He, S. Preparative isolation and purification of lignans from Justicia procumbens using high-speed counter-current chromatography in stepwise elution mode. Molecules 2015, 20, 7048–7058. [Google Scholar] [CrossRef]
- Baker, G.H.; Brown, P.J.; Dorgan, R.J.J.; Everett, J.R.; Ley, S.V.; Slawin, A.M.Z.; Williams, D.J. A conformational study of bafilomycin A1 by X-ray crystallography and nmr techniques. Tetrahedron Lett. 1987, 28, 5565–5568. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Volume Ratio | Phase Formation | Phase Ratio (%) | Baf A1 Distribution (UP/MP/LP %) | System Settling Time (Second) |
|---|---|---|---|---|
| 3:7:5:5 | 2 | 68.6/31.4 | / | / |
| 5:5:5:5 | 2 | 67.6/32.4 | / | / |
| 6:4:5:5 | 3 | 31.4/34.3/34.3 | 15.1/84.9/0 | >180 |
| 7:3:5:5 | 3 | 38.9/25/36.1 | 15.6/84.4/0 | 50 |
| 8:2:5:5 | 3 | 45.7/17.1/37.2 | 15.9/82.5/1.56 | <20 |
| 9:1:5:5 | 2 | 47.2/52.8 | / | / |
| Solvent Systems | UP (g) | MP (g) | Control (g) 1 | Total Soybean Oil Elimination (%) |
|---|---|---|---|---|
| 6:4:5:5 | 4.41 | 0.11 | 4.67 | 94.43% |
| 7:3:5:5 | 3.94 | 0.02 | 4.10 | 96.09% |
| 8:2:5:5 | 4.42 | 0.02 | 4.52 | 97.78% |
| Volume Ratio | KD (MP Extract) | Volume Ratio (UP/LP) |
|---|---|---|
| 15:6:6 | 0.06 | 1.2/1 |
| 15:8:8 | 0.08 | 1/1 |
| 15:10:10 | 0.12 | 1/1.3 |
| 15:9:11 | 0.28 | 1.3/1 |
| 15:8:12 | 0.84 | 1.1/1 |
| 15:7:13 | 1.74 | 1/1.3 |
| 15:6:14 | 2.45 | 1/1.4 |
| HSCCC | HPLC | |
|---|---|---|
| Stationary phase | Upper phase n-hexane–acetonitrile–water (15:8:12, v/v/v) | YMC-Pack C18 column (250 × 10 mm ID, 5 μm) |
| Mobile phase | Lower phase | Acetonitrile–water (80:20, v/v) |
| Sample capacity per run (mg) | 480 | 5 |
| Run time (min) | 90 | 30 |
| Productivity (mg/min) | 0.81 | 0.03 |
| Purity of isolated compound | >95% | 98% |
| Organic solvent consumption (L/g) | 2.29 | 68.57 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; He, X.; Wang, T.; Zhang, X.; Li, Z.; Xu, X.; Zhang, W.; Yan, X.; Li, S.; He, S. Efficient Preparation of Bafilomycin A1 from Marine Streptomyces lohii Fermentation Using Three-Phase Extraction and High-Speed Counter-Current Chromatography. Mar. Drugs 2020, 18, 332. https://doi.org/10.3390/md18060332
Yuan Y, He X, Wang T, Zhang X, Li Z, Xu X, Zhang W, Yan X, Li S, He S. Efficient Preparation of Bafilomycin A1 from Marine Streptomyces lohii Fermentation Using Three-Phase Extraction and High-Speed Counter-Current Chromatography. Marine Drugs. 2020; 18(6):332. https://doi.org/10.3390/md18060332
Chicago/Turabian StyleYuan, Ye, Xiaoping He, Tingting Wang, Xingwang Zhang, Zhong Li, Xiaoqing Xu, Weiyan Zhang, Xiaojun Yan, Shengying Li, and Shan He. 2020. "Efficient Preparation of Bafilomycin A1 from Marine Streptomyces lohii Fermentation Using Three-Phase Extraction and High-Speed Counter-Current Chromatography" Marine Drugs 18, no. 6: 332. https://doi.org/10.3390/md18060332
APA StyleYuan, Y., He, X., Wang, T., Zhang, X., Li, Z., Xu, X., Zhang, W., Yan, X., Li, S., & He, S. (2020). Efficient Preparation of Bafilomycin A1 from Marine Streptomyces lohii Fermentation Using Three-Phase Extraction and High-Speed Counter-Current Chromatography. Marine Drugs, 18(6), 332. https://doi.org/10.3390/md18060332