Highly Substituted Phenol Derivatives with Nitric Oxide Inhibitory Activities from the Deep-Sea-Derived Fungus Trichobotrys effuse FS524

 and
and
Abstract
1. Introduction
2. Results and Discussion
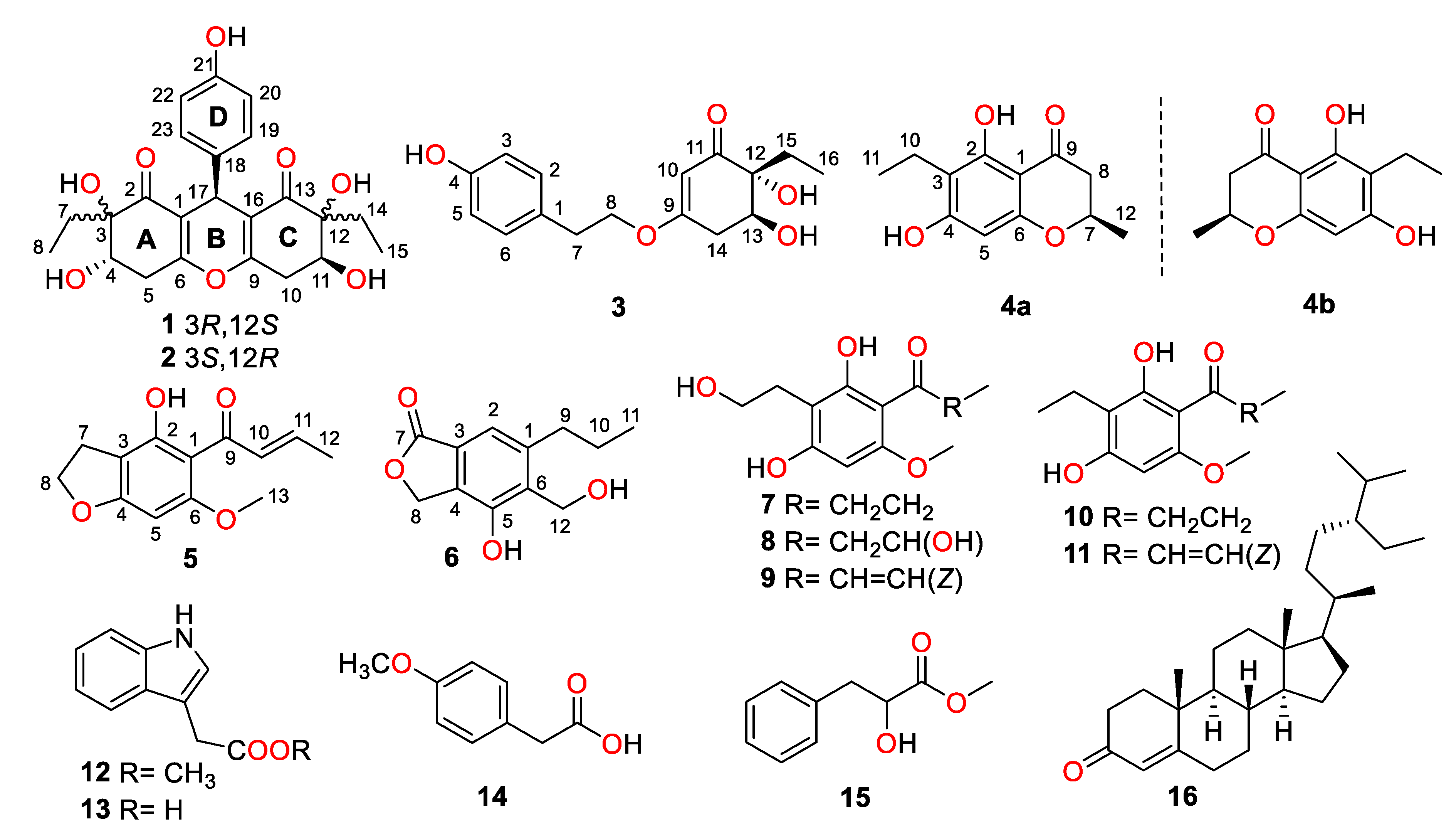
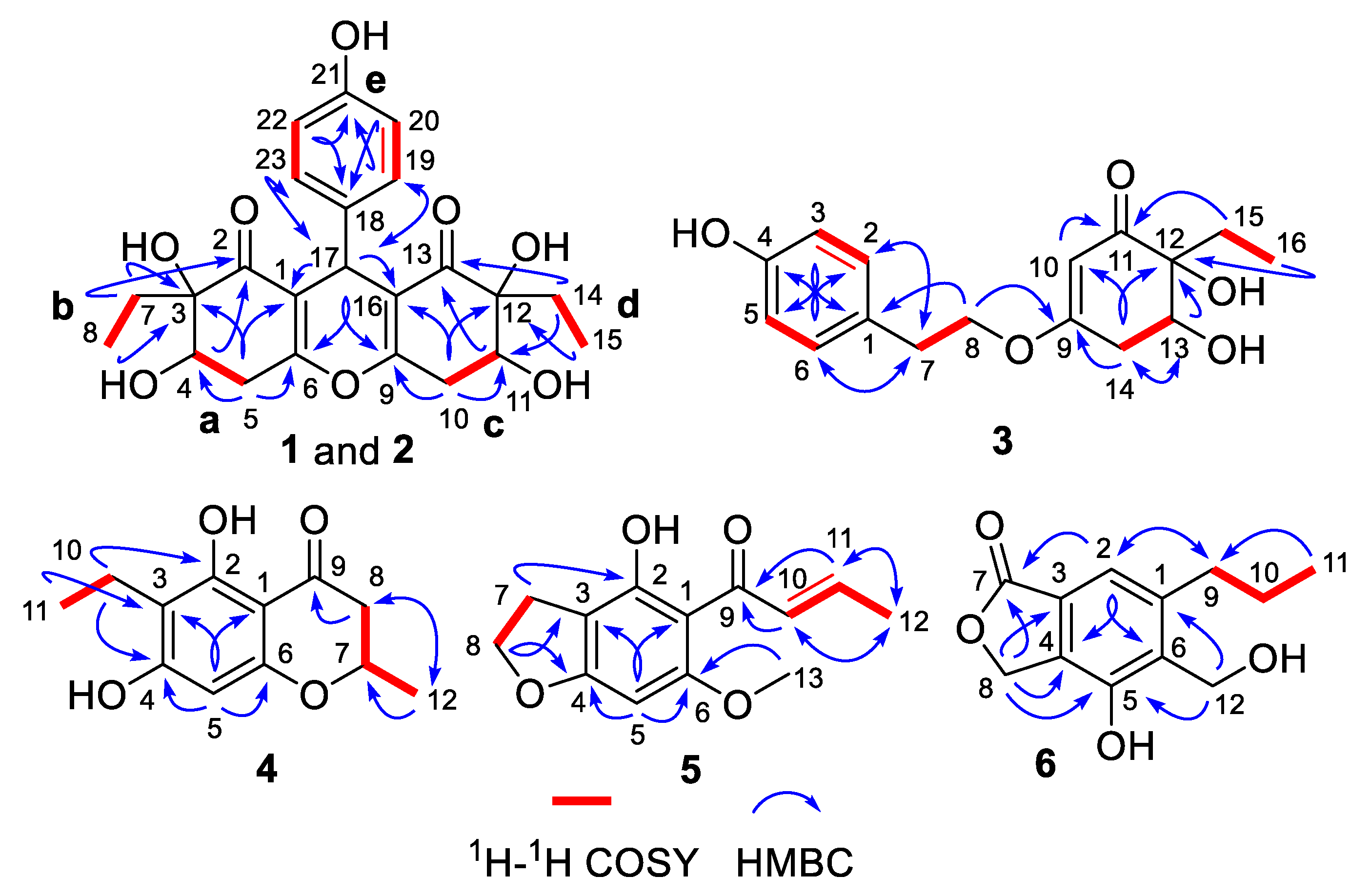
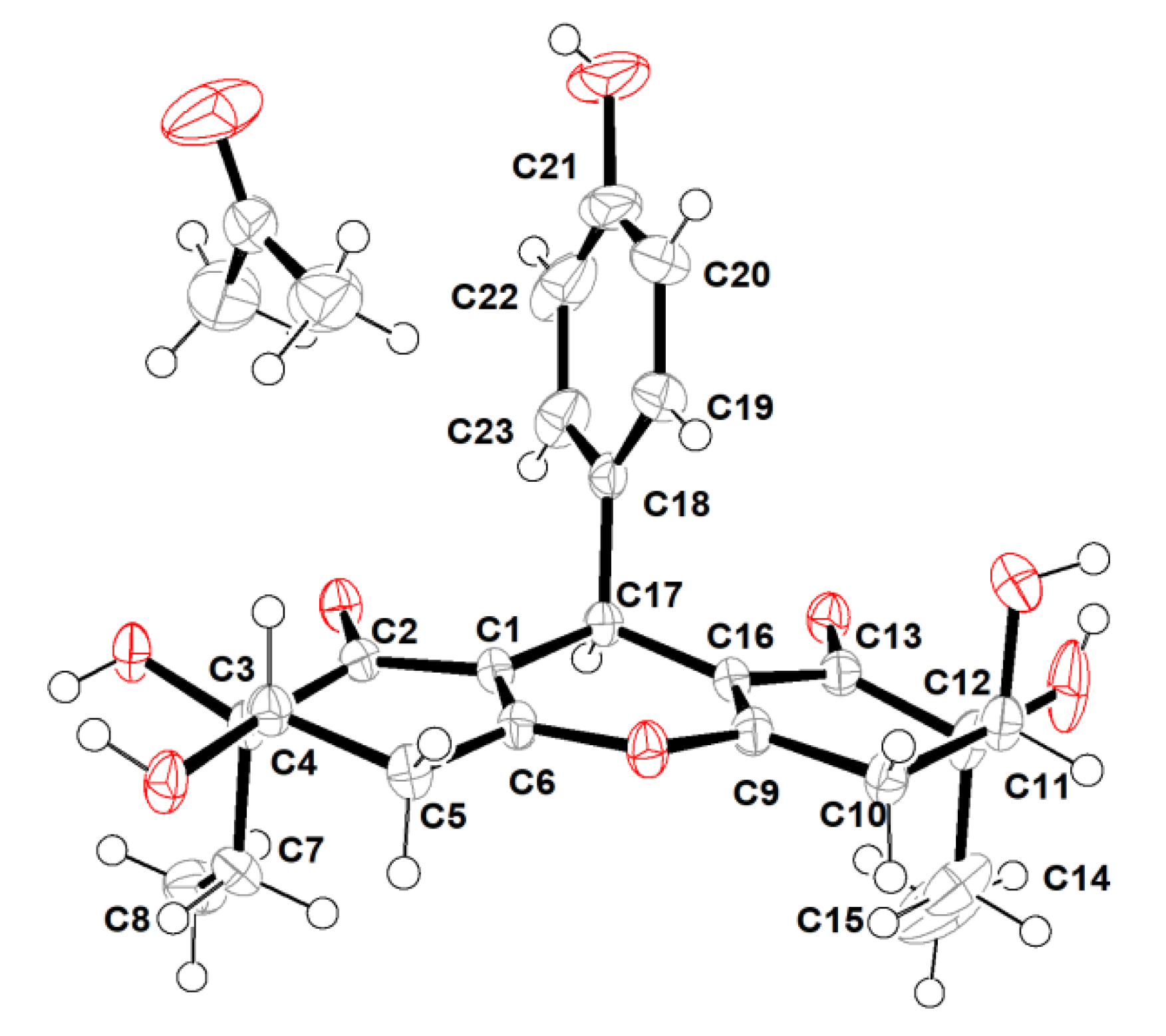
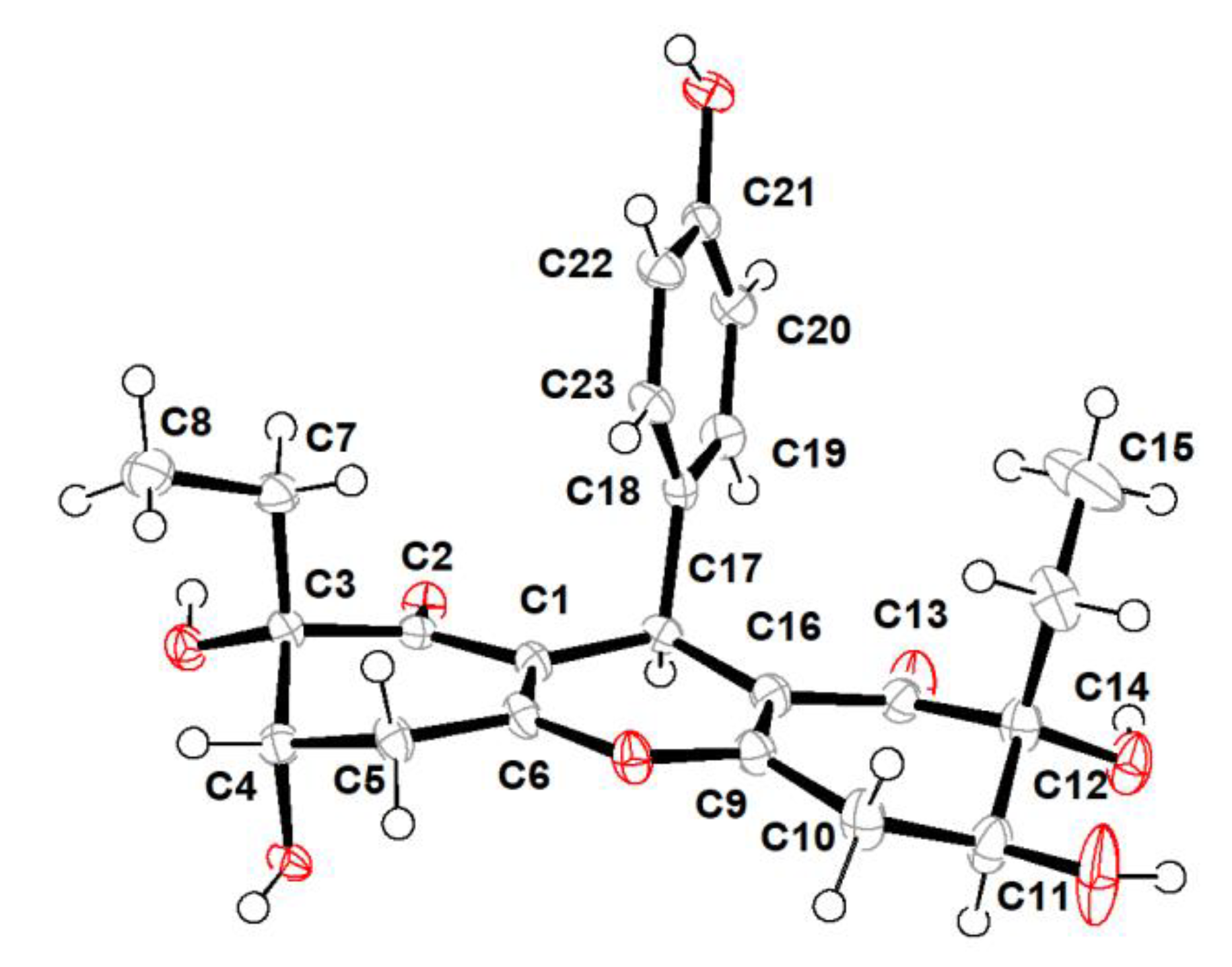
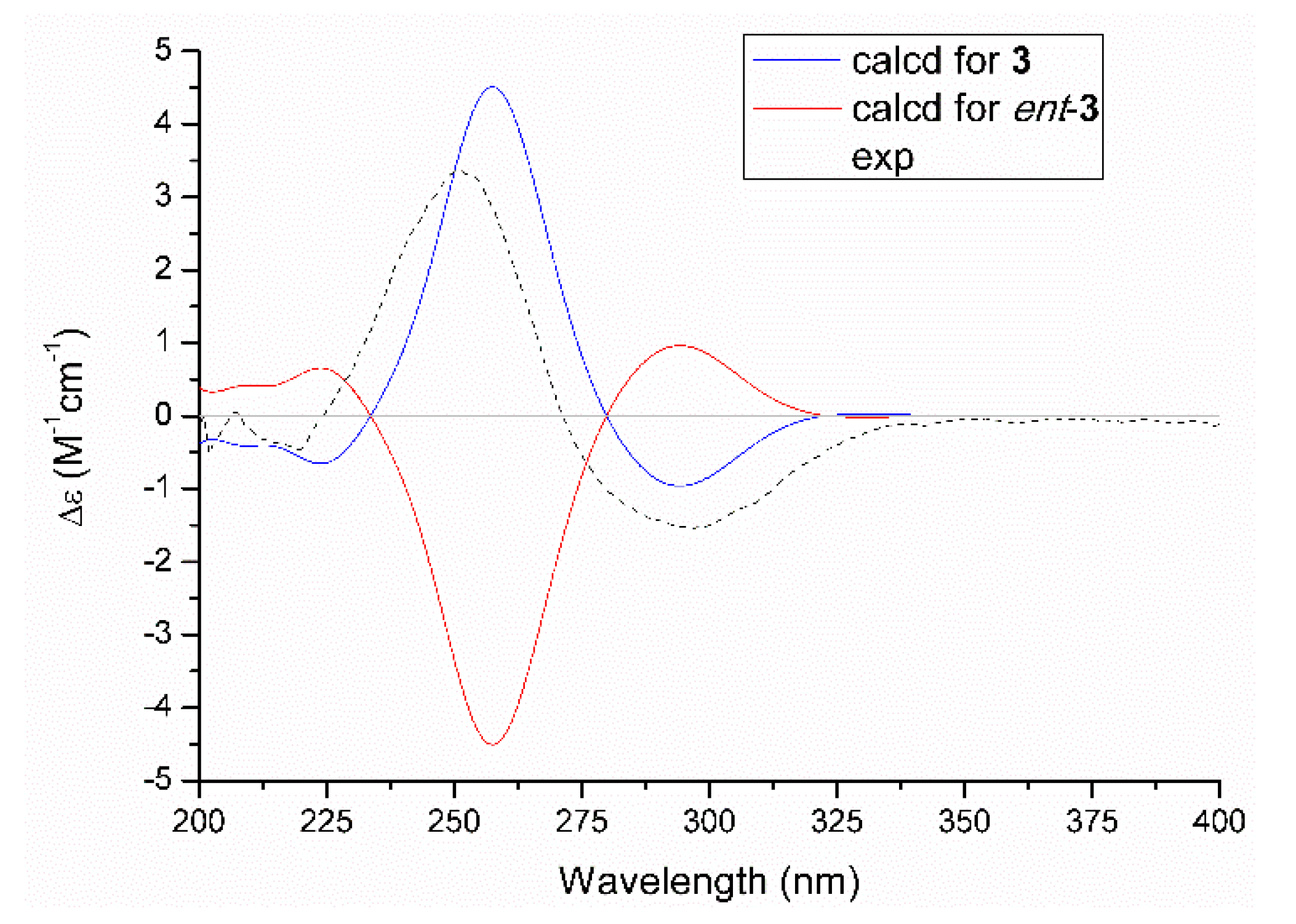
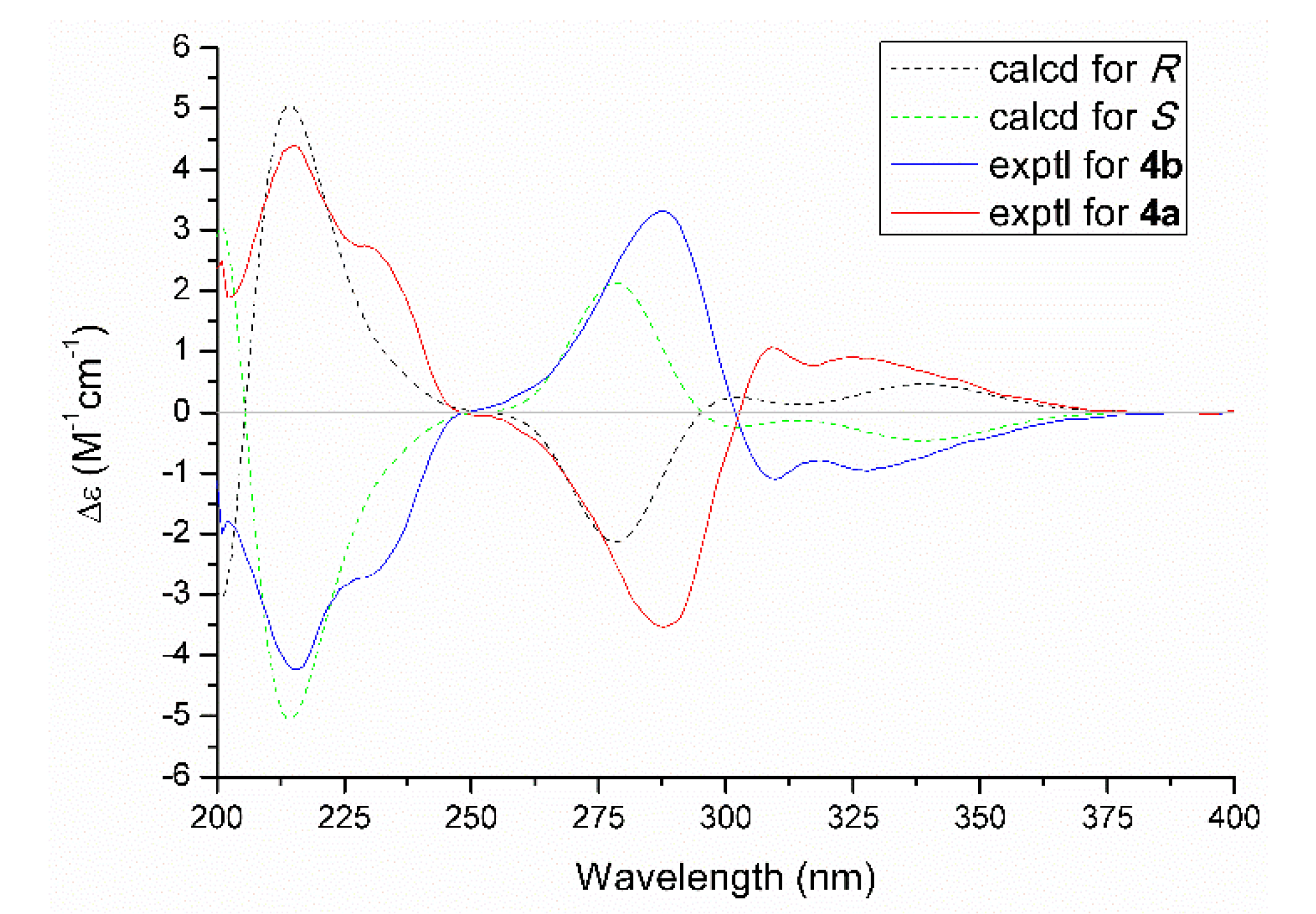
2.1. Structure Elucidation
2.2. Biological Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. X-ray Crytallographic Data of Compounds 1 and 2
3.5. Quantum Chemical Calculations
3.6. Nitric Oxide Inhibitory Activities Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jimenez, C. Marine natural products in medicinal chemistry. ACS Med. Chem. Lett. 2018, 9, 959–961. [Google Scholar] [CrossRef] [PubMed]
- El-Kashef, D.H.; Daletos, G.; Plenker, M.; Hartmann, R.; Mandi, A.; Kurtan, T.; Weber, H.; Lin, W.; Ancheeva, E.; Proksch, P. Polyketides and a dihydroquinolone alkaloid from a marine-derived strain of the fungus Metarhizium marquandii. J. Nat. Prod. 2019, 82, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.D.; Fan, P.; Zhou, L.M.; Ma, Q.Y.; Xie, Q.Y.; Zheng, H.Z.; Zheng, Z.H.; Zhang, R.S.; Yuan, J.Z.; Dai, H.F.; et al. Penerpenes A–D, four indole terpenoids with potent protein tyrosine phosphatase inhibitory activity from the marine-derived fungus Penicillium sp. KFD28. Org. Lett. 2019, 21, 4864–4867. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wang, J.; Wei, X.; Chen, Y.; Fu, T.; Xiang, Y.; Huang, X.; Tian, X.; Xiao, Z.; Zhang, W.; et al. Variecolortins A−C, three pairs of spirocyclic diketopiperazine enantiomers from the marine-derived fungus Eurotium sp. SCSIO F452. Org. Lett. 2018, 20, 4593–4596. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, Y.; Chen, S.; Liu, Y.; Lu, Y.; Chen, D.; Lin, Y.; Huang, X.; She, Z. Aspterpenacids A and B, two sesterterpenoids from a mangrove endophytic fungus Aspergillus terreus H010. Org. Lett. 2016, 18, 1406–1409. [Google Scholar] [CrossRef]
- Zhao, D.L.; Cao, F.; Wang, C.Y.; Yang, L.J.; Shi, T.; Wang, K.L.; Shao, C.L.; Wang, C.Y. Alternatone A, an unusual perylenequinone-related compound from a soft-coral-derived strain of the fungus Alternaria alternate. J. Nat. Prod. 2019, 82, 3201–3204. [Google Scholar] [CrossRef]
- Limbadri, S.; Luo, X.; Lin, X.; Wang, J.; Yang, B.; Zhou, X.; Liu, Y. Versispiroketal A, an unusual tetracyclic bridged spiroketal from the sponge-associated fungus Aspergillus versicolor SCSIO 41013. Org. Biomol. Chem. 2019, 17, 2182–2186. [Google Scholar]
- Jiao, W.H.; Li, J.; Wang, D.; Zhang, M.M.; Liu, L.Y.; Sun, F.; Li, J.Y.; Capon, R.J.; Lin, H.W. Cinerols, nitrogenous meroterpenoids from the marine sponge Dysidea cinerea. J. Nat. Prod. 2019, 82, 2586–2593. [Google Scholar] [CrossRef]
- Luo, M.; Cui, Z.; Huang, H.; Song, X.; Sun, A.; Dang, Y.; Lu, L.; Ju, J. Amino acid conjugated anthraquinones from the marine-derived fungus Penicillium sp. SCSIO sof101. J. Nat. Prod. 2017, 80, 1668–1673. [Google Scholar] [CrossRef]
- Luo, X.; Lin, X.; Tao, H.; Wang, J.; Li, J.; Yang, B.; Zhou, X.; Liu, Y. Isochromophilones A−F, cytotoxic chloroazaphilones from the marine mangrove endophytic fungus Diaporthe sp. SCSIO 41011. J. Nat. Prod. 2018, 81, 934–941. [Google Scholar] [CrossRef]
- Wang, J.; Cong, Z.; Huang, X.; Hou, C.; Chen, W.; Tu, Z.; Huang, D.; Liu, Y. Soliseptide A, a cyclic hexapeptide possessing piperazic acid groups from Streptomyces solisilvae HNM30702. Org. Lett. 2018, 20, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tang, X.; Luo, X.; Wang, Q.; Liu, K.; Zhang, Y.; de Voogd, N.J.; Yang, J.; Li, P.; Li, G. Agelanemoechine, a dimeric bromopyrrole alkaloid with a pro-angiogenic effect from the south china sea sponge Agelas nemoechinata. Org. Lett. 2019, 21, 9483–9486. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.R.; Wang, S.W.; Chang, F.R.; Cheng, Y.B. Anti-lymphangiogenic alkaloids from the zoanthid Zoanthus vietnamensis collected in Taiwan. J. Nat. Prod. 2019, 82, 2790–2799. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lee, J.; Kim, K.J.; Sung, Y.; Park, K.H.; Oh, E.; Park, C.; Son, Y.J.; Kang, H. Austalides, osteoclast differentiation inhibitors from a marine-derived strain of the fungus Penicillium rudallense. J. Nat. Prod. 2019, 82, 3083–3088. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, Q.; Li, S.; Cui, H.; Sun, Z.; Chen, D.; Lu, Y.; Liu, H.; Zhang, W. Polypropionate derivatives with mycobacterium tuberculosis protein tyrosine phosphatase B inhibitory activities from the deep-sea-derived fungus Aspergillus fischeri FS452. J. Nat. Prod. 2019, 82, 3440–3449. [Google Scholar] [CrossRef]
- Xu, J.; Tan, H.; Chen, Y.; Li, S.; Huang, Z.; Guo, H.; Li, H.; Gao, X.; Liu, H.; Zhang, W. Lithocarpins A–D: Four tenellone-macrolide conjugated [4 + 2] hetero-adducts from the deep-sea derived fungus Phomopsis lithocarpus FS508. Org. Chem. Front. 2018, 5, 1792–1797. [Google Scholar] [CrossRef]
- Chen, S.C.; Liu, Z.M.; Tan, H.B.; Chen, Y.C.; Li, S.N.; Li, H.H.; Guo, H.; Zhu, S.; Liu, H.X.; Zhang, W.M. Tersone A-G, new pyridone alkaloids from the deep-dea fungus Phomopsis tersa. Mar. Drugs 2019, 17, 394. [Google Scholar] [CrossRef]
- Kim, J.W.; Ko, W.; Kim, E.; Kim, G.S.; Hwang, G.J.; Son, S.; Jeong, M.H.; Hur, J.S.; Oh, H.; Ko, S.K.; et al. Anti-inflammatory phomalichenones from an endolichenic fungus Phoma sp. J. Antibiot. 2018, 71, 753–756. [Google Scholar] [CrossRef]
- Kang, U.; Ryu, S.M.; Lee, D.; Seo, E.K. Chemical constituents of the leaves of Brassica oleracea var. acephala. Chem. Nat. Compd. 2018, 54, 1023–1026. [Google Scholar] [CrossRef]
- Rathnayake, G.R.N.; Kumar, N.S.; Jayasinghe, L.; Araya, H.; Fujimoto, Y. Chemical investigation of metabolites produced by an endophytic fungi Phialemonium curvatum from the leaves of Passiflora edulis. Nat. Prod. Res. 2018, 32, 2483–2486. [Google Scholar] [CrossRef]
- Li, X.J.; Gao, J.M.; Chen, H.; Zhang, A.L.; Tang, M. Toxins from a symbiotic fungus, Leptographium qinlingensis associated with Dendroctonus armandi and their in vitro toxicities to Pinus armandi seedlings. Eur. J. Plant. Pathol. 2012, 134, 239–247. [Google Scholar] [CrossRef]
- Valerio, F.; Masi, M.; Cimmino, A.; Moeini, S.A.; Lavermicocca, P.; Evidente, A. Antimould microbial and plant metabolites with potential use in intelligent food packaging. Nat. Prod. Res. 2018, 2, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Liu, Z.Z.; Kim, K.W.; Wang, X.; Li, Z.; Kim, Y.C.; Yook, C.S.; Liu, X.Q. Chemical constituents from leaves of Pileostegia viburnoides Hook.f.et Thoms. Nat. Prod. Sci. 2016, 22, 154–161. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. D.01 ed; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Wu, P.; Xue, J.; Yao, L.; Xu, L.; Li, H.; Wei, X. Bisacremines E-G, three polycyclic dimeric acremines produced by Acremonium persicinum SC0105. Org. Lett. 2015, 17, 4922–4925. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumloffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef]
- Miranda, K.M.; Espey, M.G.; Wink, D.A. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide 2001, 5, 62–71. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 115.2, C | 113.6, C | ||
| 2 | 198.8, C | 199.8, C | ||
| 3 | 79.2, C | 79.6, C | ||
| 4 | 4.03 (m) | 71.3, CH | 4.13 (m) | 72.3, CH |
| 5α 5β | 2.63 (dd, 18.1, 8.7) 3.00 (dd, 18.1, 5.7) | 33.9, CH2 | 2.63 (dd, 18.5, 1.9) 3.20 (dd, 18.5, 4.1) | 33.7, CH2 |
| 6 | 162.8, C | 158.9, C | ||
| 7a 7b | 1.56 (m) 1.92 (dq, 15.0, 7.6) | 23.5, CH2 | 1.50 (m) | 28.8, CH2 |
| 8 | 0.77 (t, 7.6) | 7.0, CH3 | 0.32 (t, 7.4) | 6.9, CH3 |
| 9 | 161.8, C | 160.3, C | ||
| 10α 10β | 3.06 (dd, 18.8, 3.9) 2.83 (dd, 18.8, 2.6) | 33.5, CH2 | 2.83 (m) 2.75 (dd, 17.6, 9.9) | 34.3, CH2 |
| 11 | 4.16 (m) | 70.9, CH | 4.09 (m) | 72.2, CH |
| 12 | 79.3, C | 80.1, C | ||
| 13 | 199.2, C | 199.7, C | ||
| 14a 14b | 1.63 (m) | 29.4, CH2 | 1.45 (m) 1.81 (m) | 23.3, CH2 |
| 15 | 0.83 (t, 7.4) | 7.8, CH3 | 0.20 (t, 7.5) | 6.2, CH3 |
| 16 | 113.5, C | 114.3, C | ||
| 17 | 4.42 (s) | 32.8, CH | 4.70 (s) | 32.8, CH |
| 18 | 136.2, C | 135.6, C | ||
| 19 | 7.11 (d, 8.6) | 130.2, CH | 7.07 (d, 8.5) | 130.5, CH |
| 20 | 6.63 (d, 8.6) | 115.4, CH | 6.66 (d, 8.5) | 115.4, CH |
| 21 | 156.7, C | 156.9, C | ||
| 22 | 6.63 (d, 8.6) | 115.4, CH | 6.66 (d, 8.5) | 115.4, CH |
| 23 | 7.11 (d, 8.6) | 130.2, CH | 7.07 (d, 8.5) | 130.5, CH |
| 21-OH | 8.09 (s) | 8.14 (s) | ||
| No. | 3 | 4 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 129.7, C | 103.1, C | ||
| 2 | 7.07 (d, 8.5) | 131.0, CH | 162.4, C | |
| 3 | 6.72 (d, 8.5) | 116.3, CH | 111.6, C | |
| 4 | 157.2, C | 165.8, C | ||
| 5 | 6.72 (d, 8.5) | 116.3, CH | 5.89 (s) | 95.2, CH |
| 6 | 7.07 (d, 8.5) | 131.0, CH | 162.5, C | |
| 7 | 2.94 (t, 6.8) | 35.1, CH2 | 4.47 (dqd, 12.3, 6.3, 3.2) | 75.2, CH |
| 8 | 4.07 (td, 6.8, 1.6) | 71.5, CH2 | 2.58 (d, 3.2) 2.65 (17.1, 12.3) | 44.2, CH2 |
| 9 | 176.5, C | 198.0, C | ||
| 10 | 5.34 (s) | 100.8, CH | 2.53 (q, 7.4) | 16.0, CH2 |
| 11 | 202.1, C | 1.04 (t, 7.4) | 13.8, CH3 | |
| 12 | 80.0, C | 1.43 (d, 6.3) | 21.0, CH3 | |
| 13 | 3.98 (dd, 9.9, 5.9) | 72.6, CH | ||
| 14a | 2.53 (dd, 17.7, 9.9) | 36.8, CH2 | ||
| 14b | 2.68 (dq, 17.7, 5.9) | |||
| 15a | 1.58 (dq, 14.3, 7.5) | 24.1, CH2 | ||
| 15b | 1.89 (dd, 14.3, 7.5) | |||
| 16 | 0.81 (t, 7.5) | 7.2, CH3 | ||
| No. | 5 | 6 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 106.8, C | 145.5, C | ||
| 2 | 163.0, C | 7.21 (s) | 117.7, CH | |
| 3 | 106.5, C | 126.7, C | ||
| 4 | 169.1, C | 132.7, C | ||
| 5 | 6.07 (s) | 87.2, CH | 152.6, C | |
| 6 | 165.5, C | 131.6, C | ||
| 7 | 3.07 (t, 8.7) | 26.7, CH2 | 173.8, C | |
| 8 | 4.66 (t, 8.7) | 74.4, CH2 | 5.27 (s) | 69.7, CH2 |
| 9 | 194.6, C | 2.73 (m) | 36.0, CH2 | |
| 10 | 7.21 (dd, 15.1, 1.6) | 133.5, CH | 1.64 (m) | 25.5, CH2 |
| 11 | 6.98 (dq, 15.1, 6.9) | 142.9, CH | 1.00 (t, 7.3) | 14.3, CH3 |
| 12 | 1.94 (dd, 6.9, 1.6) | 18.6, CH3 | 4.89 (s) | 58.2, CH2 |
| 13 | 3.86 (s) | 56.4, CH3 | ||
| Compounds | Inhibition of NO Production (IC50/μM) a | Cytotoxicity (IC50/μM) |
|---|---|---|
| 1 | >200 | >200 |
| 2 | 108.1 ± 3.0 | >200 |
| 3 | 51.9 ± 1.4 | >200 |
| 4a | 54.3 ± 2.2 | >200 |
| 4b | 55.9 ± 1.6 | >200 |
| 5 | 65.5 ± 1.3 | >200 |
| 6 | 111.2 ± 4.6 | >200 |
| Aminoguanidine | 24.8 ± 0.8 | >200 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Liu, Z.; Chen, Y.; Tan, H.; Li, S.; Liu, H.; Zhang, W.; Zhu, S. Highly Substituted Phenol Derivatives with Nitric Oxide Inhibitory Activities from the Deep-Sea-Derived Fungus Trichobotrys effuse FS524. Mar. Drugs 2020, 18, 134. https://doi.org/10.3390/md18030134
Chen S, Liu Z, Chen Y, Tan H, Li S, Liu H, Zhang W, Zhu S. Highly Substituted Phenol Derivatives with Nitric Oxide Inhibitory Activities from the Deep-Sea-Derived Fungus Trichobotrys effuse FS524. Marine Drugs. 2020; 18(3):134. https://doi.org/10.3390/md18030134
Chicago/Turabian StyleChen, Shanchong, Zhaoming Liu, Yuchan Chen, Haibo Tan, Saini Li, Hongxin Liu, Weimin Zhang, and Shuang Zhu. 2020. "Highly Substituted Phenol Derivatives with Nitric Oxide Inhibitory Activities from the Deep-Sea-Derived Fungus Trichobotrys effuse FS524" Marine Drugs 18, no. 3: 134. https://doi.org/10.3390/md18030134
APA StyleChen, S., Liu, Z., Chen, Y., Tan, H., Li, S., Liu, H., Zhang, W., & Zhu, S. (2020). Highly Substituted Phenol Derivatives with Nitric Oxide Inhibitory Activities from the Deep-Sea-Derived Fungus Trichobotrys effuse FS524. Marine Drugs, 18(3), 134. https://doi.org/10.3390/md18030134