
Contemporary Strategies for the Synthesis of Tetrahydropyran Derivatives: Application to Total Synthesis of Neopeltolide, a Marine Macrolide Natural Product


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
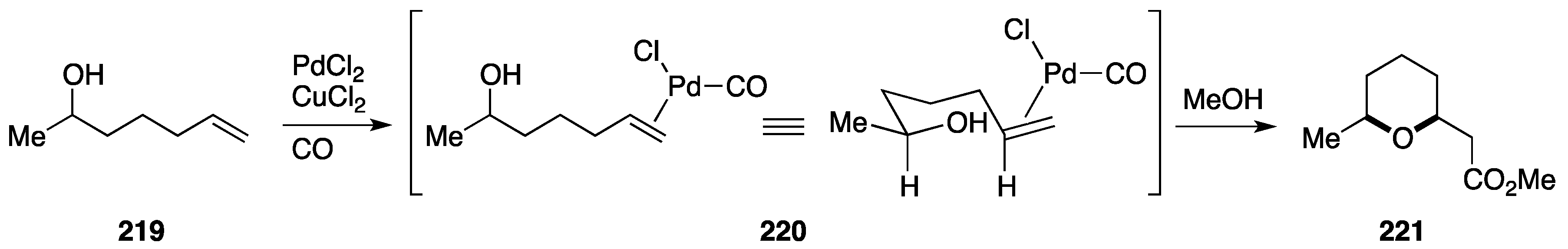{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
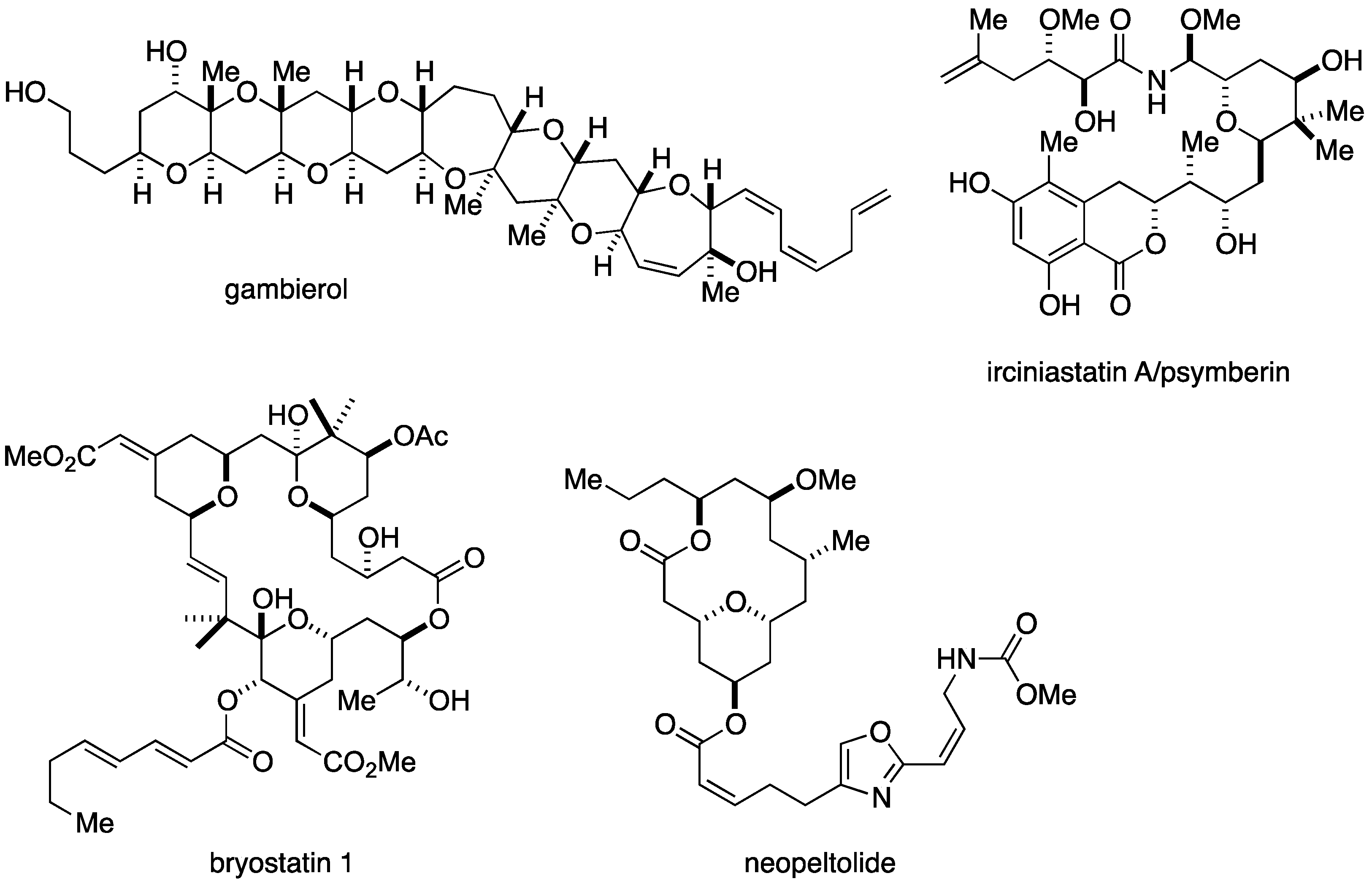
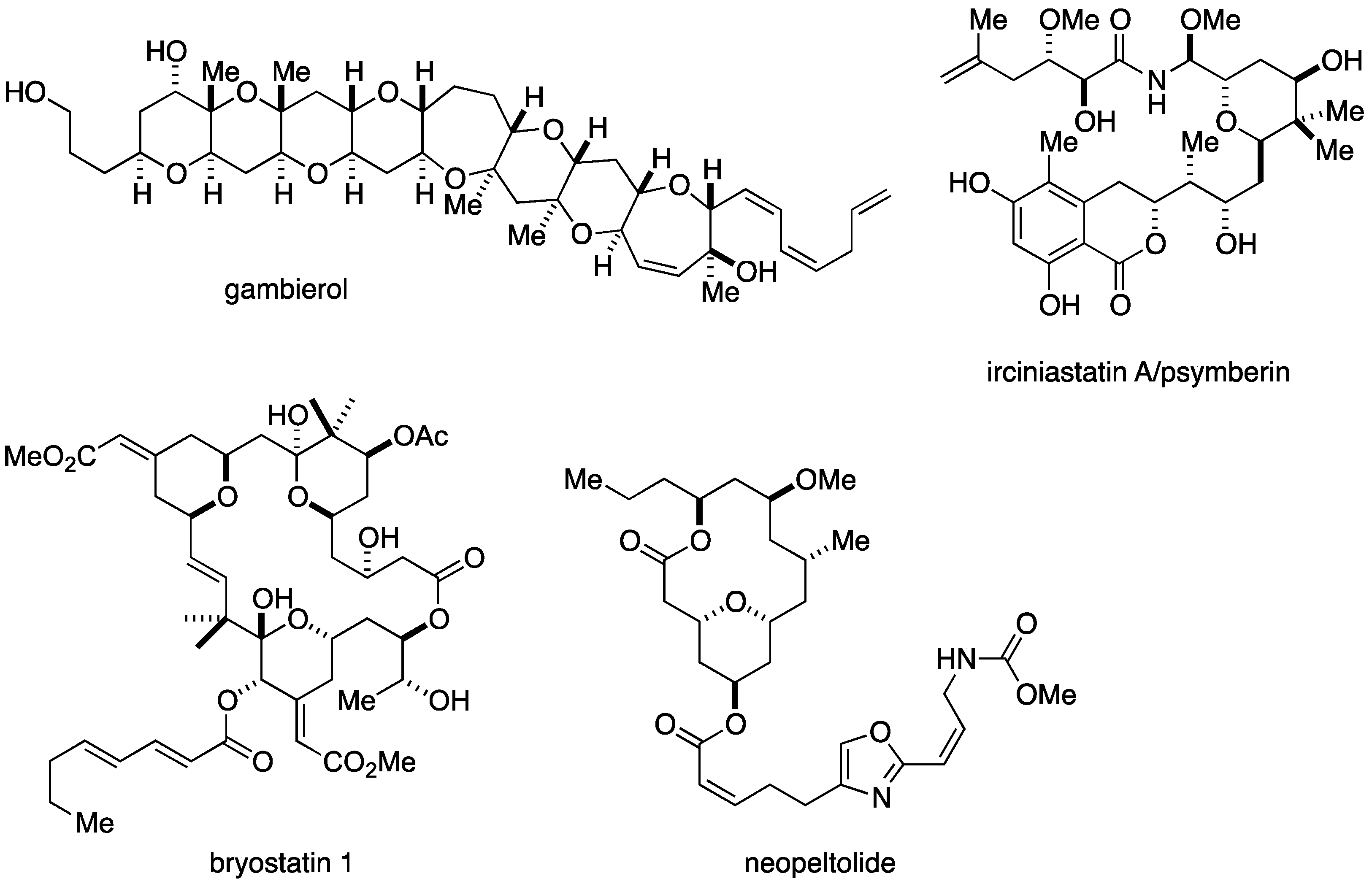
:1. Introduction
2. Synthesis of Tetrahydropyrans via Oxocarbenium Ions
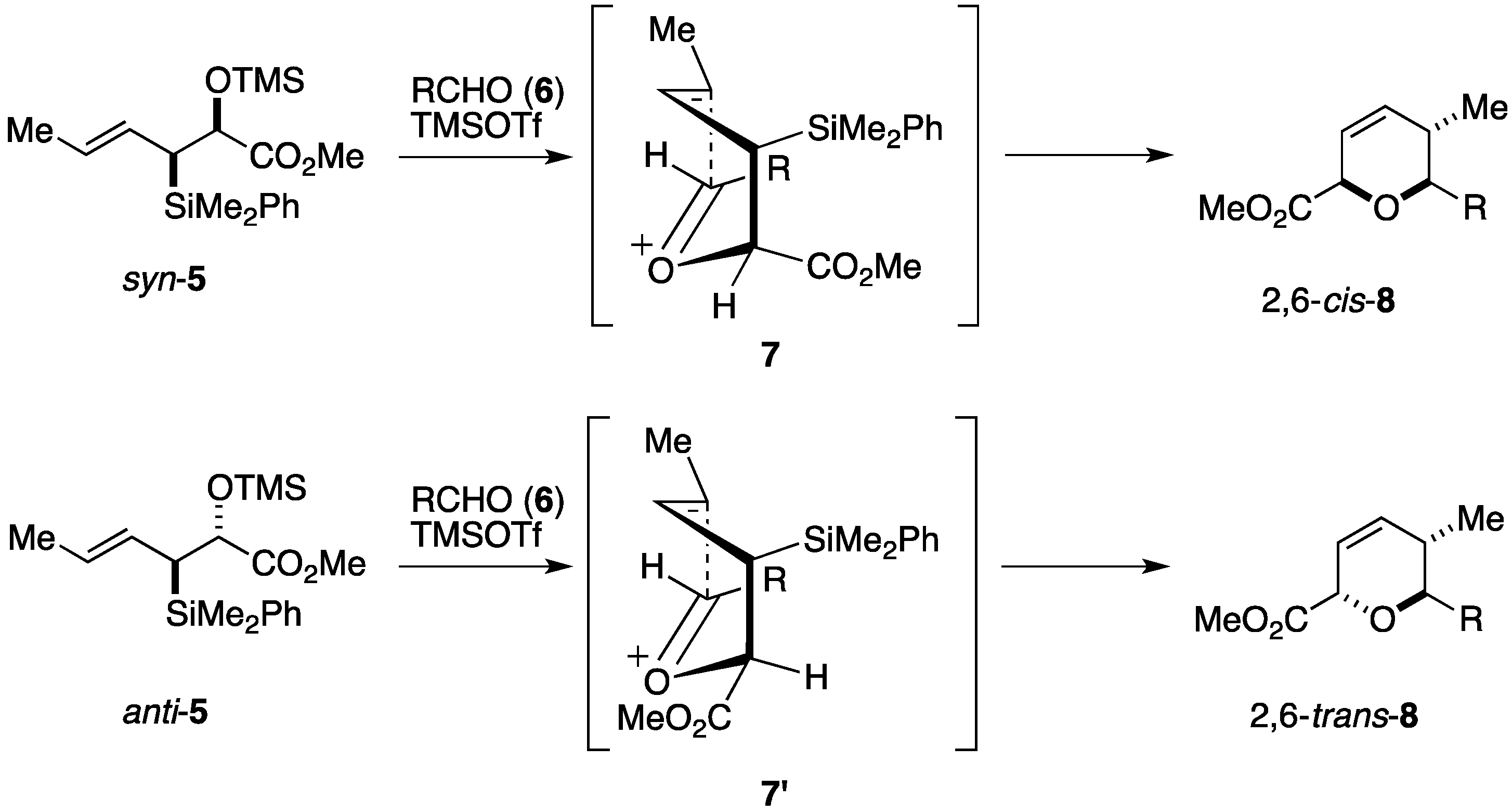
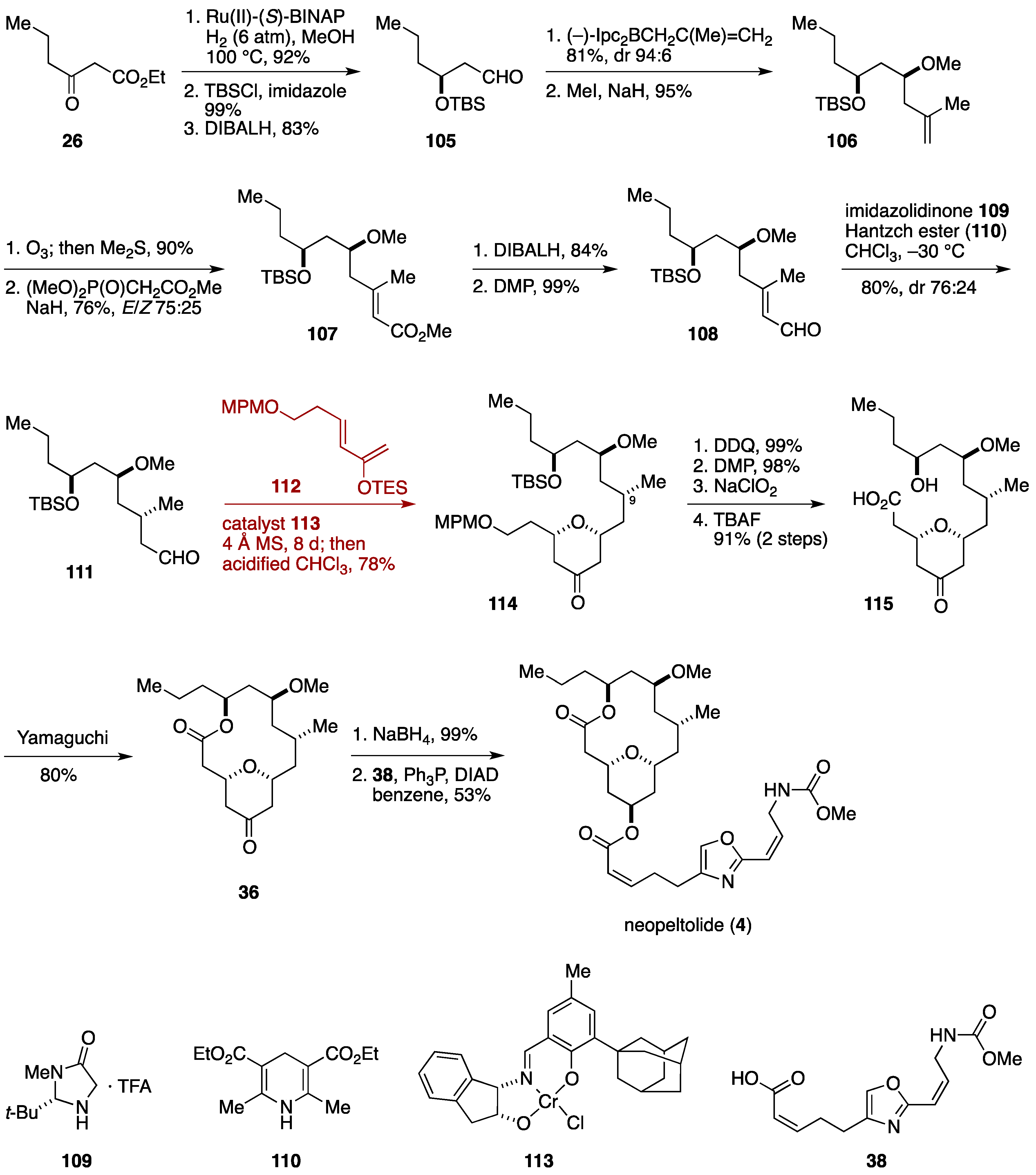
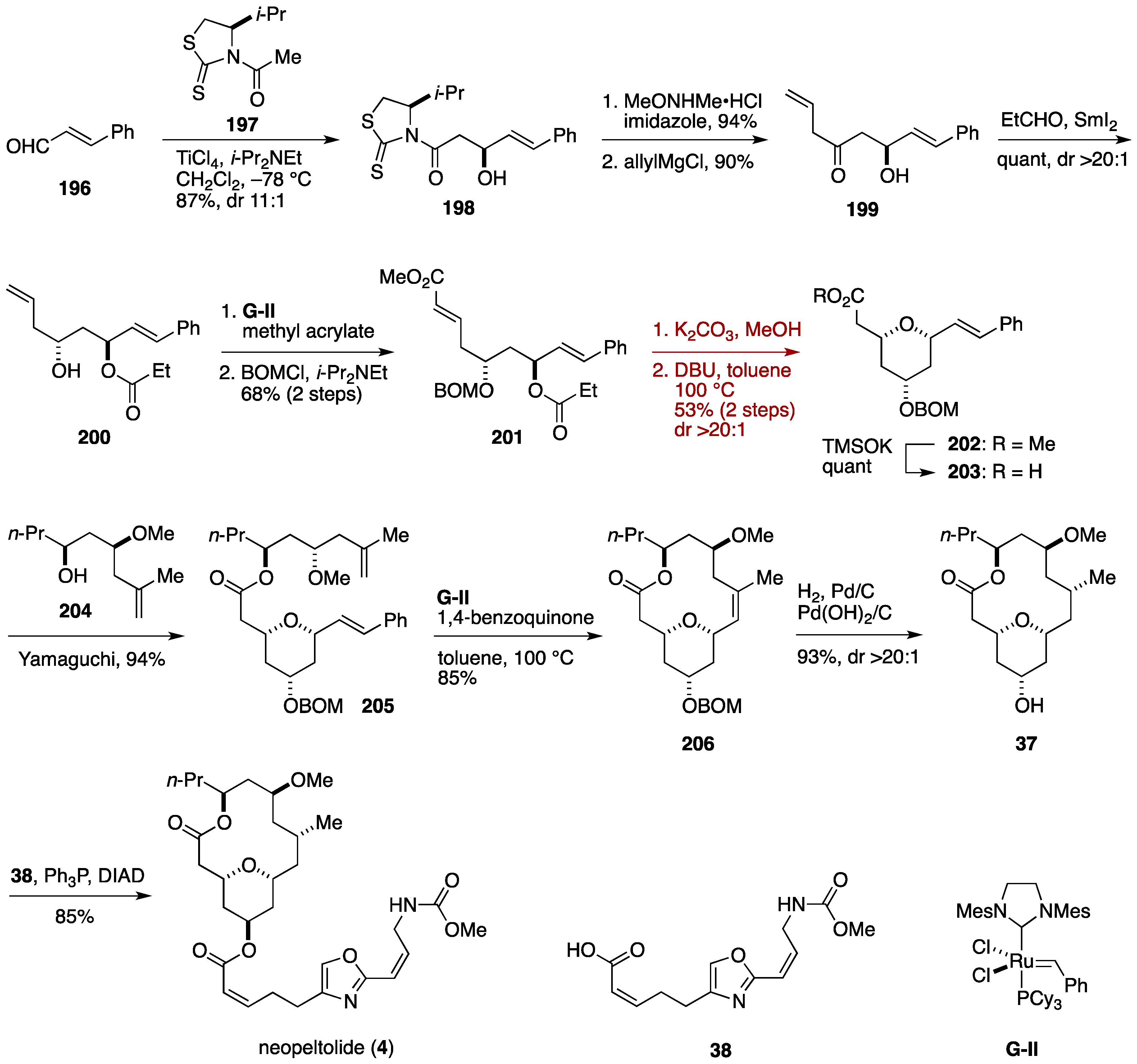
2.1. Total Synthesis by the Panek Group
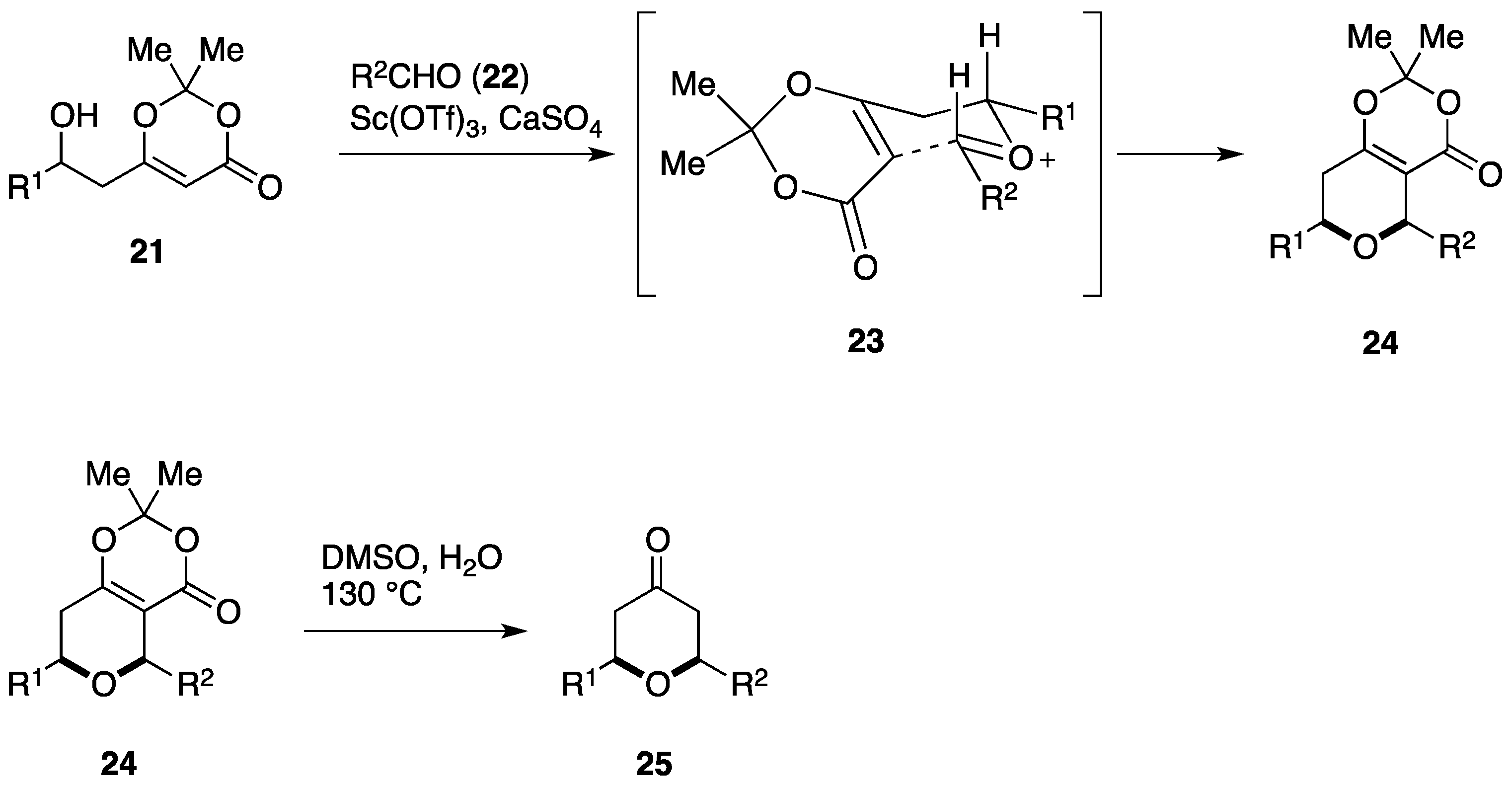
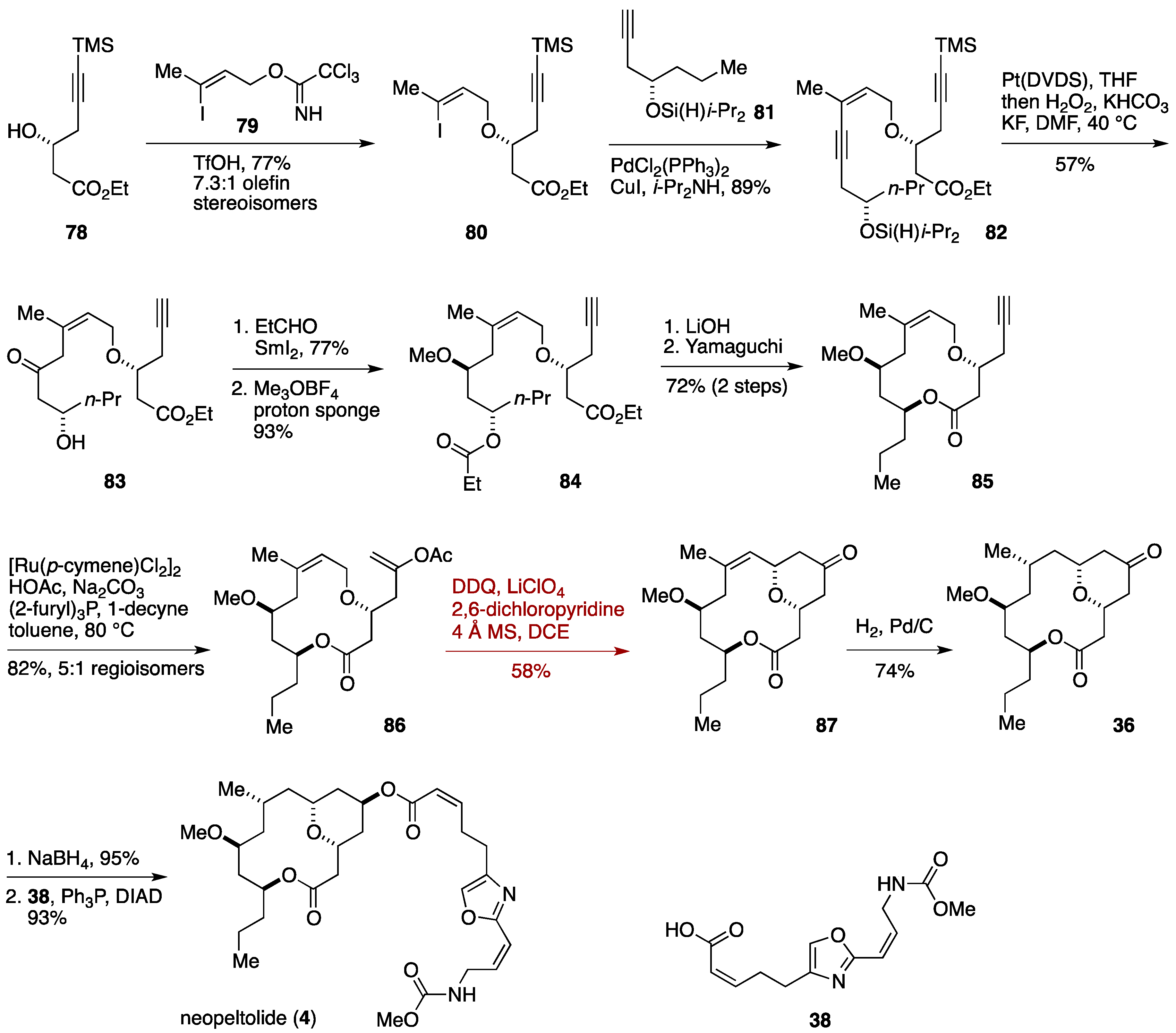
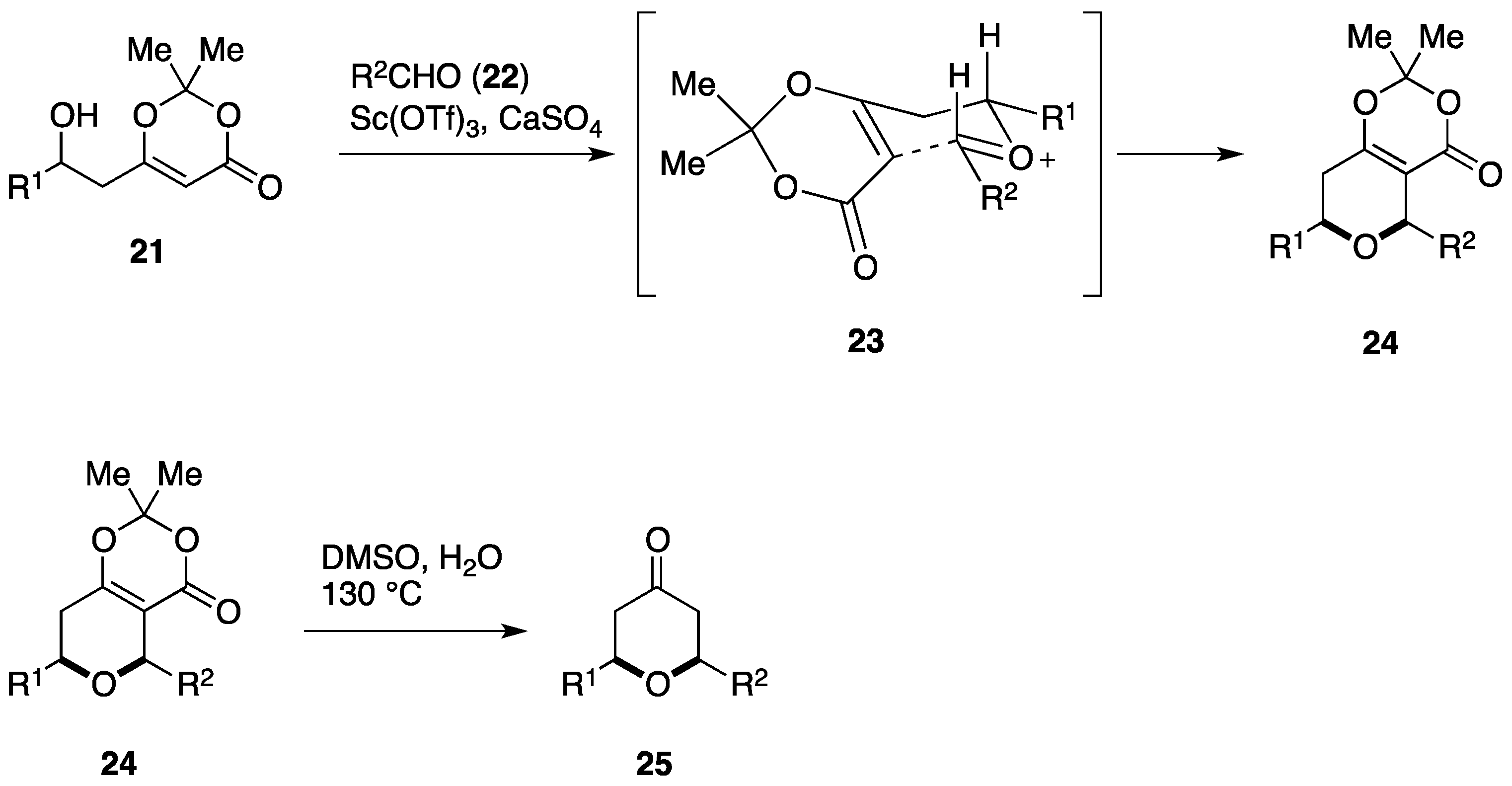
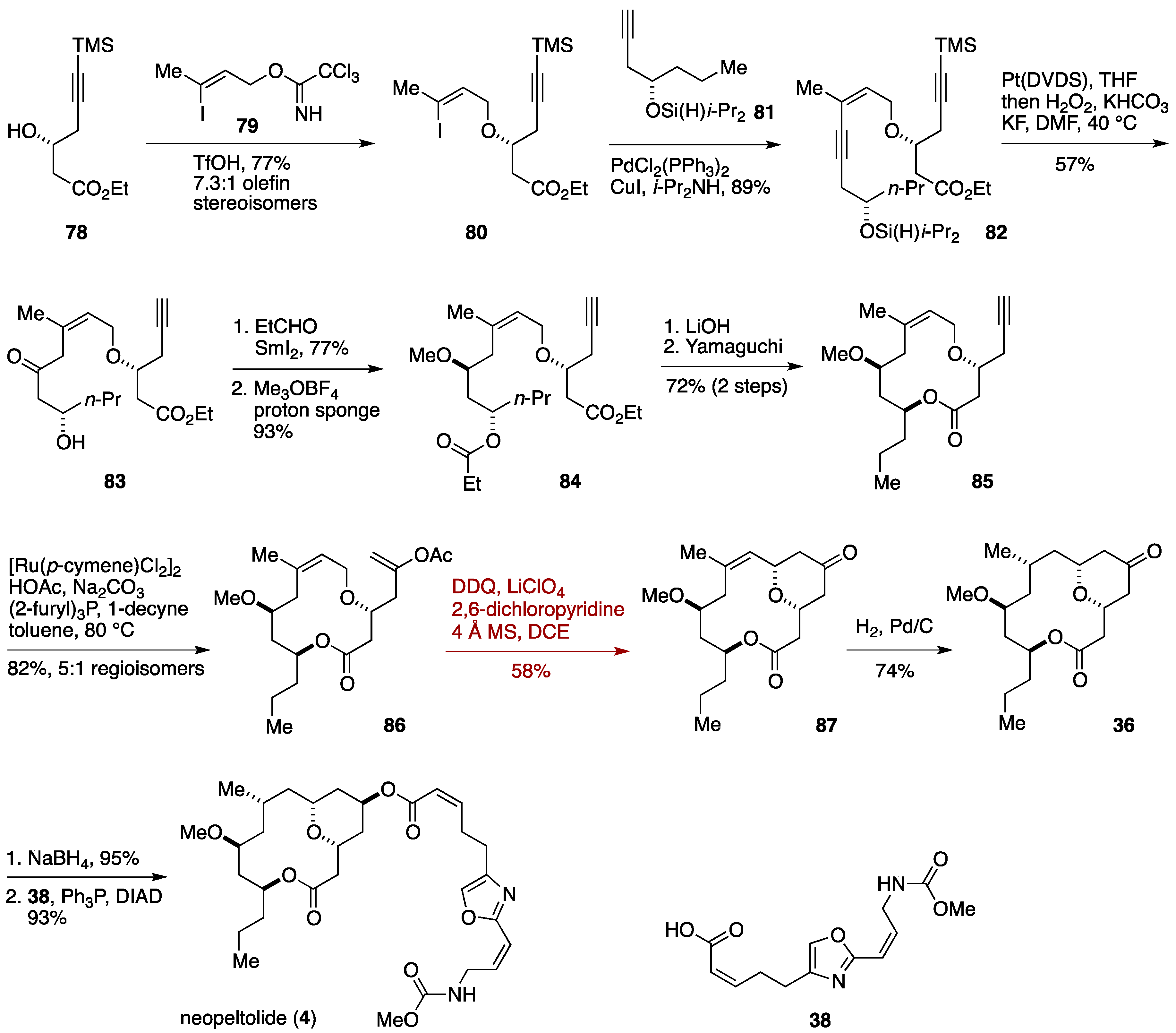
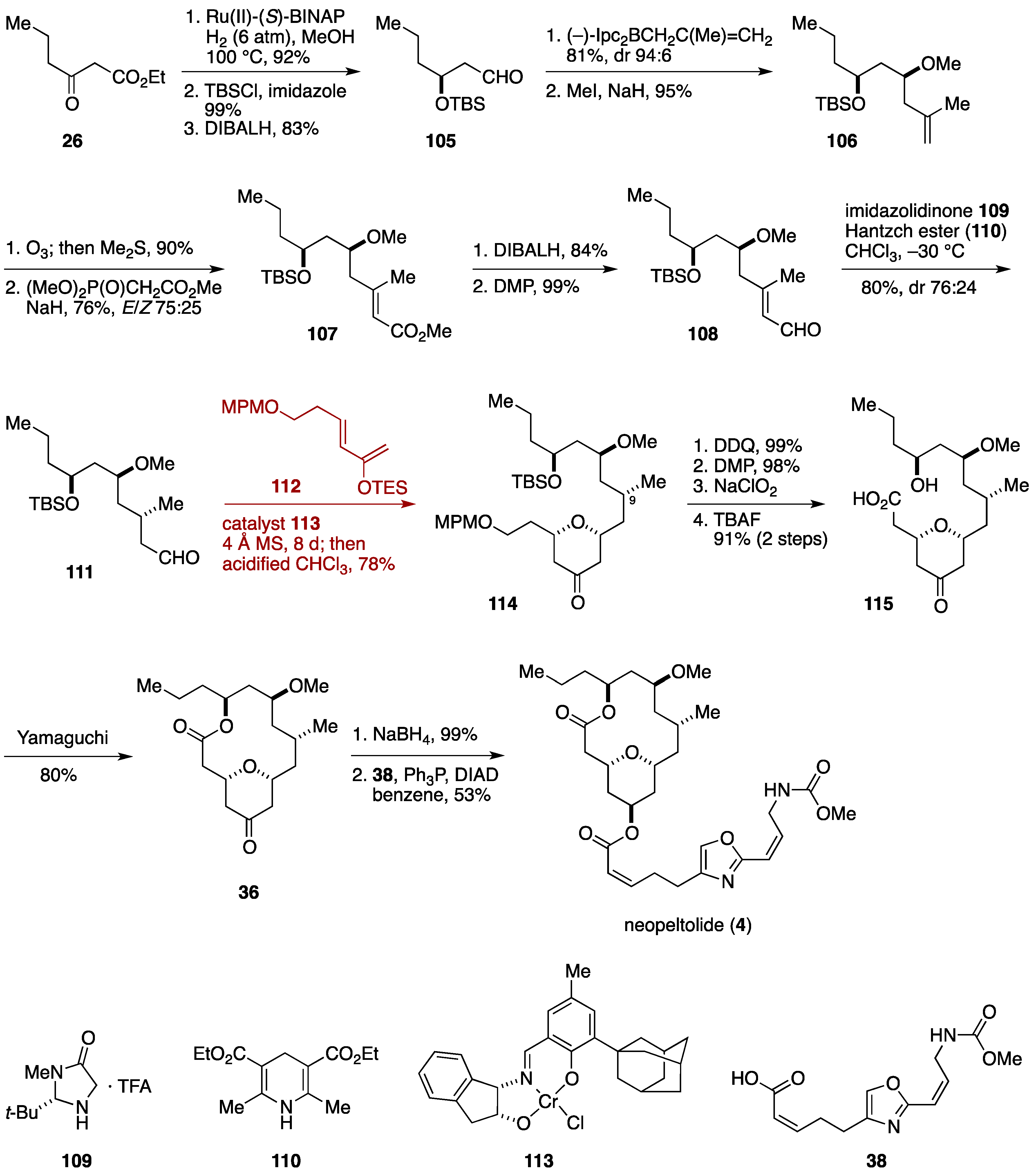
2.2. Total Synthesis by the Scheidt Group
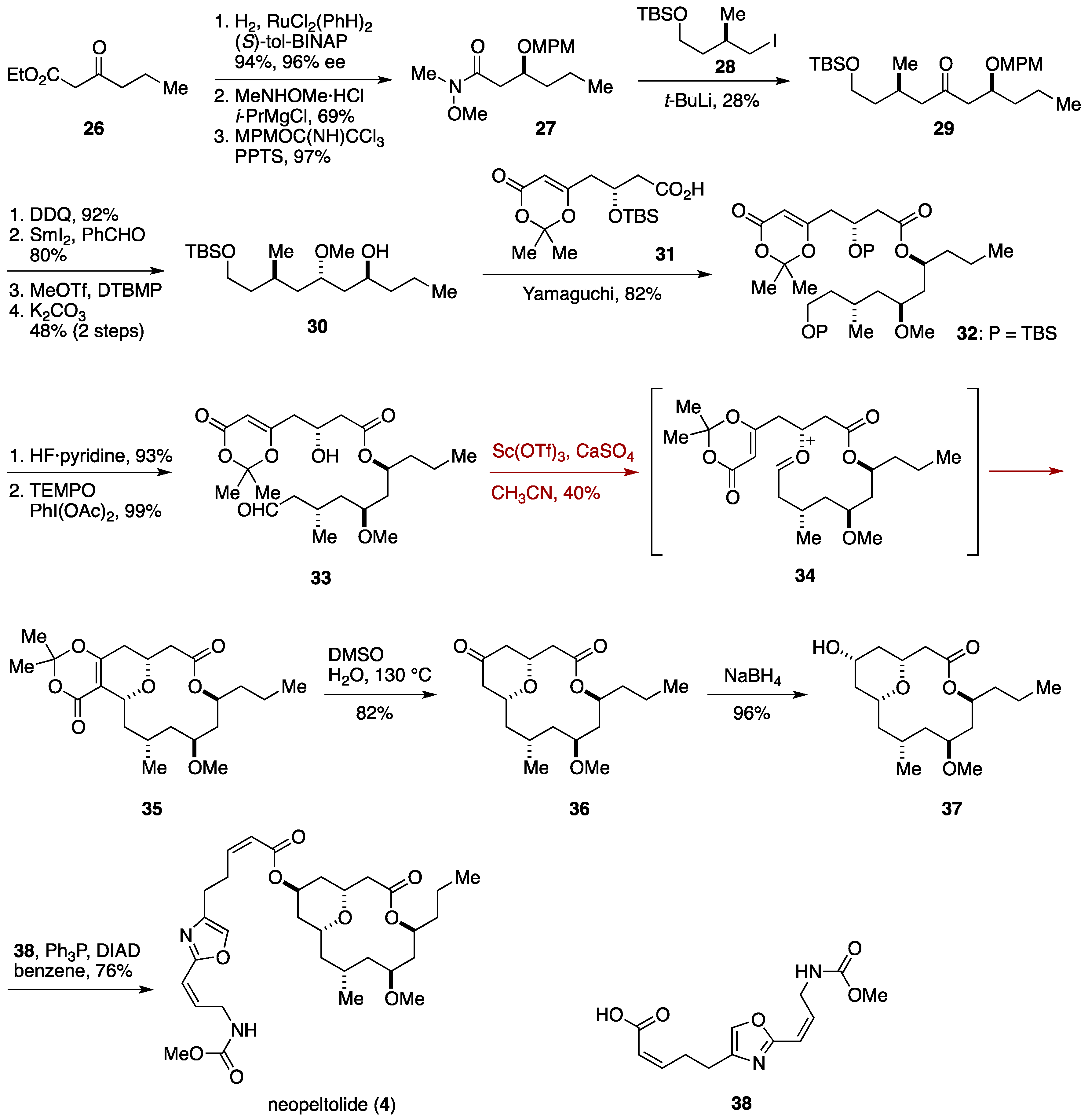
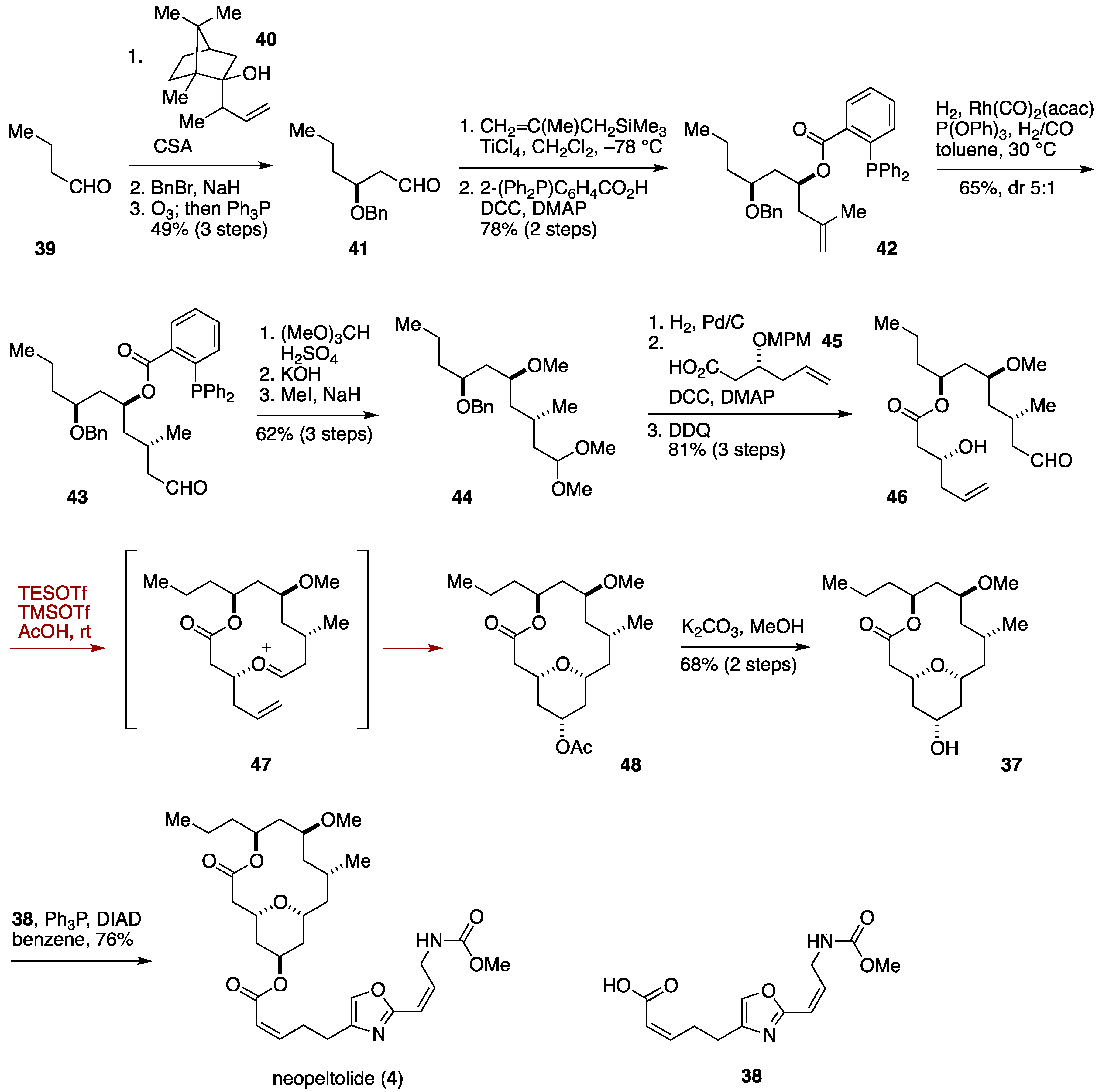
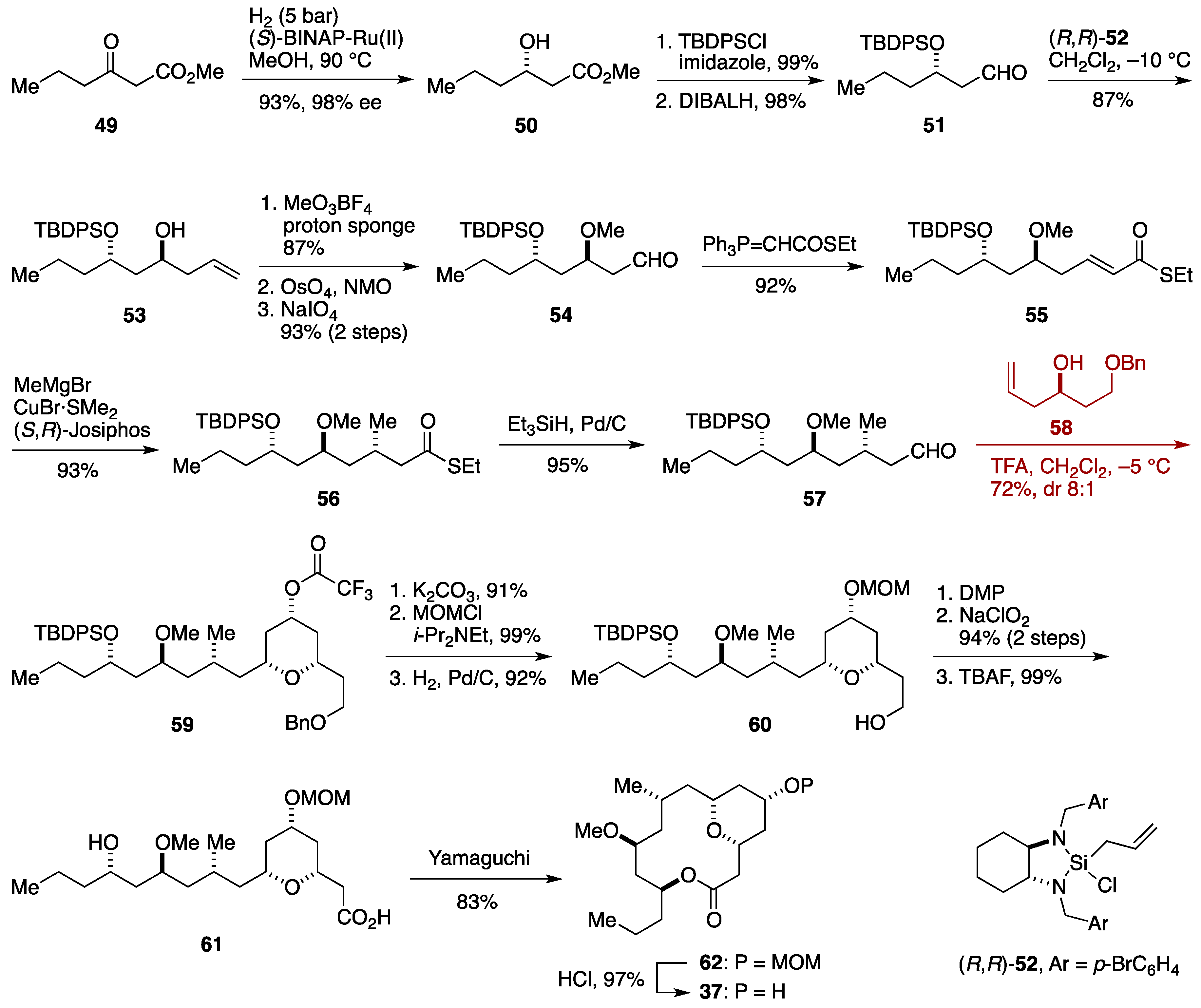
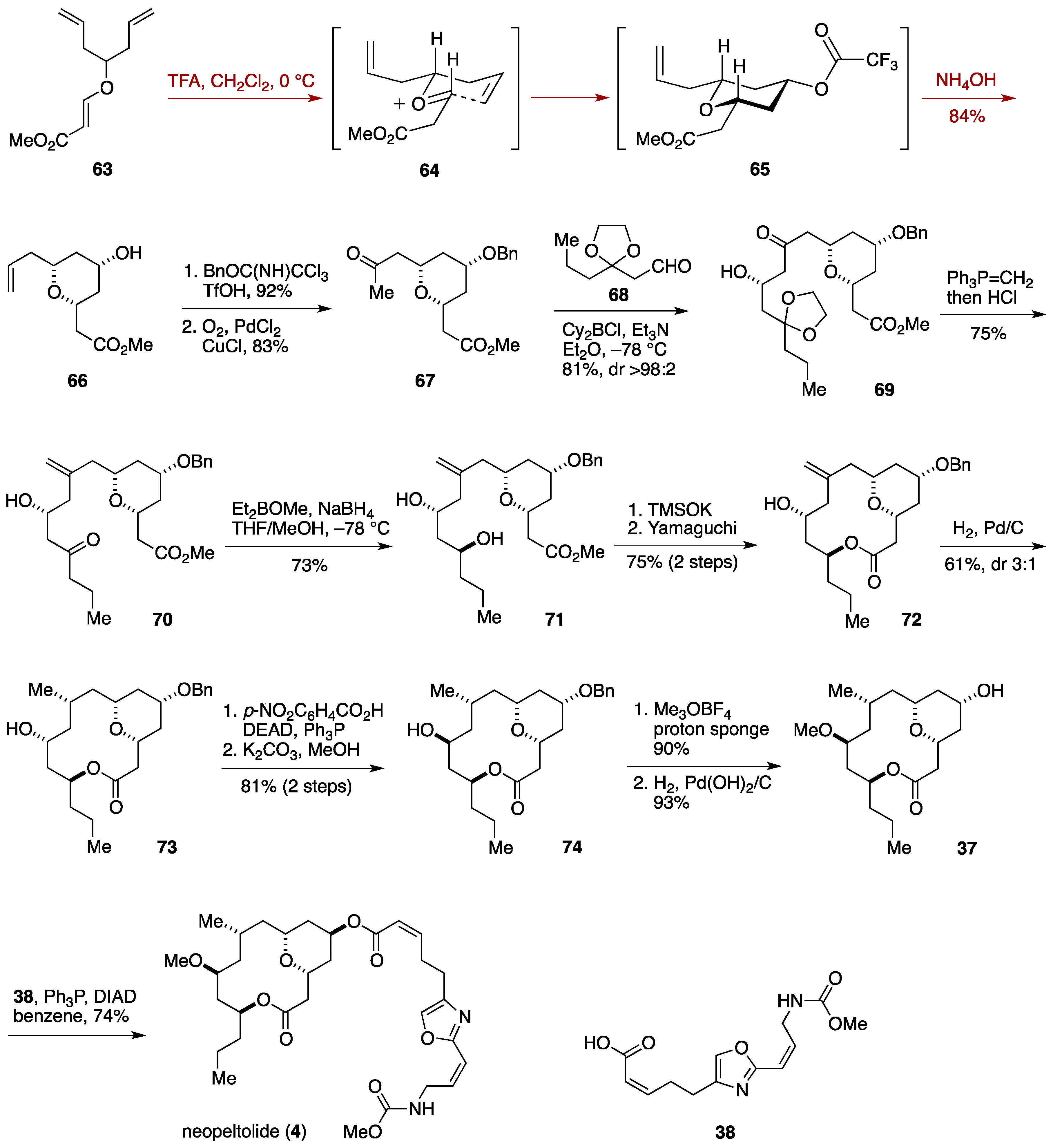
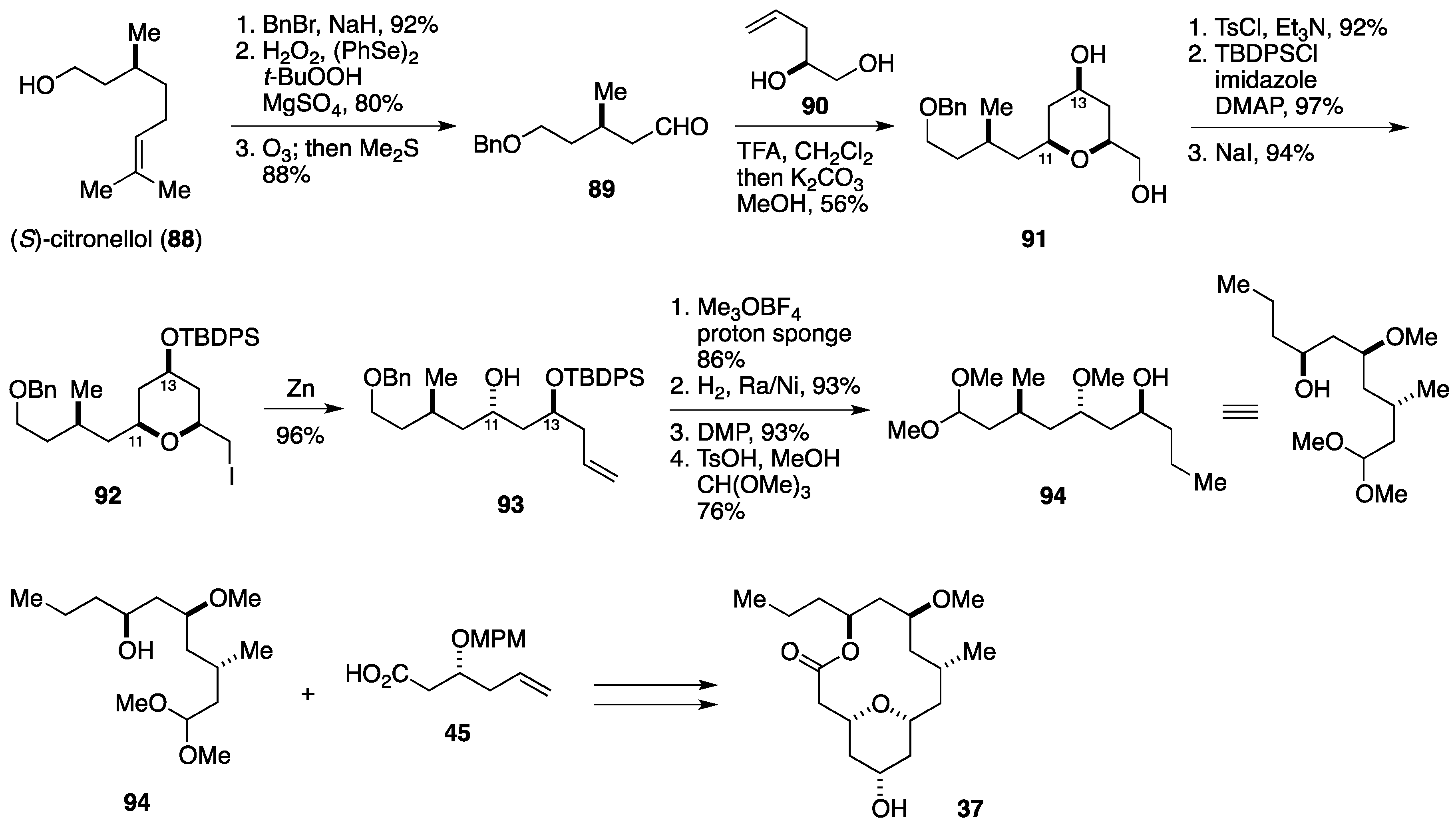
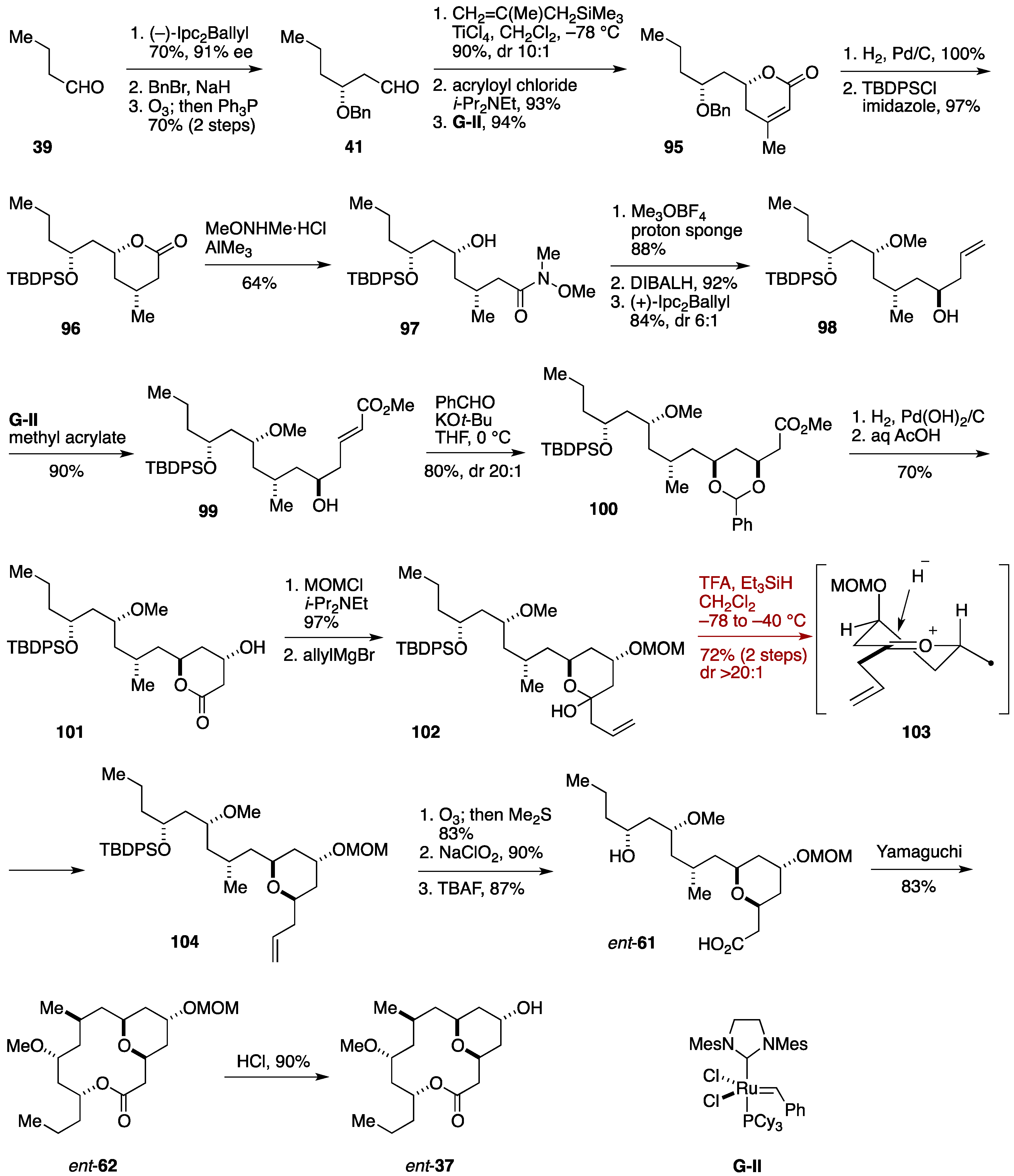
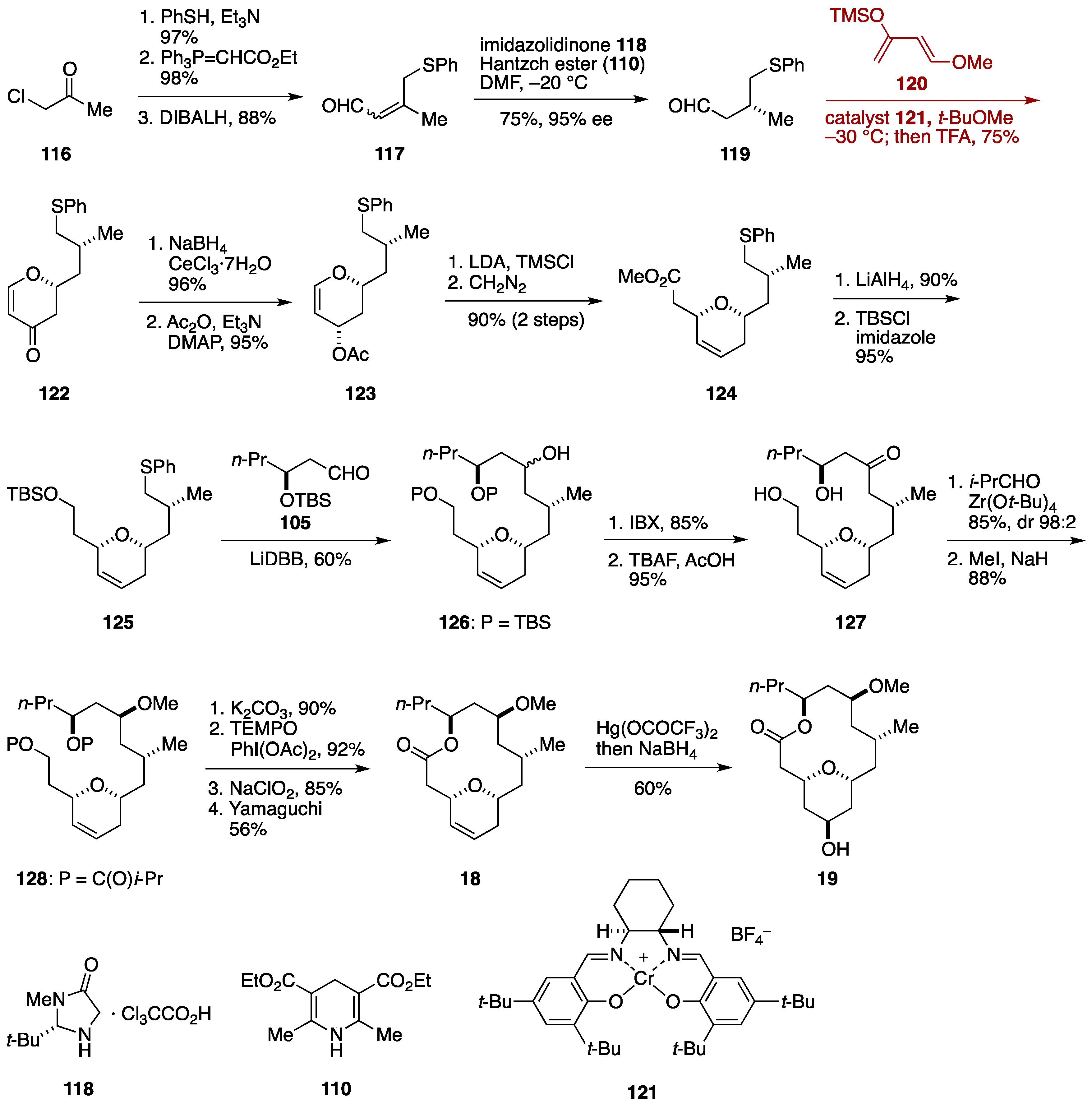
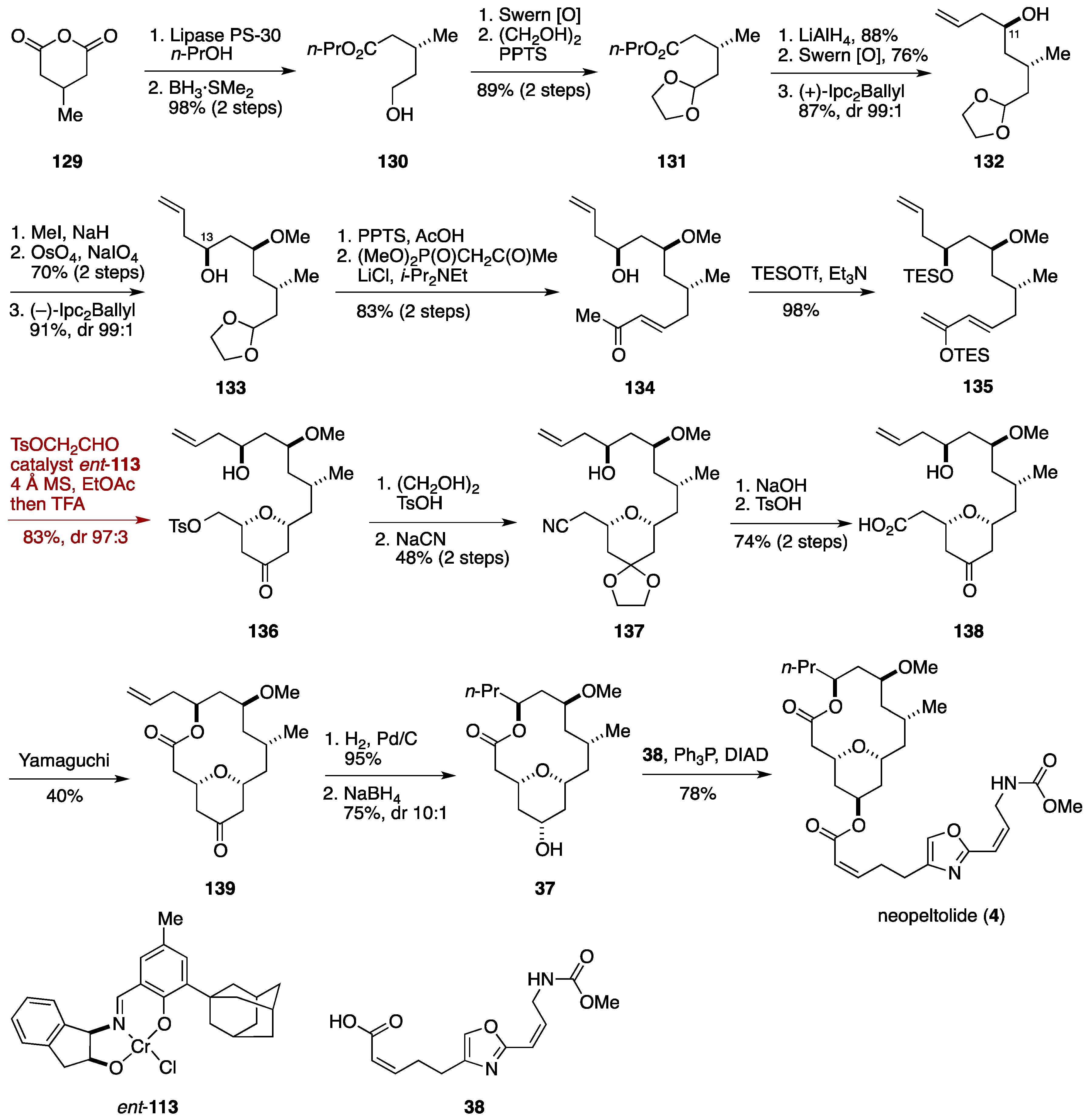
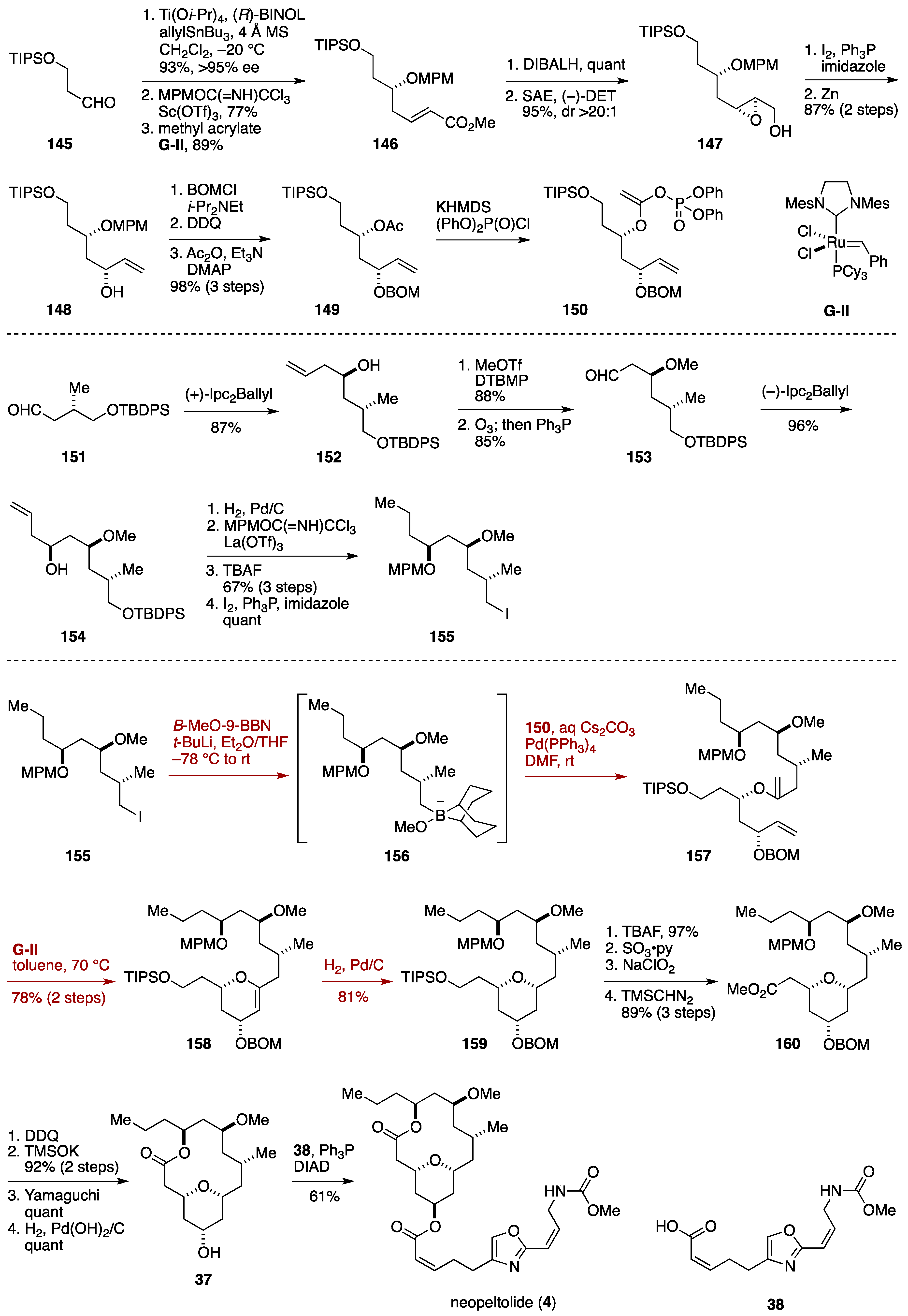
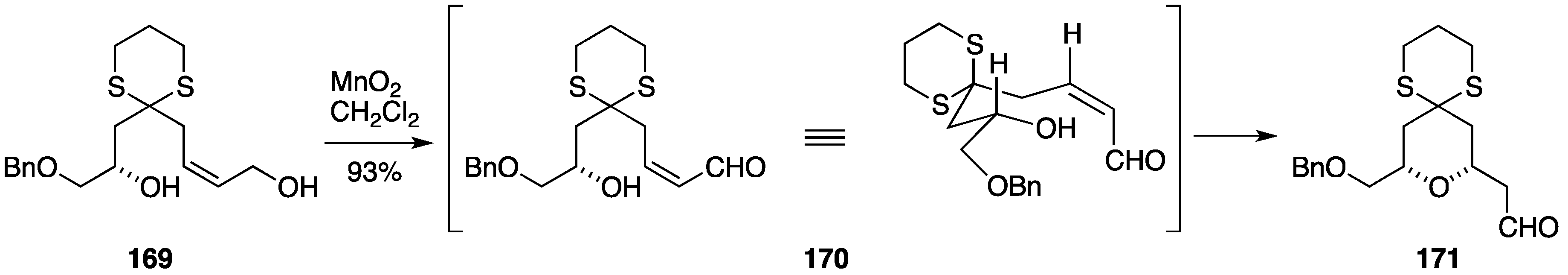
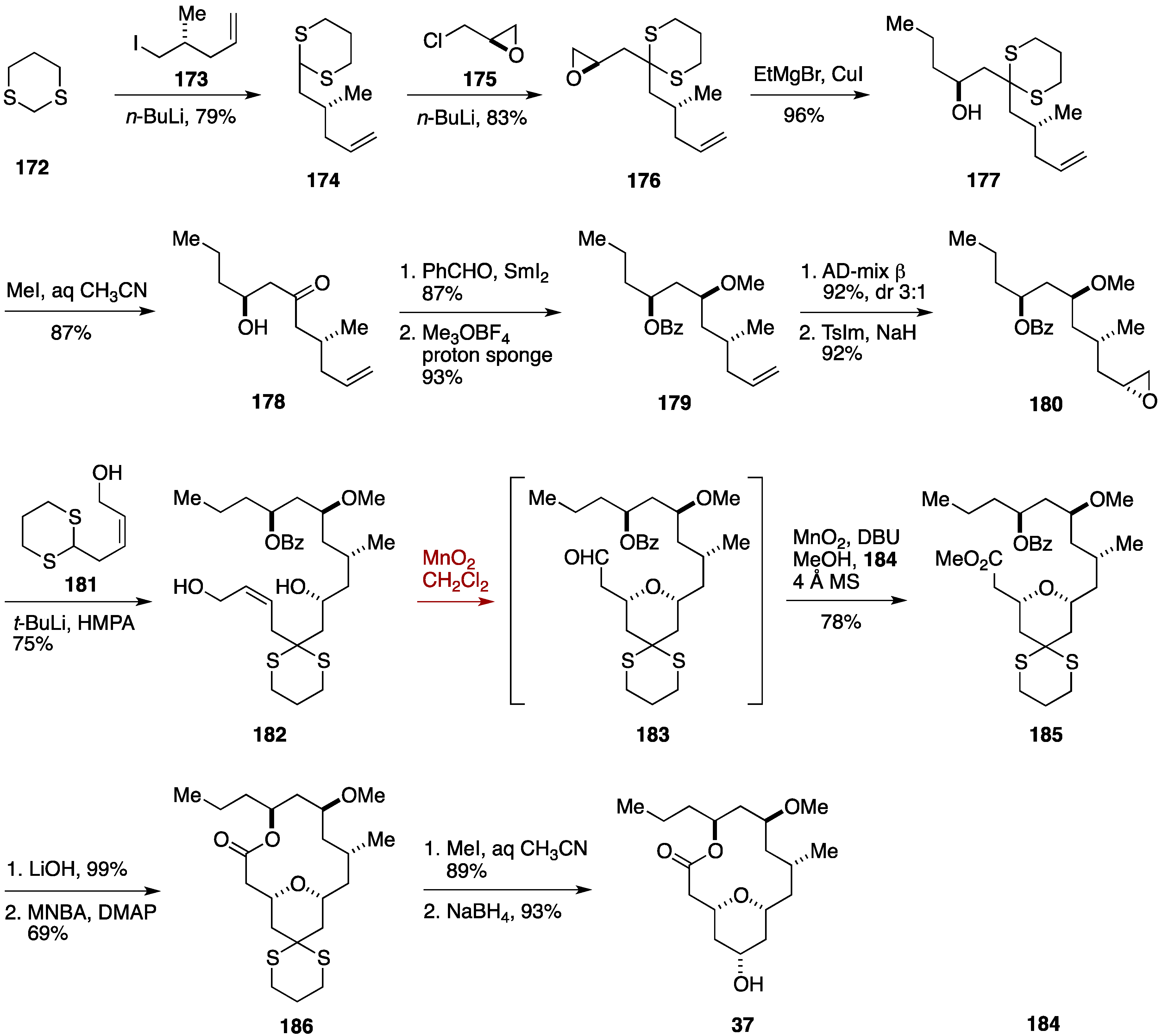
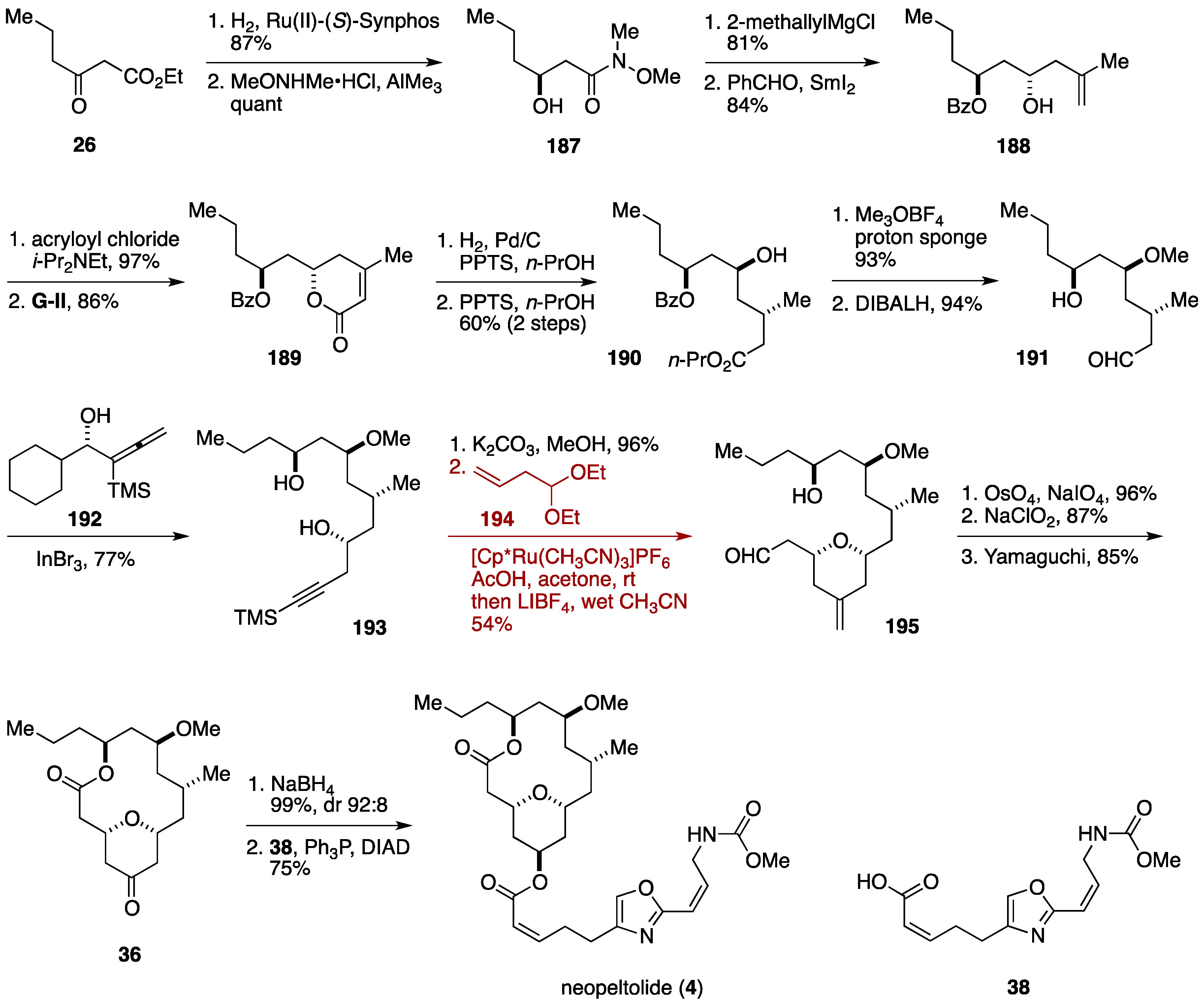
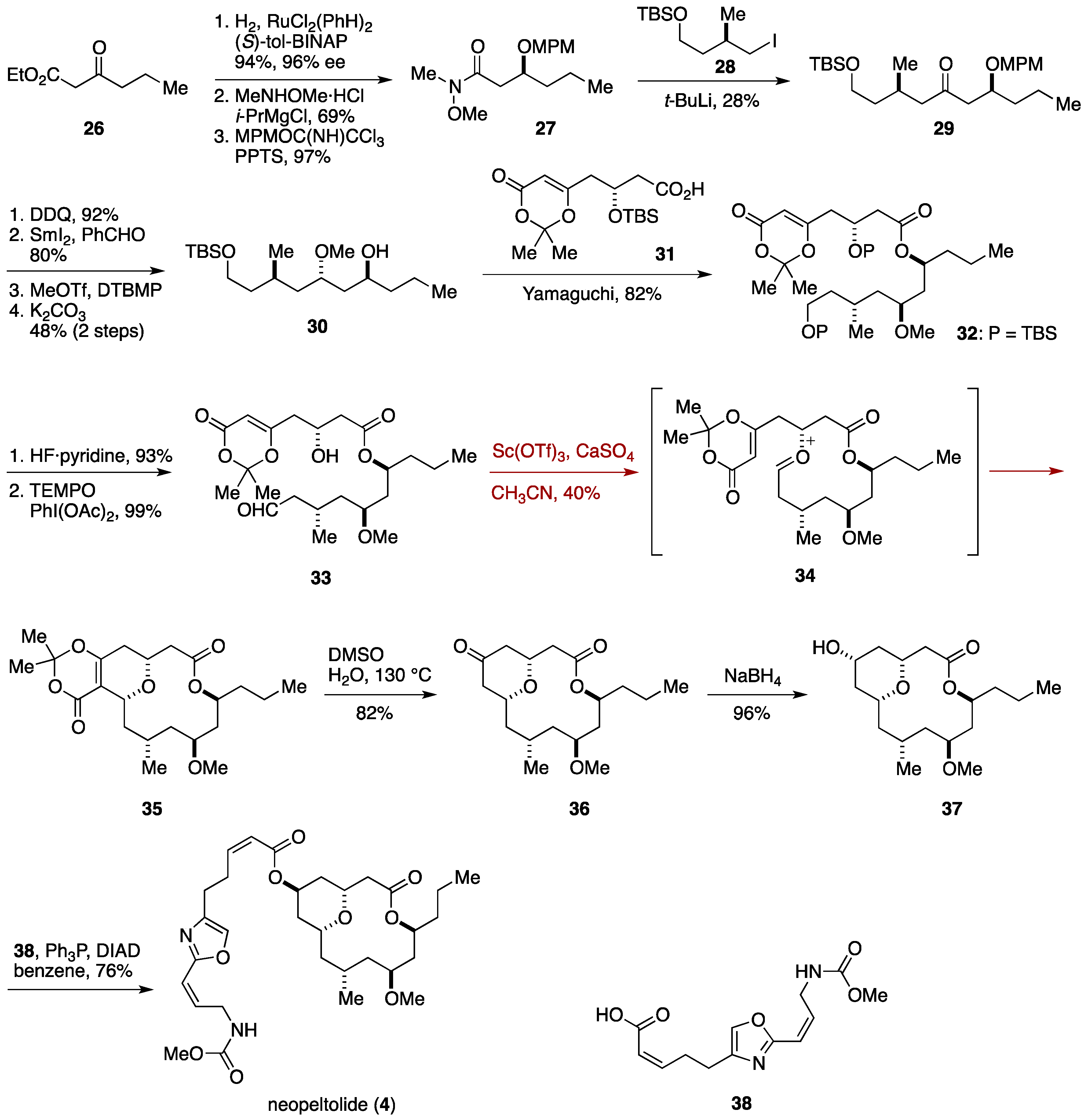
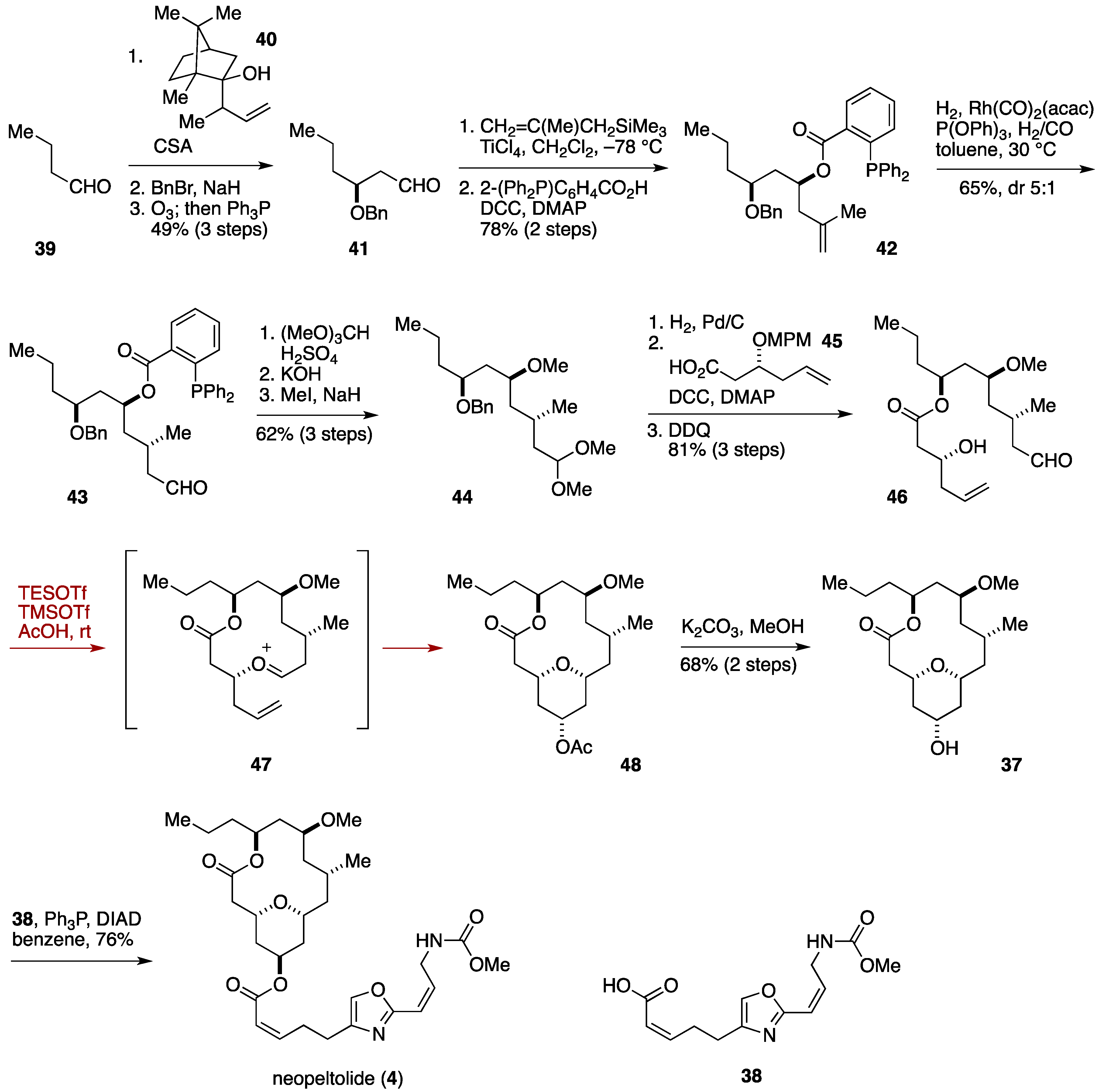
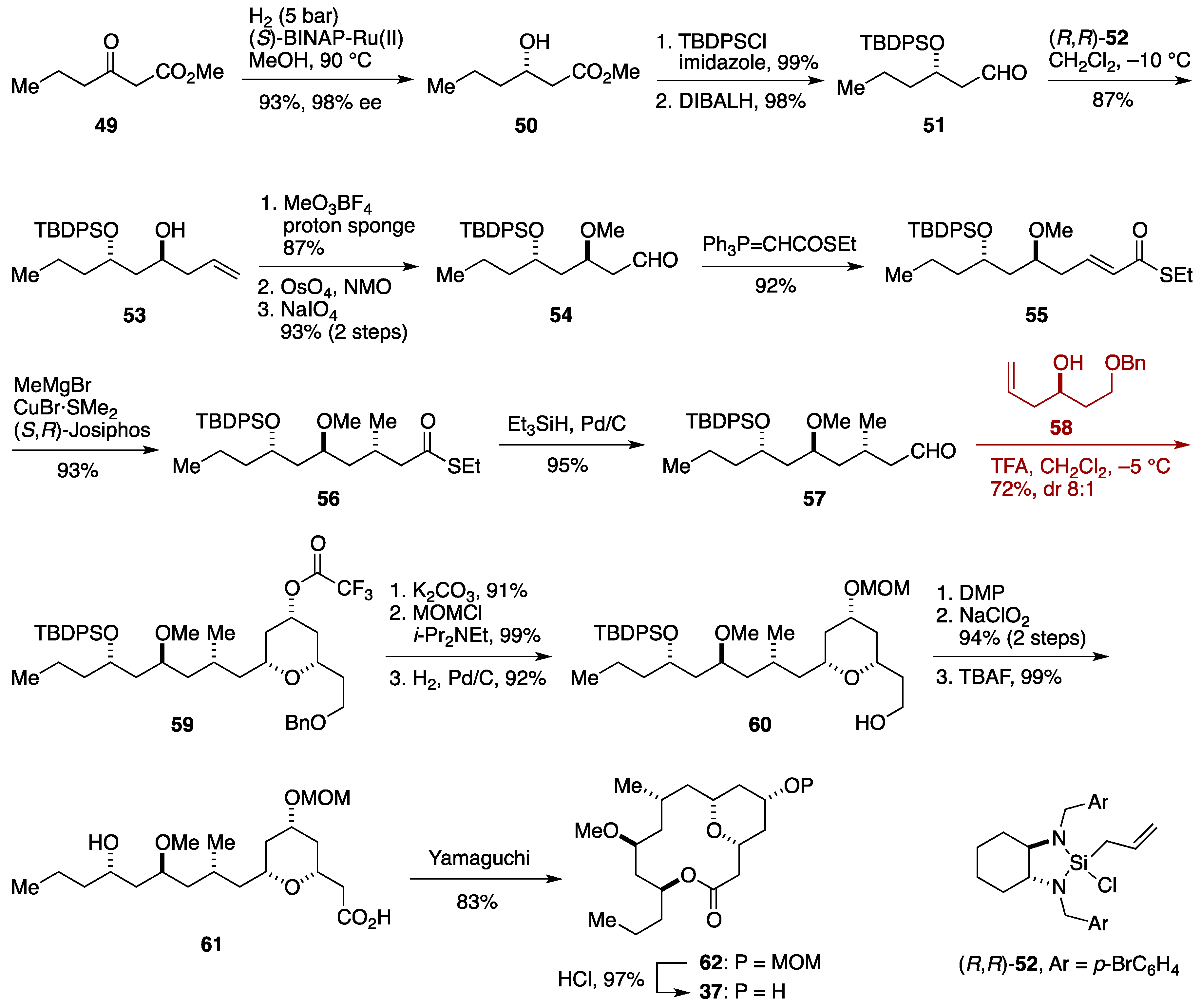
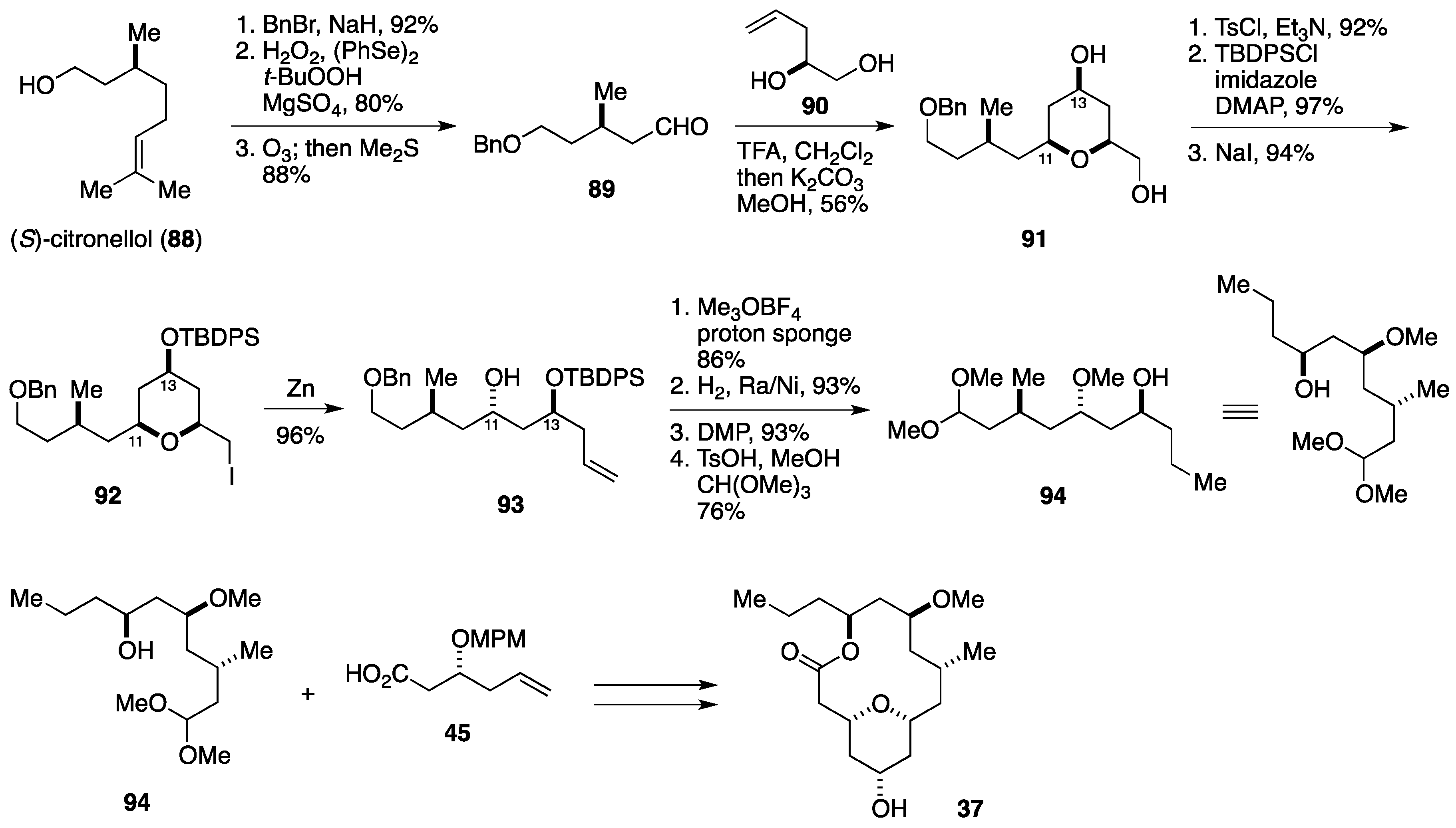
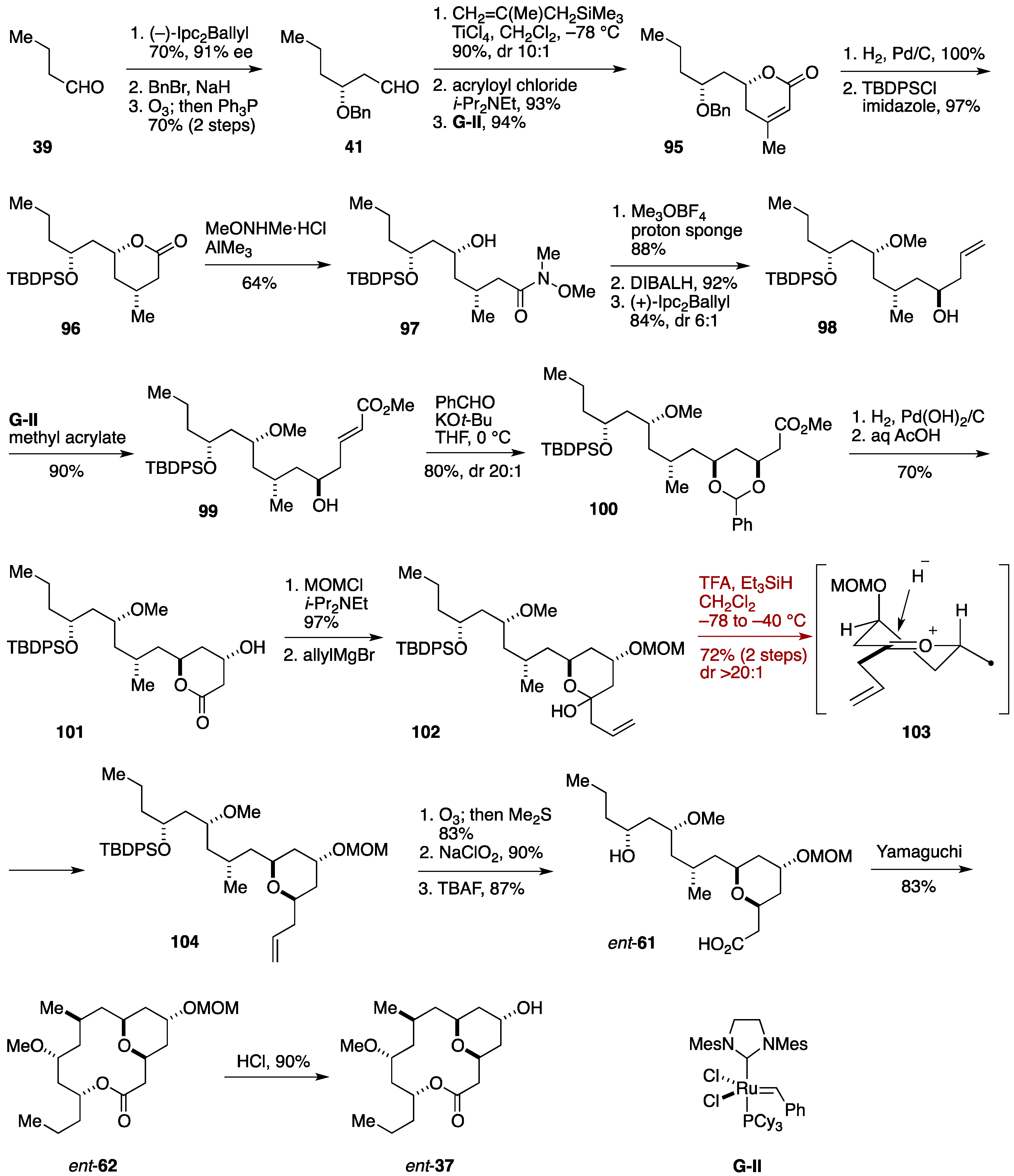
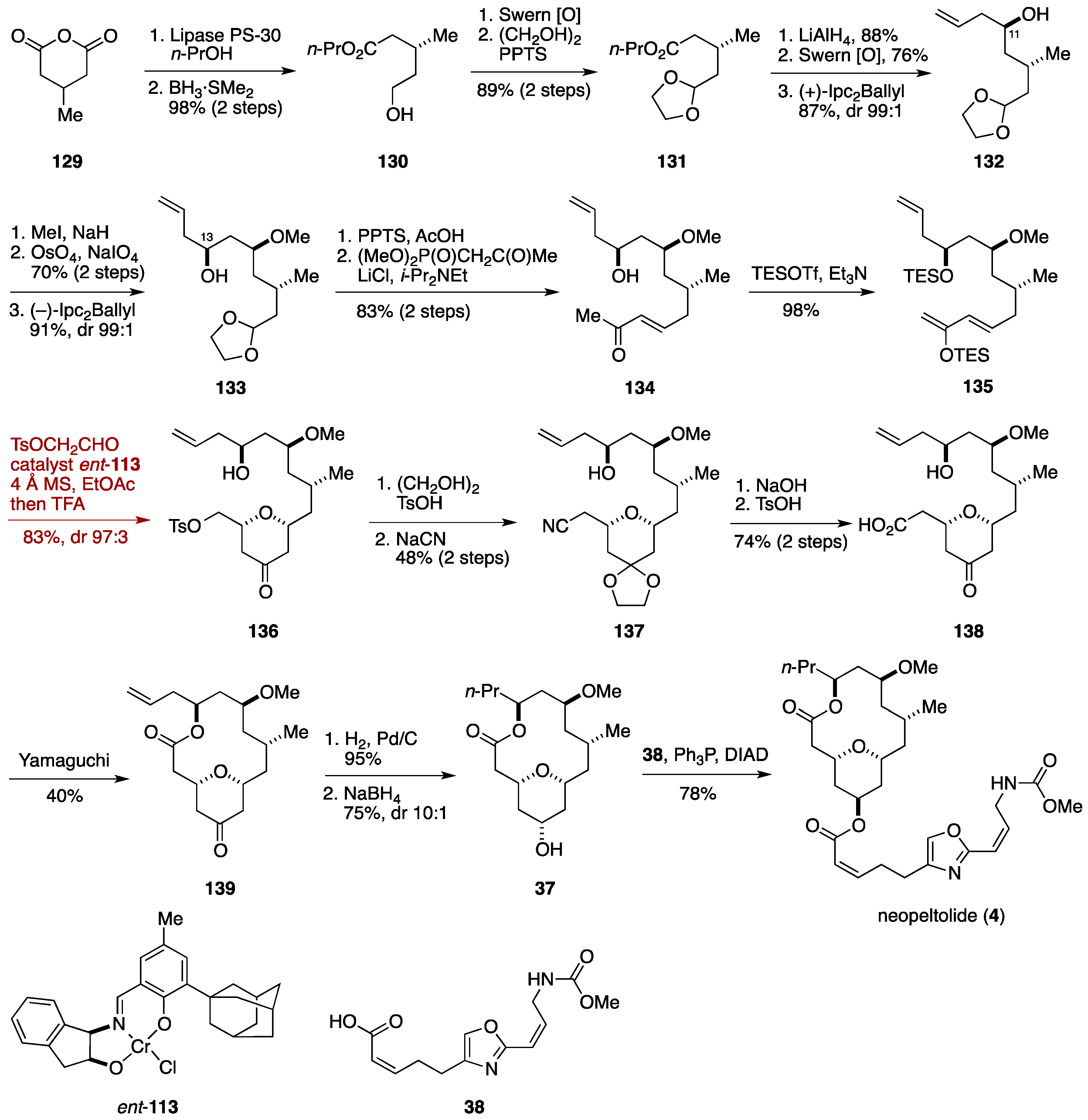
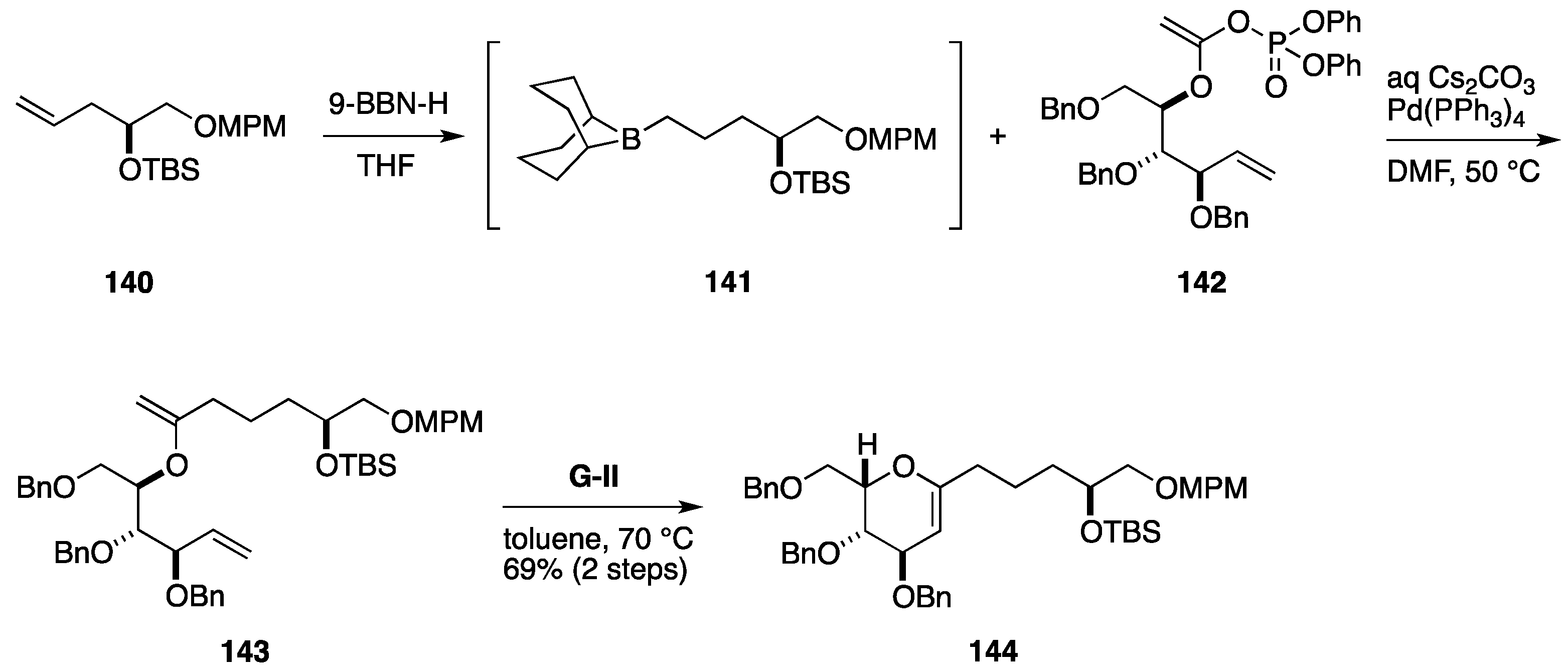
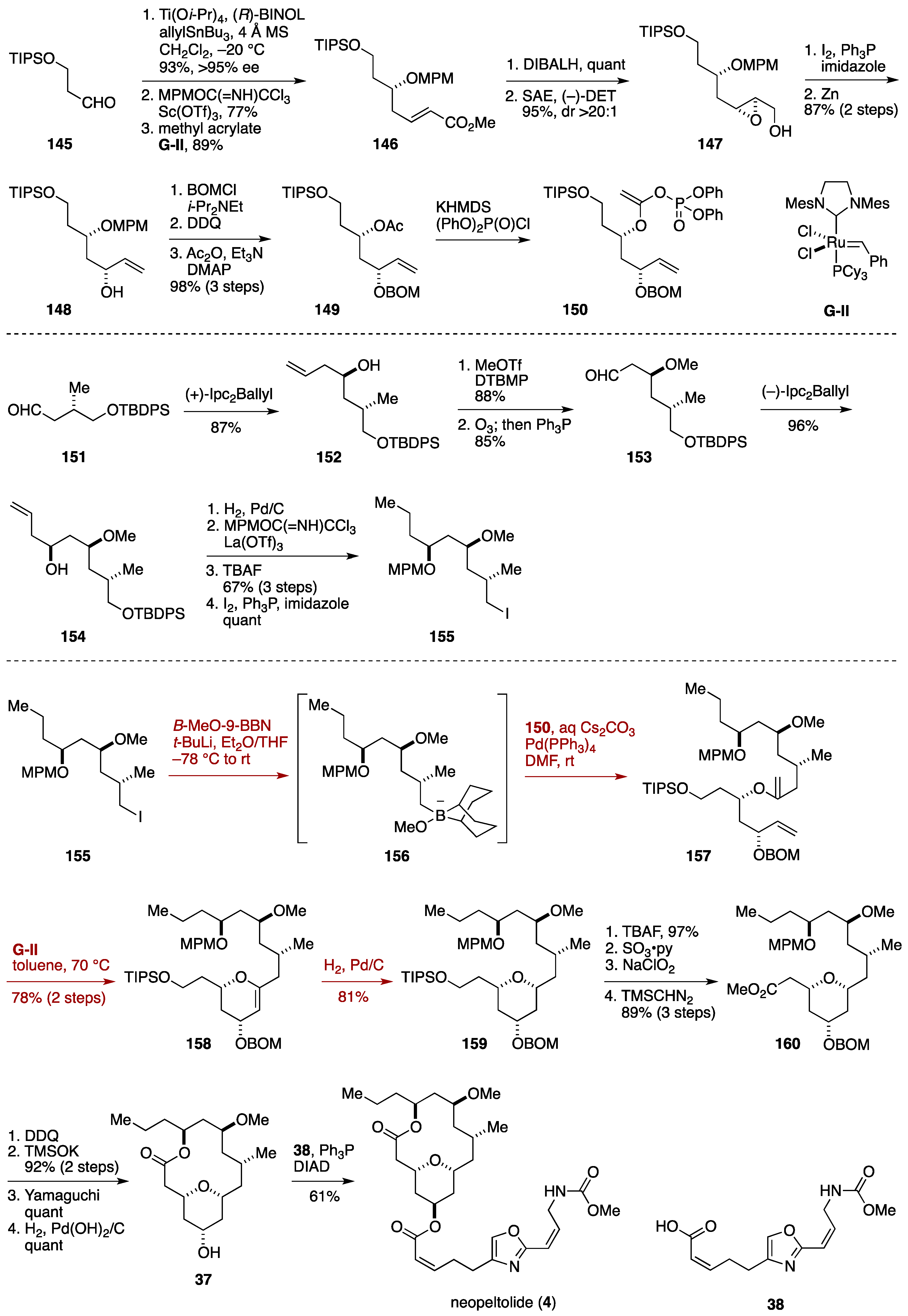
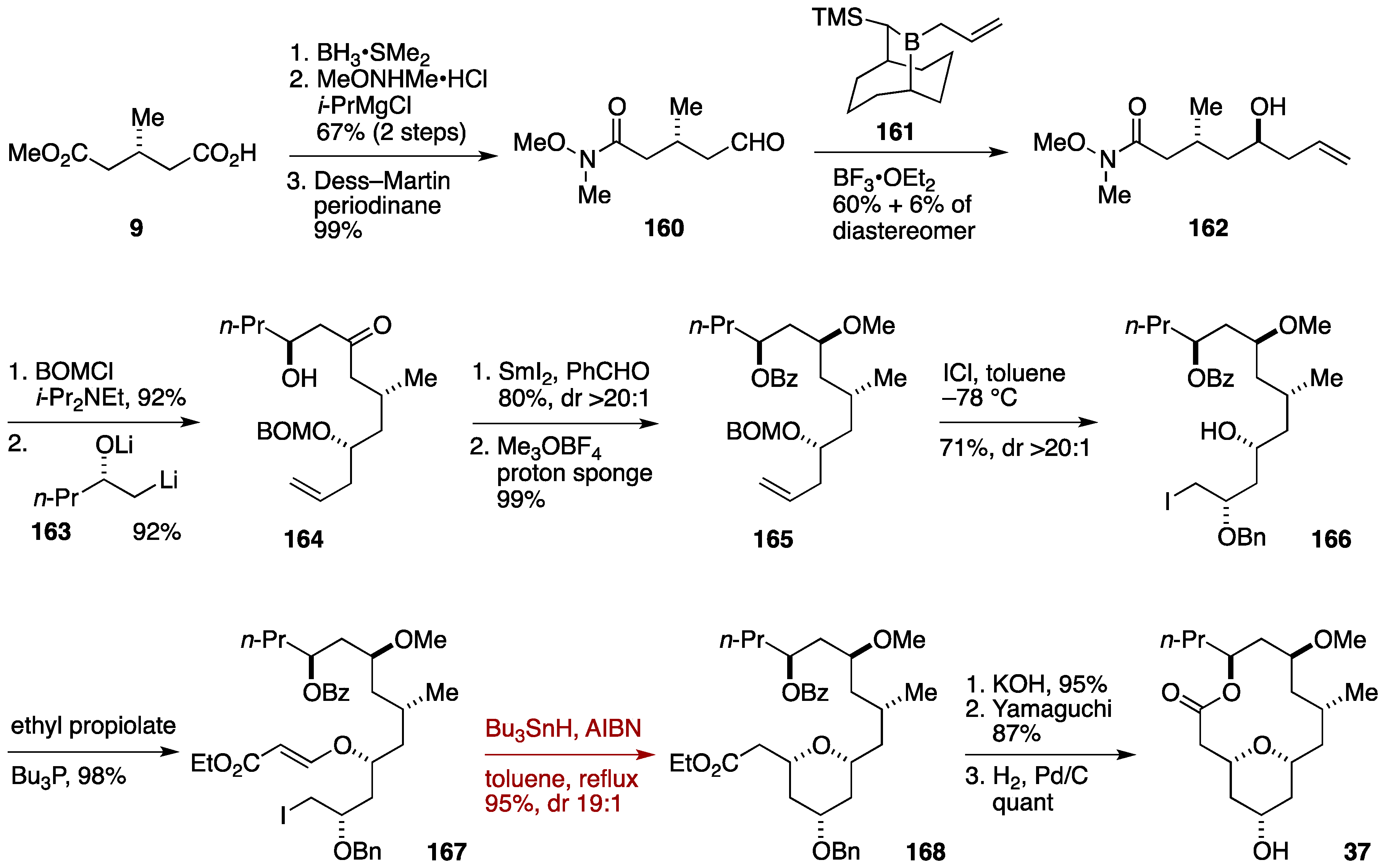
2.3. Total Synthesis by the Lee Group
2.4. Formal Synthesis by the Maier Group
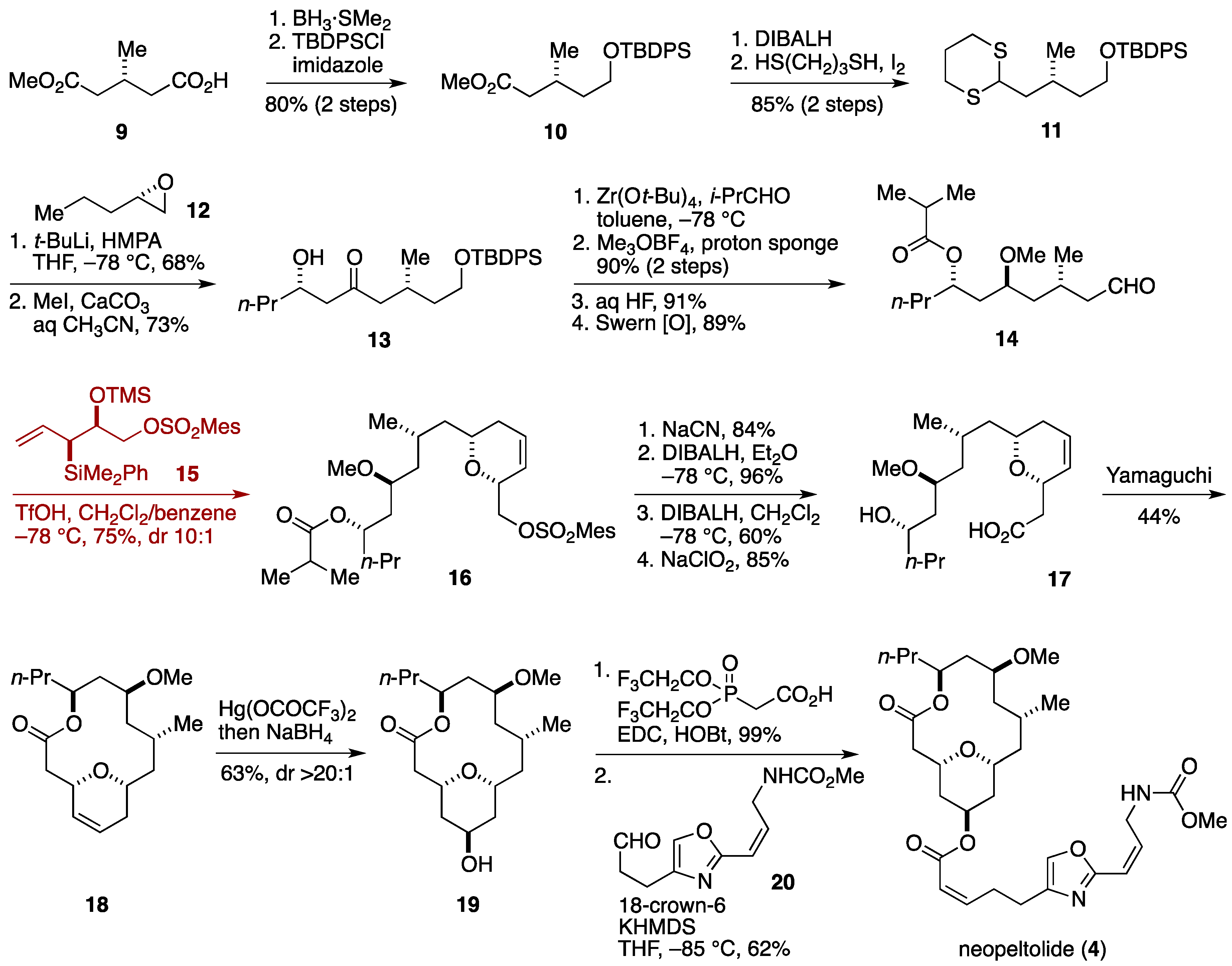
2.5. Total Synthesis by the Kozmin Group
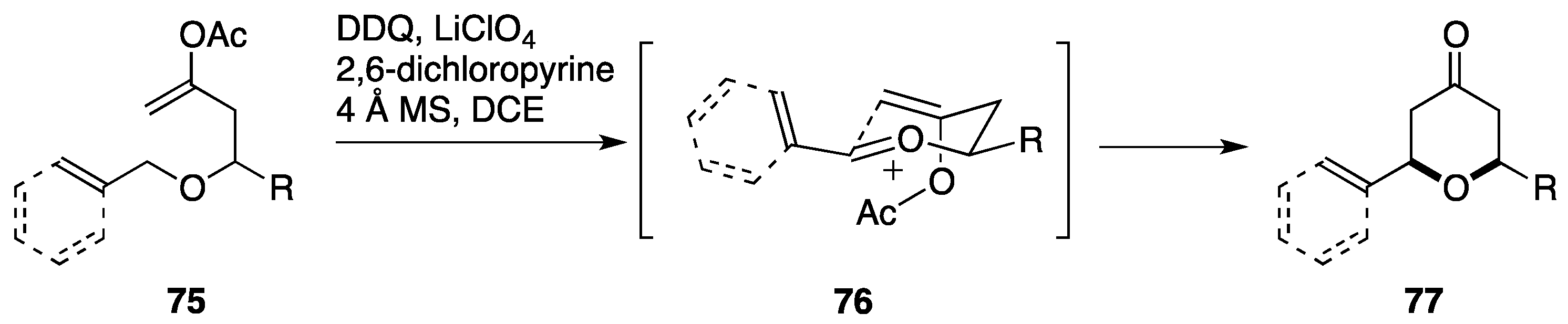
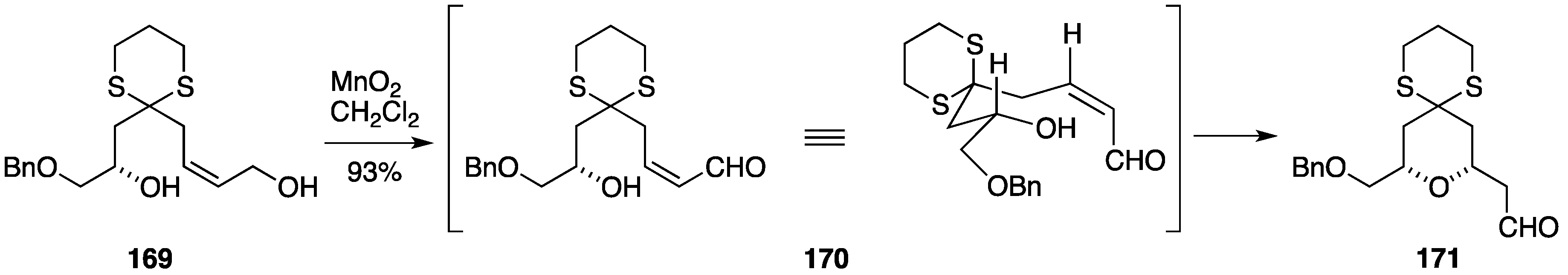
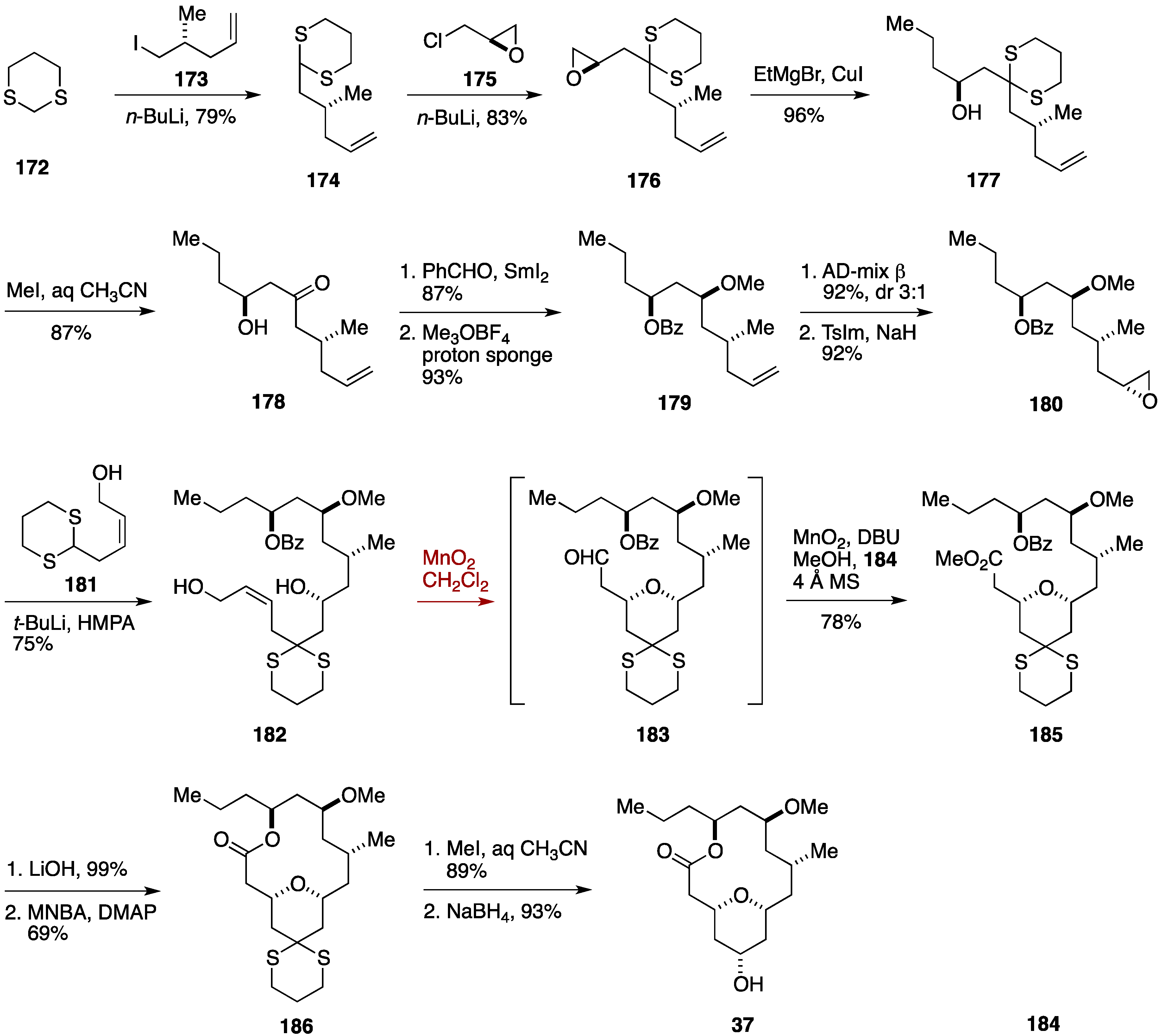
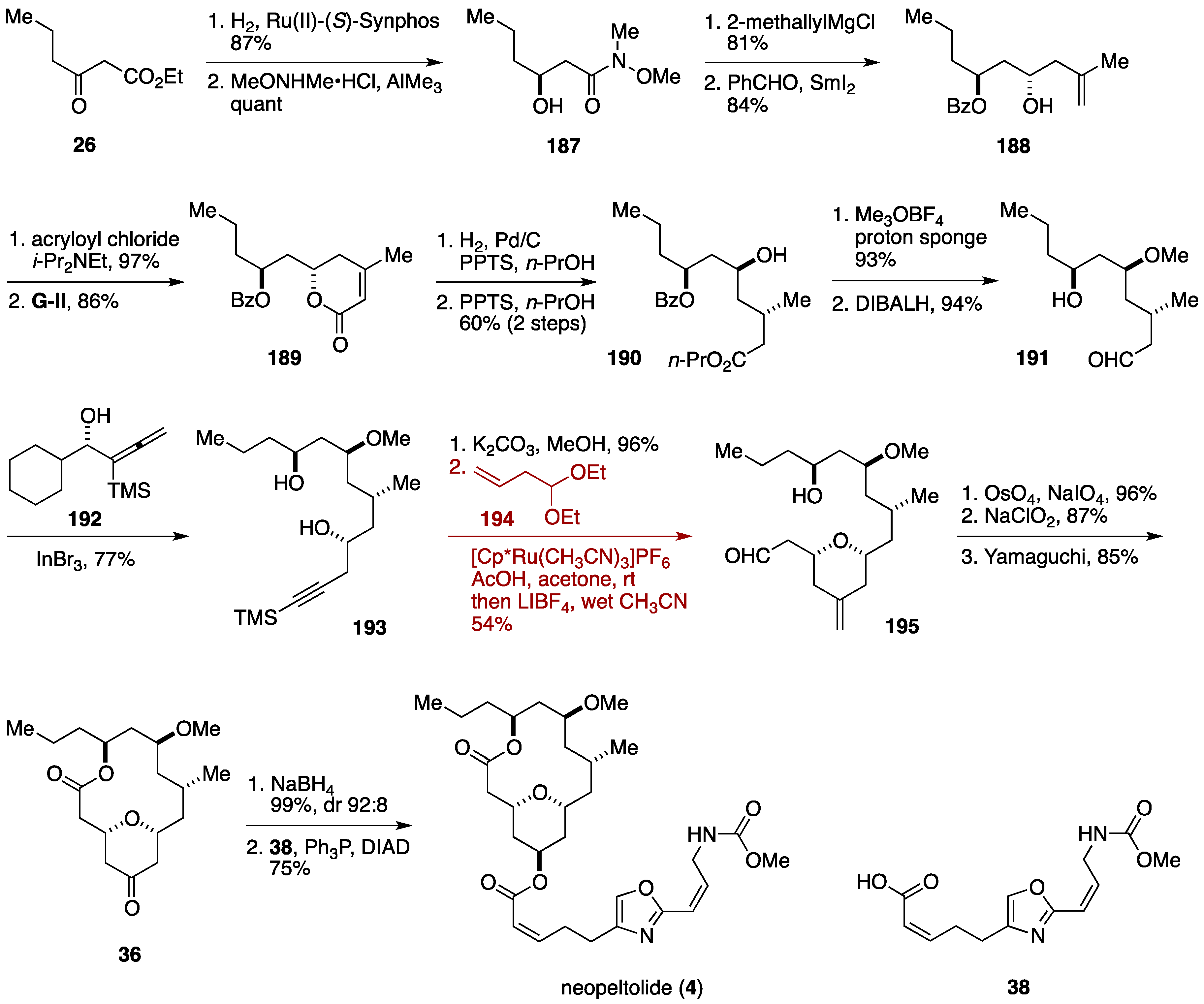
2.6. Formal Synthesis by the Floreancig Group
2.7. Formal Synthesis by the Yadav Group
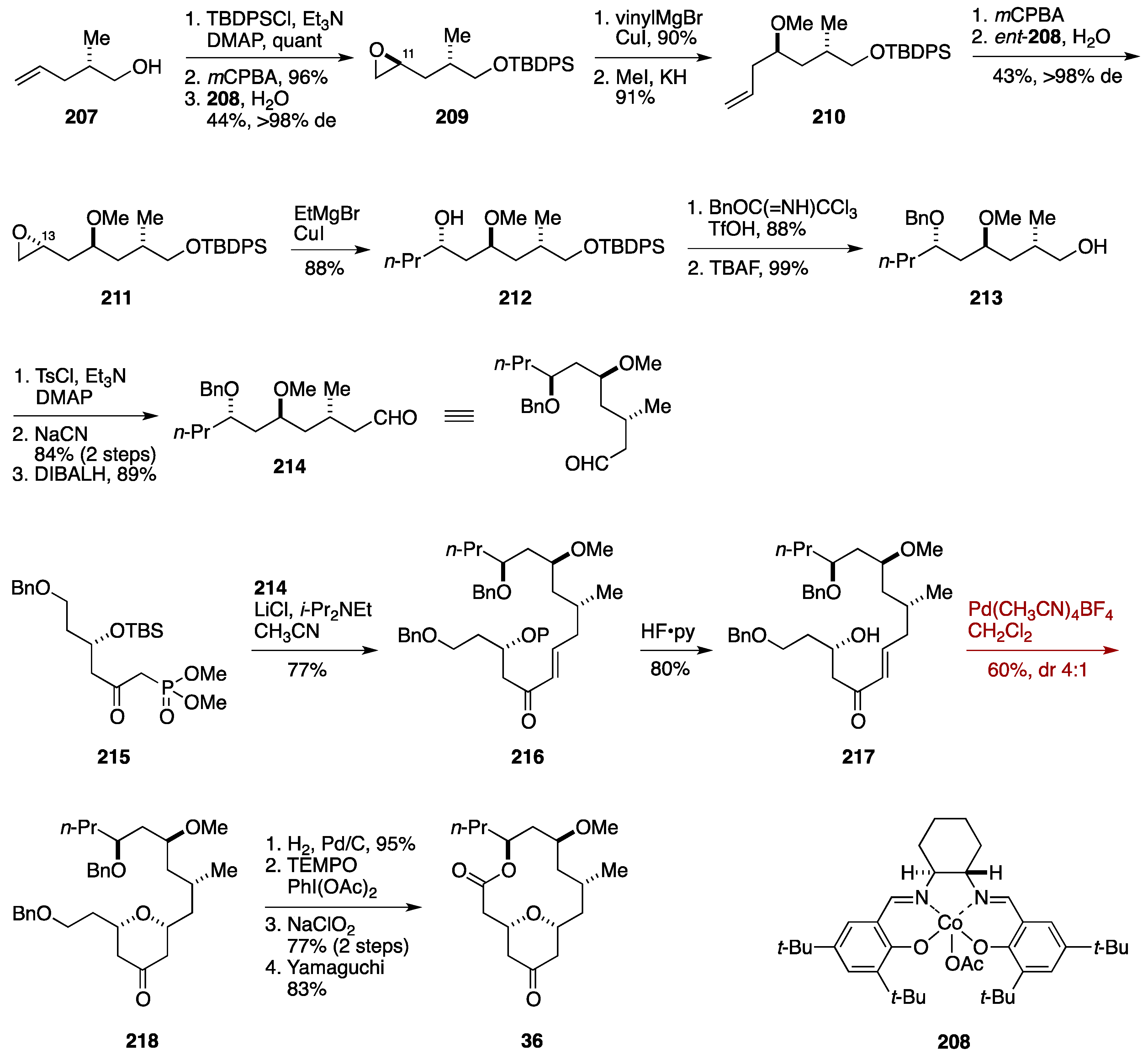
2.8. Formal Synthesis by the Jennings Group
3. Synthesis of Tetrahydropyrans via Hetero-Diels-Alder Cycloaddition
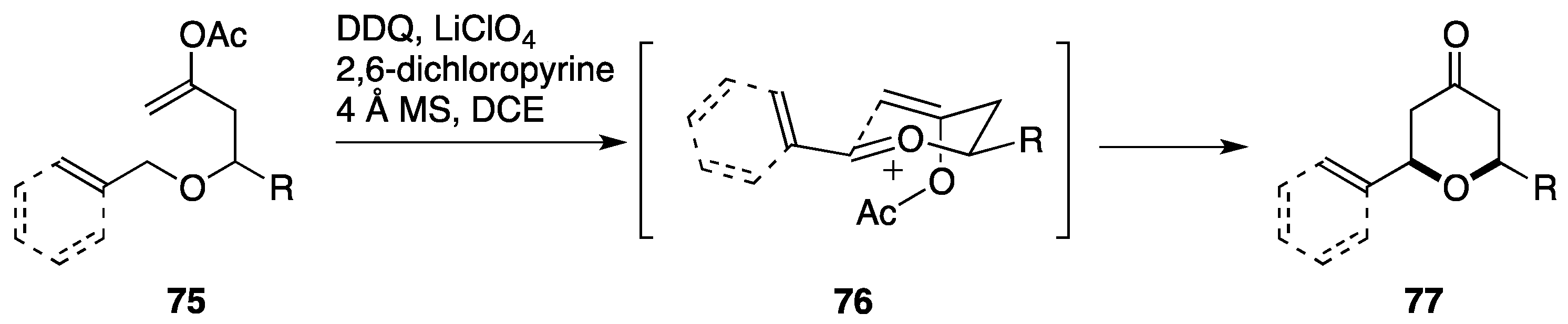
3.1. Total Synthesis by the Paterson Group
3.2. Formal Synthesis by the Raghavan Group
3.3. Total Synthesis by the Arun K. Ghosh Group
4. Synthesis of Tetrahydropyrans via Ring-Closing Metathesis
Total Synthesis by the Fuwa Group (First-Generation Synthesis)
5. Synthesis of Tetrahydropyrans via Intramolecular Radical Cyclizations
Formal Synthesis by the Taylor Group
6. Synthesis of Tetrahydropyrans via Intramolecular Oxa-Michael Reaction
6.1. Formal Synthesis by the Hong Group
6.2. Total Synthesis by the Roulland Group
6.3. Total Synthesis by the Fuwa Group (Second-Generation Synthesis)
6.4. Formal Synthesis by the Subhash Ghosh Group
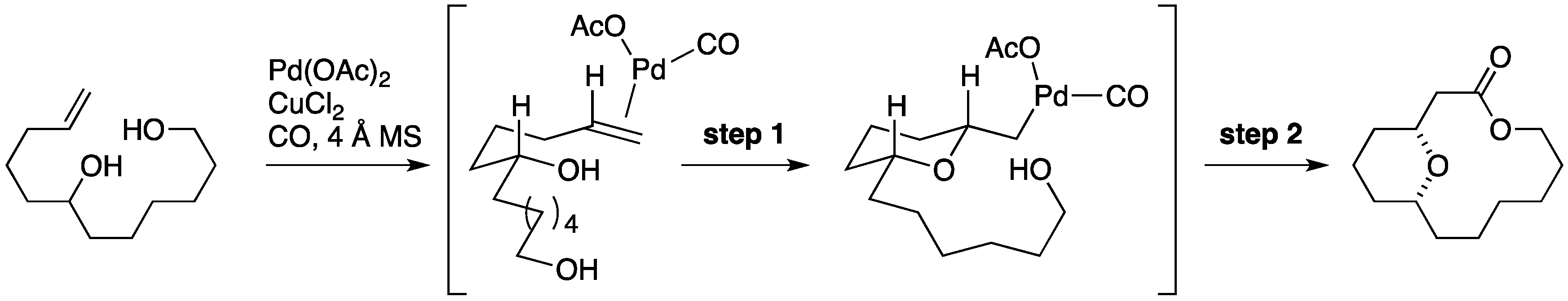
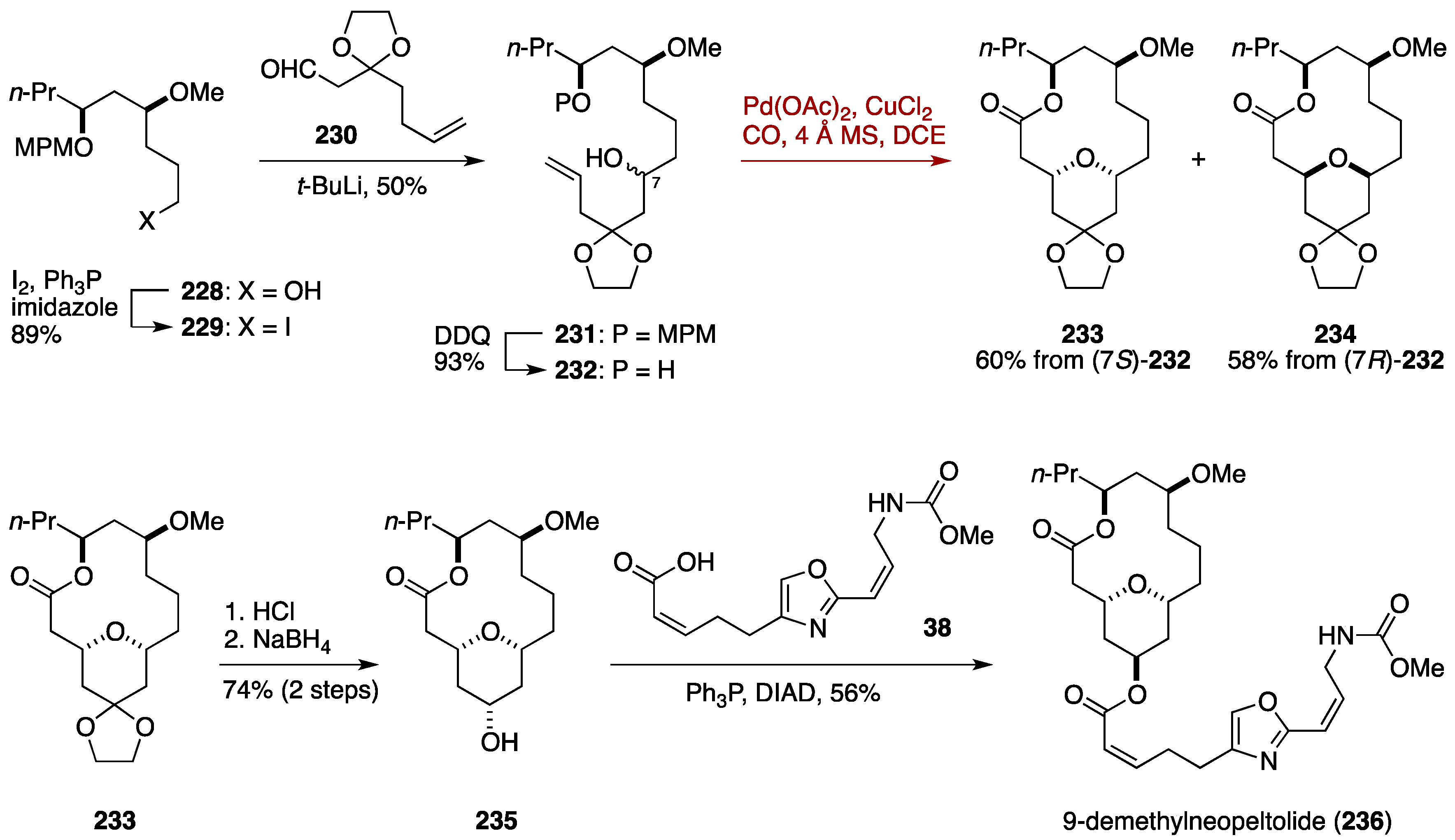
7. Synthesis of Tetrahydropyrans via Palladium-Catalyzed Intramolecular Alkoxycarbonylation
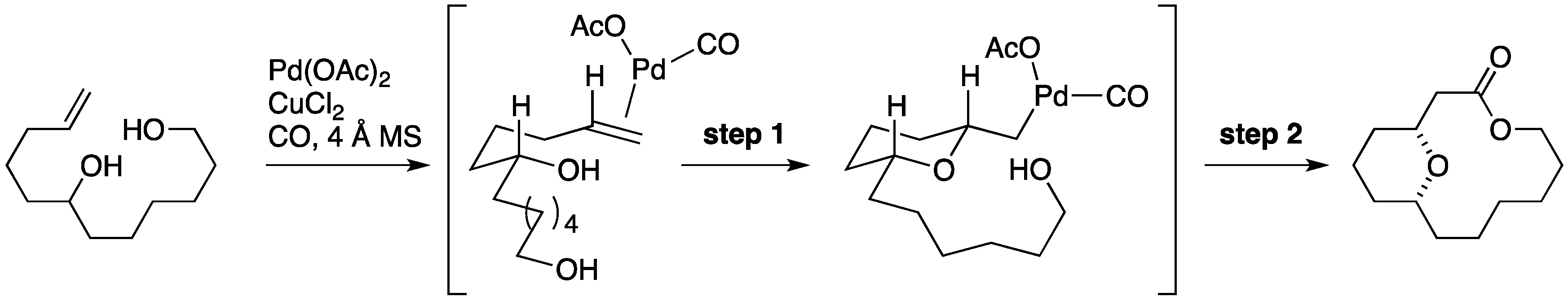
7.1. Formal Synthesis by the She Group
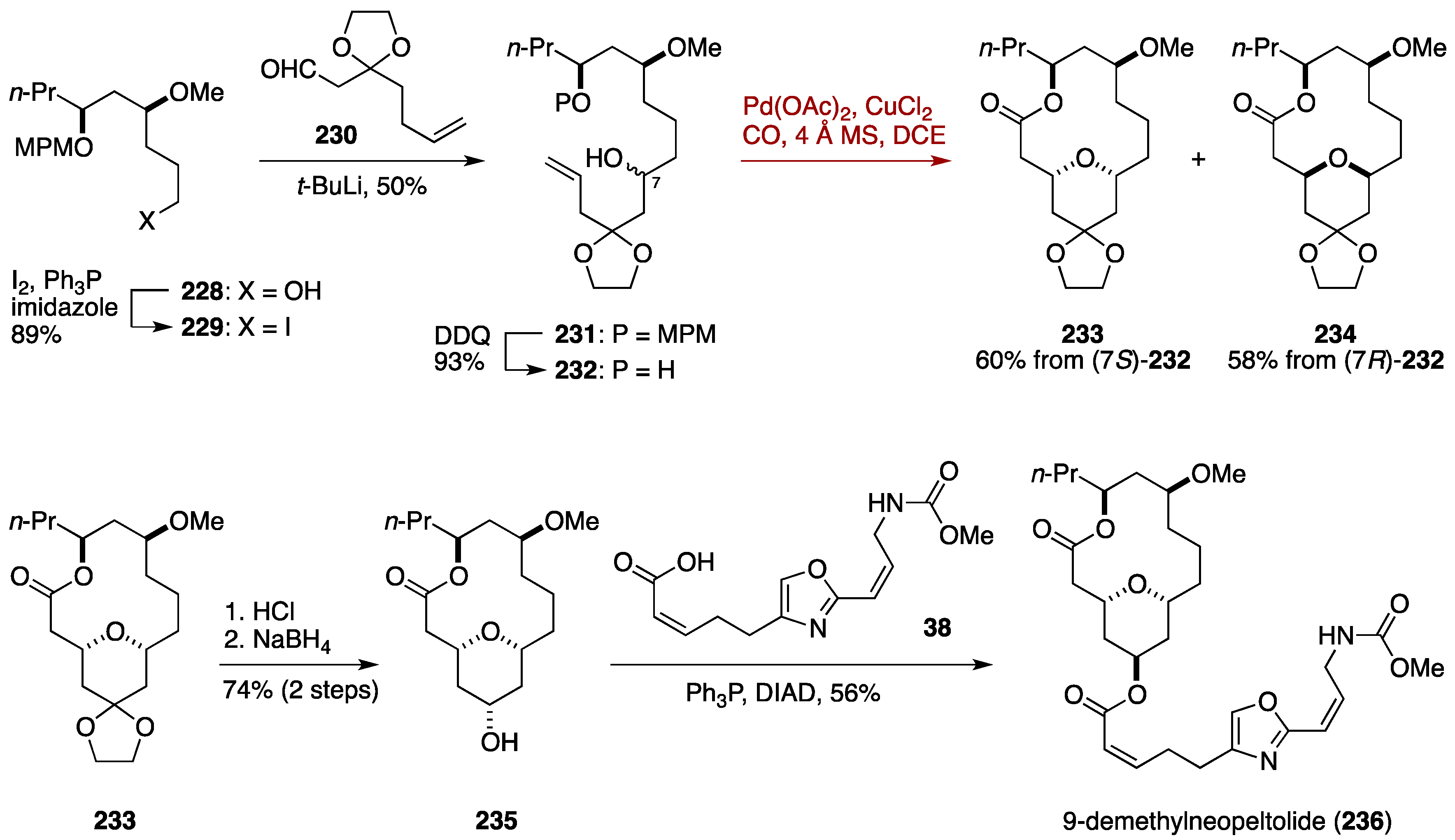
7.2. Total Synthesis of 9-Demethylneopeltolide by the Dai Group
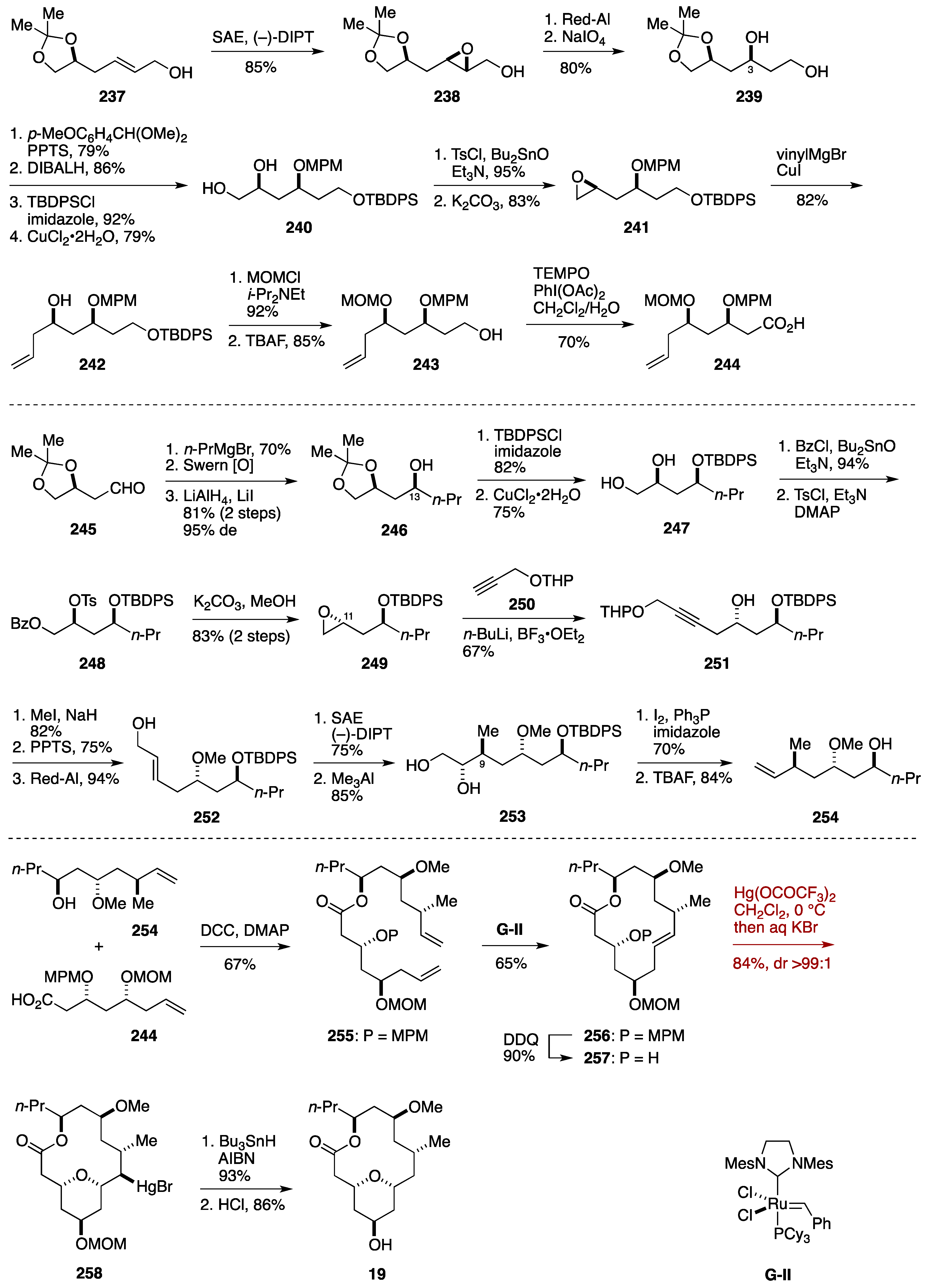
8. Synthesis of Tetrahydropyrans via Transannular Oxymercuration
Formal Synthesis by the Sharma Group
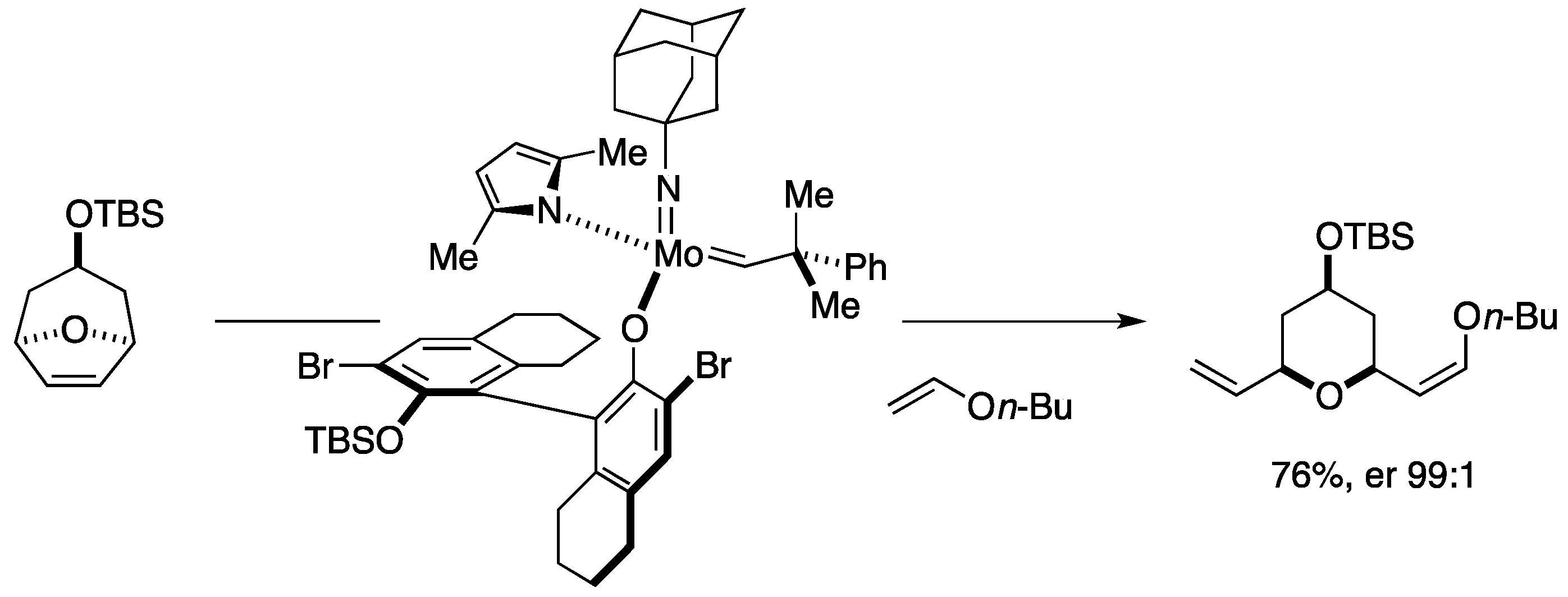
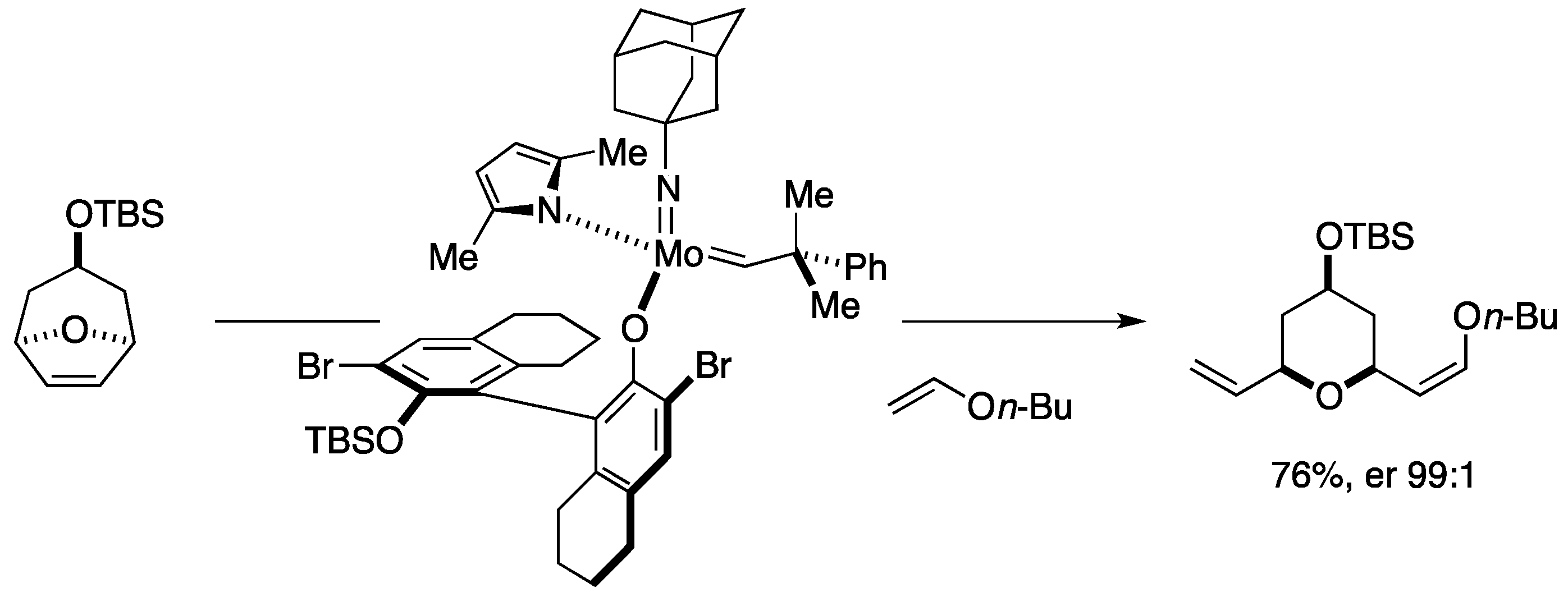
9. Synthesis of Tetrahydropyrans via Desymmetrizing Ring-Opening/Cross-Metathesis Cascade
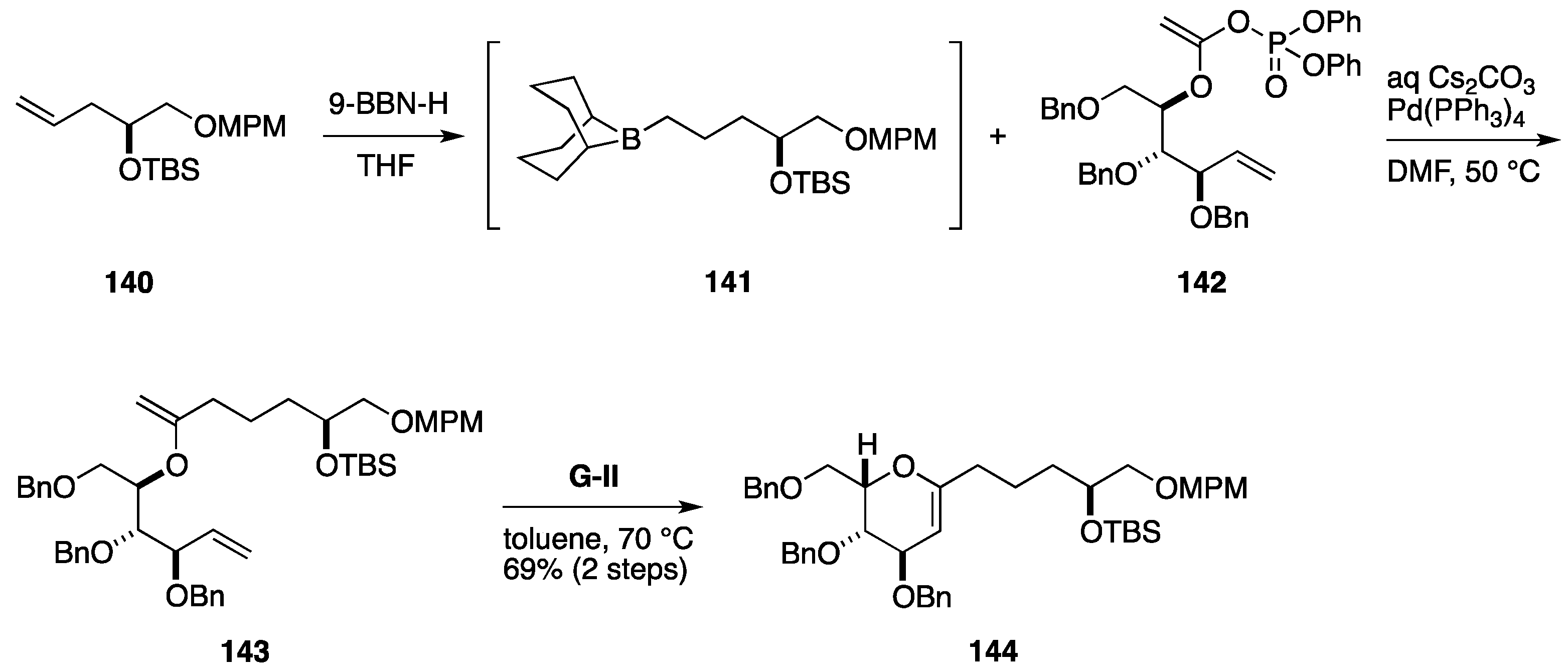
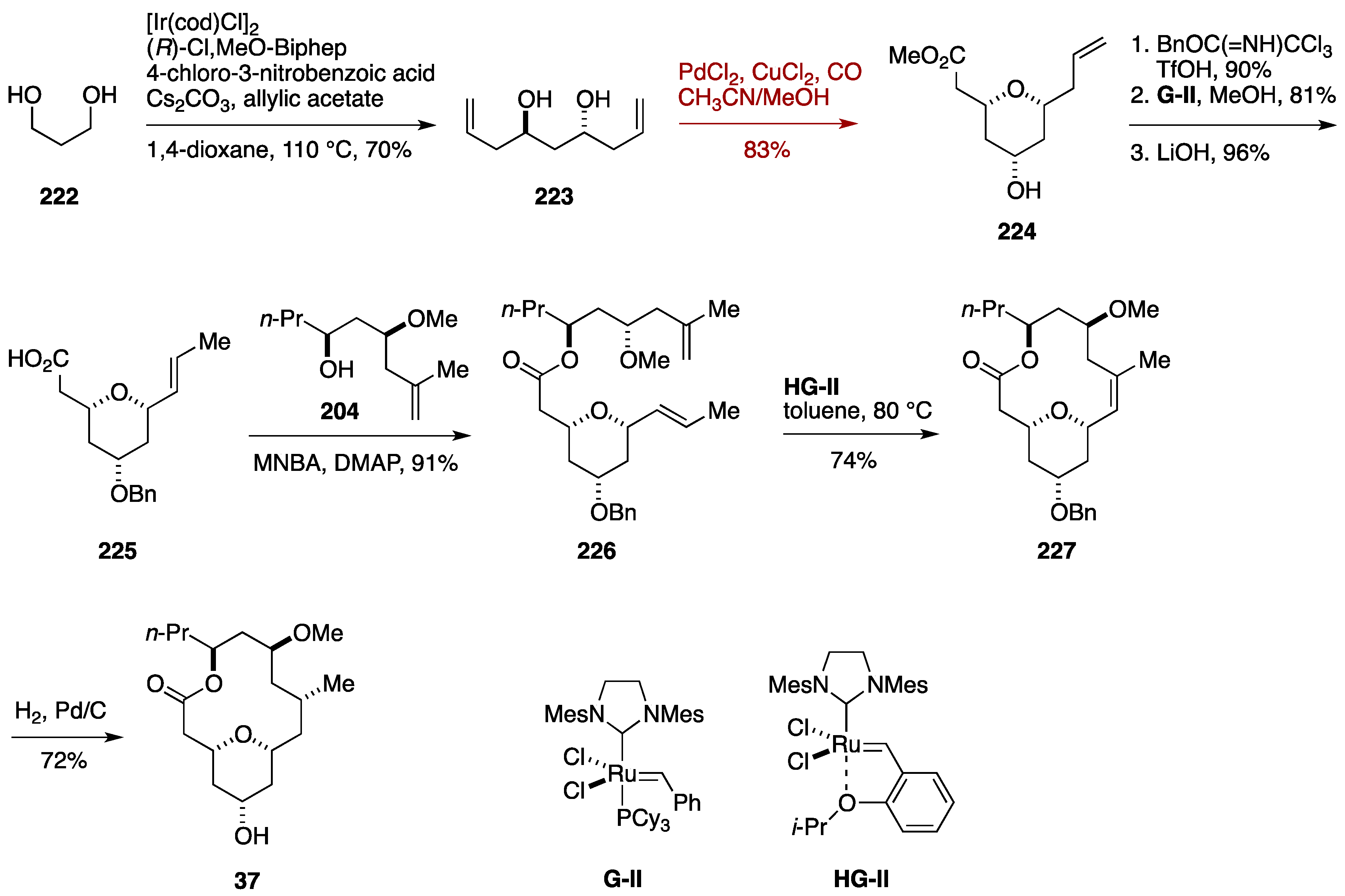
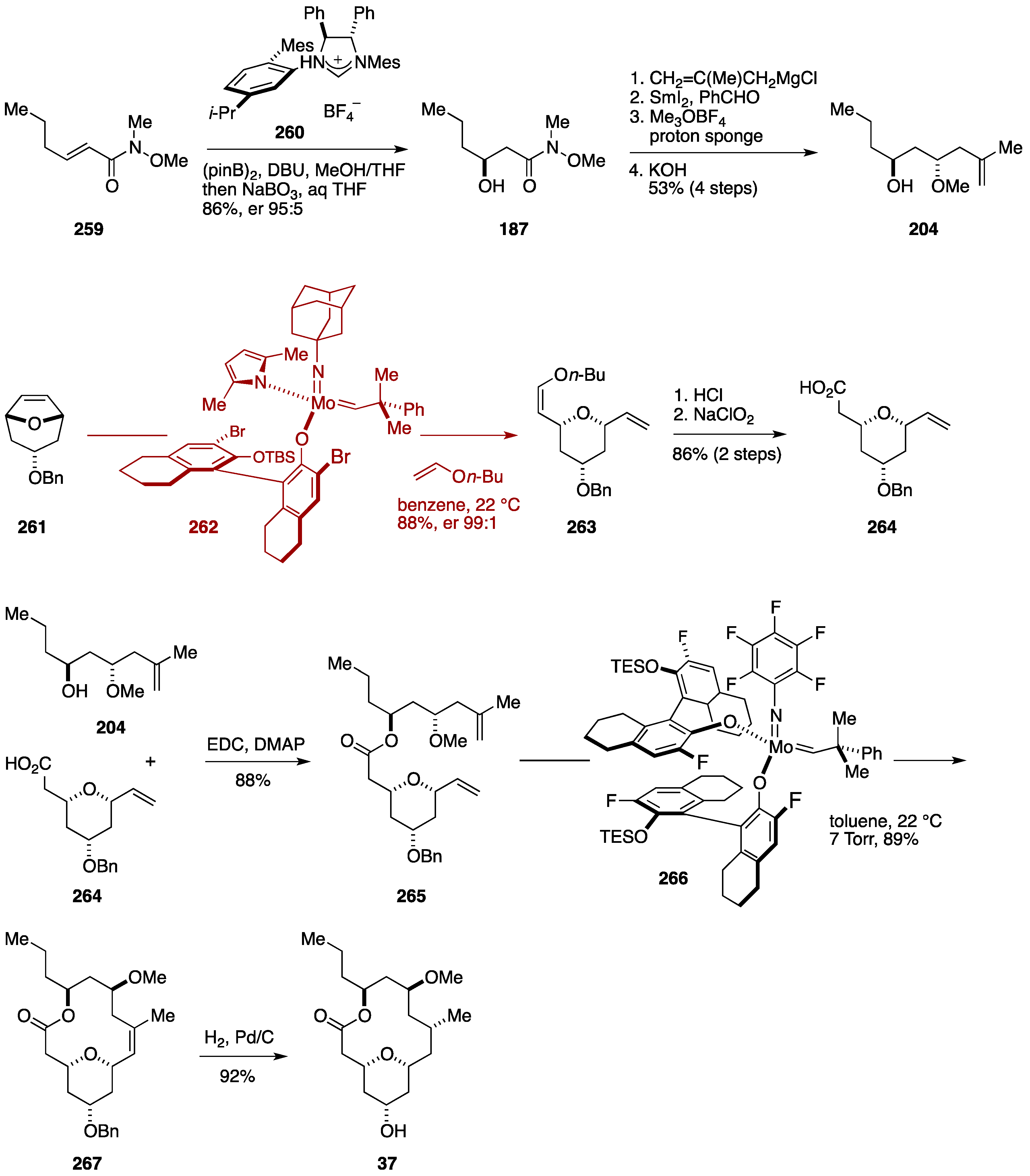
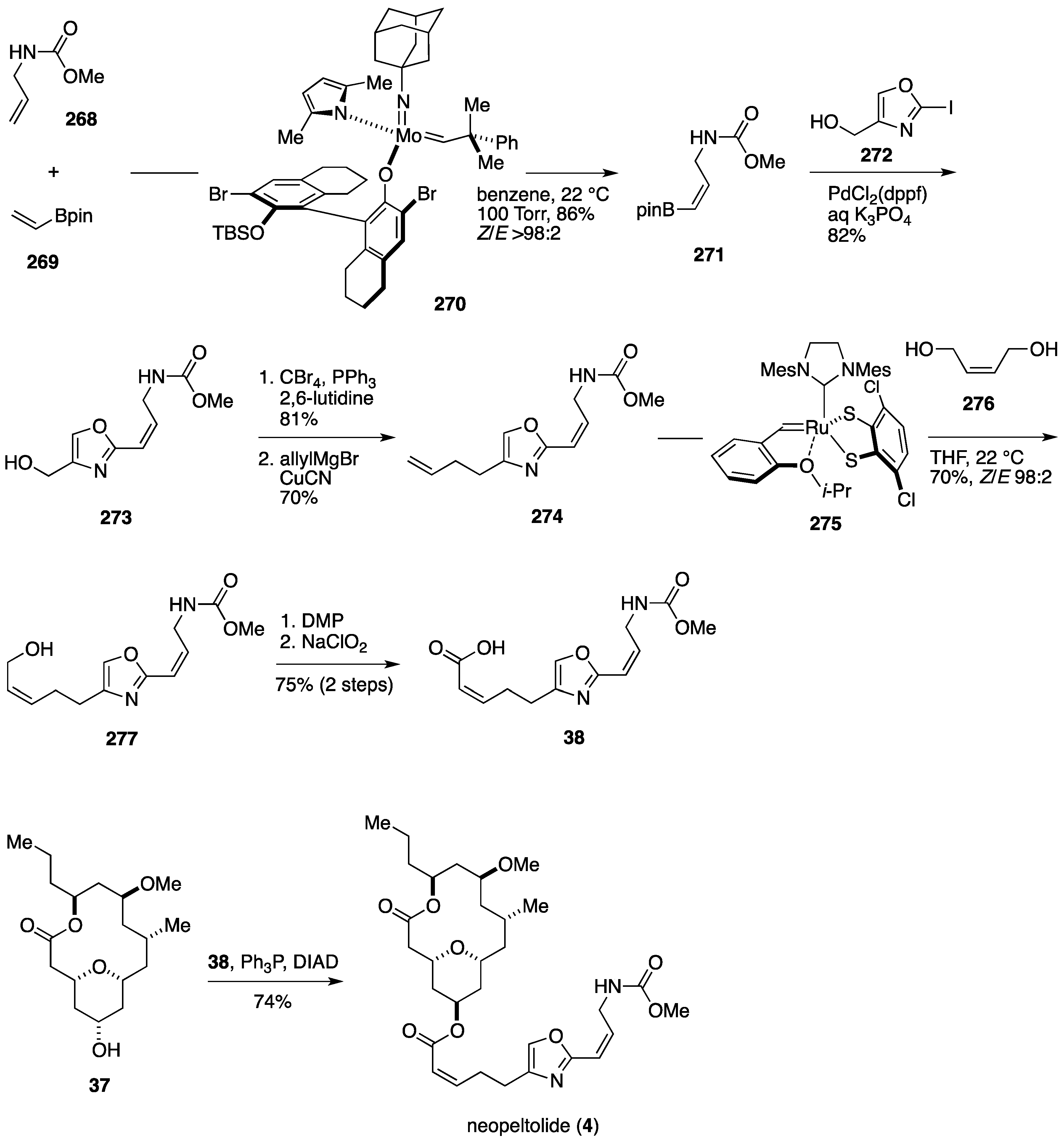
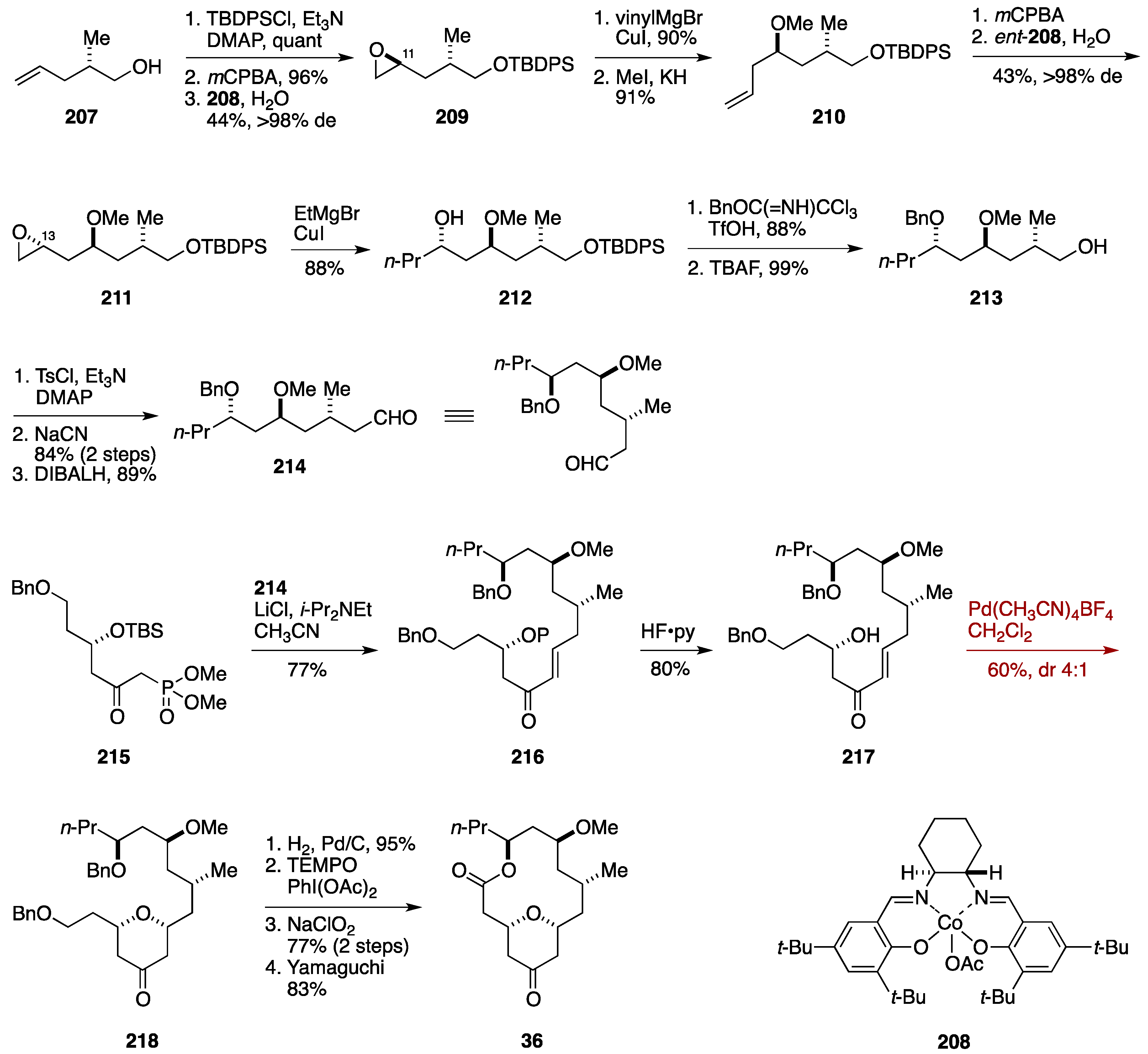
Total Synthesis by the Hoveyda Group
10. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| 9-BBN | 9-borabicyclo[3.3.1]nonyl |
| acac | acetylacetone |
| AIBN | 2,2′-azobis(isobutyronitrile) |
| BINAP | 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl |
| BINOL | 1,1′-bi-2-naphthol |
| Biphep | 2,2′-bis(diphenylphosphino)-1,1′-biphenyl |
| Bn | benzyl |
| BOM | benzyloxymethyl |
| Bz | benzoyl |
| cod | 1,5-cyclooctadienyl |
| Cp* | 1,2,3,4,5-pentamethylcyclopentadienyl |
| CSA | 10-camphorsulfonic acid |
| Cy | cyclohexyl |
| DBB | 4,4′-di-t-butylbiphenyl |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| DCC | dicyclohexylcarbodiimide |
| DCE | 1,2-dichloroethane |
| DDQ | 2,3-dichloro-5,6-dicyanobenzoquinone |
| DEAD | diethyl azodicarboxylate |
| DET | diethyl tartrate |
| DIAD | diisopropyl azodicarboxylate |
| DIBALH | diisobutylaluminum hydride |
| DIPT | diisopropyl tartrate |
| DMAP | 4-dimethylaminopyridine |
| DMF | N,N-dimethylformamide |
| DMP | Dess-Martin periodinane |
| DMSO | dimethylsulfoxide |
| dppf | 1,1′-bis(diphenylphosphino)ferrocene |
| DTBMP | 2,6-di-t-butyl-4-methylpyridine |
| DVDS | 1,3-divinyl-1,1,3,3-tetramethyldisiloxane |
| EDC | 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride |
| HMPA | hexamethylphosphoramide |
| HOBt | 1-hydroxybenzotriazole |
| IBX | 2-iodoxybenzoic acid |
| Ipc | isopinocampheyl |
| (S,R)-Josiphos | (S)-1-[(Rp)-2-(diphenylphosphino)ferrocenyl]ethyldicyclohexylphosphine |
| KHMDS | potassium hexamethyldisilazide |
| LDA | lithium diisopropylamide |
| mCPBA | m-chloroperbenzoic acid |
| Mes | mesityl |
| MNBA | 2-methyl-6-nitrobenzoic anhydride |
| MOM | methoxymethyl |
| MPM | p-methoxyphenylmethyl |
| MS | molecular sieves |
| NMO | N-methylmorpholine N-oxide |
| pin | pinacol |
| PPTS | pyridinium p-toluenesulfonate |
| Ra/Ni | Raney-Nickel catalyst |
| Red-Al | sodium bis(2-methoxyethoxy)aluminum hydride |
| SAE | Sharpless asymmetric epoxidation |
| Synphos | [(5,6),(5′,6′)-bis(ethylenedioxy)biphenyl-2,2′-diyl]bis(diphenylphosphine) |
| TBAF | tetra-n-butylammonium fluoride |
| TBDPS | t-butyldiphenylsilyl |
| TBS | t-butyldimethylsilyl |
| TEMPO | 2,2,6,6-tetramethylpiperidin-1-oxyl radical |
| TES | triethylsilyl |
| Tf | trifluoromethanesulfonyl |
| TFA | trifluoroacetic acid |
| TMS | trimethylsilyl |
| Ts | p-toluenesulfonyl |
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.M.; Xu, R.F.; Gu, Y.C.; Wang, C.Y.; Shao, C.L. Biological and chemical diversity of coral-derived microorganisms. Curr. Med. Chem. 2015, 22, 3707–3762. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, J.; Hu, G.; Yu, J.; Zhu, X.; Lin, Y.; Chen, S.; Yuan, J. Statistical research on the bioactivity of new marine natural products discovered during the 28 Years from 1985 to 2012. Mar. Drugs 2015, 13, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Mehbub, M.F.; Lei, J.; Franco, C.; Zhang, W. Marine sponge derived natural products between 2001 and 2010: Trends and opportunities for discovery of bioactives. Mar. Drugs 2014, 12, 4539–4577. [Google Scholar] [CrossRef] [PubMed]
- Skropeta, D.; Wei, L. Recent advances in deep-sea natural products. Nat. Prod. Rep. 2014, 31, 999–1025. [Google Scholar] [CrossRef] [PubMed]
- Sakai, R.; Swanson, G.T. Recent progress in neuroactive marine natural products. Nat. Prod. Rep. 2014, 31, 273–309. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Moore, B.S. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M. Chemical biology of natural products on the basis of identification of target proteins. Chem. Lett. 2012, 41, 658–666. [Google Scholar] [CrossRef]
- Satake, M.; Murata, M.; Yasumoto, T. Gambierol: A new toxic polyether compound isolated from the marine dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1993, 115, 361–362. [Google Scholar] [CrossRef]
- Morohashi, A.; Satake, M.; Yasumoto, T. The absolute configuration of gambierol, a toxic marine polyether from the dinoflagellate, Gambierdiscus toxicus. Tetrahedron Lett. 1998, 39, 97–100. [Google Scholar] [CrossRef]
- Ghiaroni, V.; Sasaki, M.; Fuwa, H.; Rossini, G.P.; Scalera, G.; Yasumoto, T.; Pietra, P.; Bigiani, A. Inhibition of voltage-gated potassium currents by gambierol in mouse taste cells. Toxicol. Sci. 2005, 85, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Cuypers, E.; Abdel-Mottaleb, Y.; Kopljar, I.; Rainier, J.D.; Raes, A.L.; Snyders, D.J.; Tytgat, J. Gambierol, a toxin produced by the dinoflagellate Gambierdiscus toxicus, is a potent blocker of voltage-gated potassium channels. Toxicon 2008, 51, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Kopljar, I.; Labro, A.J.; Cuypers, E.; Johnson, H.W.B.; Rainier, J.D.; Tytgat, J.; Snyders, D.J. A polyether biotoxin binding site on the lipid-exposed face of the pore domain of Kv channels revealed by the marine toxin gambierol. Proc. Natl. Acad. Sci. USA 2009, 106, 9896–9901. [Google Scholar] [CrossRef] [PubMed]
- Kopljar, I.; Labro, A.J.; de Block, T.; Rainier, J.D.; Tytgat, J.; Snyders, D.J. The ladder-shaped polyether toxin gambierol anchors the gating machinery of Kv3.1 channels in the resting state. J. Gen. Physiol. 2013, 141, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Fuwa, H.; Vale, C.; Suga, Y.; Goto, T.; Konno, Y.; Sasaki, M.; LaFerla, F.M.; Vieytes, M.R.; Giménez-Llort, L.; et al. Design and synthesis of skeletal analogues of gambierol: Attenuation of amyloid-β and tau pathology with voltage-gated potassium channel and N-methyl-d-aspartate receptor implications. J. Am. Chem. Soc. 2012, 134, 7467–7479. [Google Scholar] [CrossRef] [PubMed]
- Pérez, S.; Vale, C.; Alonso, E.; Fuwa, H.; Sasaki, M.; Konno, Y.; Goto, T.; Suga, Y.; Vieytes, M.R.; Botana, L.M. Effect of gambierol and its tetracyclic and heptacyclic analogues in cultured cerebellar neurons: A structure-activity relationships study. Chem. Res. Toxicol. 2012, 25, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Xu, J.P.; Chapuis, J.C.; Pettit, R.K.; Tackett, L.P.; Doubek, D.L.; Hooper, J.N.A.; Schmidt, J.M. Antineoplastic Agents. 520. Isolation and Structure of Irciniastatins A and B from the Indo-Pacific Marine Sponge. Ircinia ramosa. J. Med. Chem. 2004, 47, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Cichewicz, R.H.; Valeriote, F.A.; Crews, P. Psymberin, a potent sponge-derived cytotoxin from Psammocinia distantly related to the Pederin family. Org. Lett. 2004, 6, 1951–1954. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; García-Fortanet, J.; de Brabander, J.K. Synthesis and complete stereochemical assignment of psymberin/irciniastatin A. J. Am. Chem. Soc. 2005, 127, 11254–11255. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Feng, Y.; Cardenas, E.R.; Williams, N.; Floreancig, P.E.; de Brabander, J.K.; Roth, M.G. Studies toward the unique pederin family member psymberin: Structure-activity relationships, biochemical studies, and genetics identify the mode-of-action of psymberin. J. Am. Chem. Soc. 2012, 134, 18998–19003. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.T.; Hirano, S.; Fukuhara, S.; Watanabe, T.; Kanoh, N.; Iwabuchi, Y.; Usui, T.; Kataoka, T. Irciniastatin A Induces Potent and Sustained Activation of Extracellular Signal-Regulated Kinase and Thereby Promotes Ectodomain Shedding of Tumor Necrosis Factor Receptor 1 in Human Lung Carcinoma A549 Cells. Biol. Pharm. Bull. 2015, 38, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Yu, H.B.; Yang, F.; Li, Y.Y.; Gan, J.H.; Jiao, W.H.; Lin, H.W. Cytotoxic Bryostatin Derivatives from the South China Sea Bryozoan Bugula neritina. J. Nat. Prod. 2015, 78, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- Hale, K.J.; Manaviazar, S. New approaches to the total synthesis of the bryostatin antitumor macrolides. Chem. Asian J. 2010, 5, 704–754. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Verma, V.A.; Paxton, T.J.; Pillow, T.H. Function-oriented synthesis, step economy, and drug design. Acc. Chem. Res. 2008, 41, 40–49. [Google Scholar] [CrossRef] [PubMed]
- DeChristopher, B.A.; Loy, B.A.; Marsden, M.D.; Schrier, A.J.; Zack, J.A.; Wender, P.A. Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat. Chem. 2012, 4, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Baryza, J.L.; Brenner, S.E.; DeChristopher, B.A.; Loy, B.A.; Schrier, A.J.; Verma, V.A. Design, synthesis, and evaluation of potent bryostatin analogs that modulate PKC translocation selectivity. Proc. Natl. Acad. Sci. USA 2011, 108, 6721–6726. [Google Scholar] [CrossRef] [PubMed]
- Wender, P.A.; Verma, V.A. The design, synthesis, and evaluation of C7 diversified bryostatin analogs reveals a hot spot for PKC affinity. Org. Lett. 2008, 10, 3331–3334. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.E.; Botelho, J.C.; Guzmán, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a macrolide from a lithistid sponge of the family Neopeltidae. J. Nat. Prod. 2007, 70, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Youngsaye, W.; Lowe, J.T.; Pohlki, F.; Ralifo, P.; Panek, J.S. Total synthesis and stereochemical reassignment of (+)-neopeltolide. Angew. Chem. Int. Ed. 2007, 46, 9211–9214. [Google Scholar] [CrossRef] [PubMed]
- Custar, D.W.; Zabawa, T.P.; Scheidt, K.A. Total synthesis and structural revision of the marine macrolide neopeltolide. J. Am. Chem. Soc. 2008, 130, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Custar, D.W.; Zabawa, T.P.; Hines, J.; Crews, C.M.; Scheidt, K.A. Total synthesis and structure-activity investigation of the marine natural product neopeltolide. J. Am. Chem. Soc. 2009, 131, 12406–12414. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, M.; Guerriero, A.; Pietra, F.; Debitus, C. Leucascandrolide A, a new type of macrolide: The first powerfully bioactive metabolite of calcareous sponges (Leucascandra caveolata, a new genus from the coral sea). Helv. Chim. Acta 1996, 79, 51–60. [Google Scholar] [CrossRef]
- Ulanovskaya, O.A.; Janjic, J.; Suzuki, M.; Sabharwal, S.S.; Schumacker, P.T.; Kron, S.J.; Kozmin, S.A. Synthesis enables identification of the cellular target of leucascandrolide A and neopeltolide. Nat. Chem. Biol. 2008, 4, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Sato, M.; Sasaki, M. Programmed cell death induced by (−)-8,9-dehydroneopeltolide in human promyelocytic leukemia HL-60 cells under energy stress conditions. Mar. Drugs 2014, 12, 5576–5589. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.K.; Kwon, M.S.; Lee, E. Total synthesis of (+)-neopeltolide by a Prins macrocyclization. Angew. Chem. Int. Ed. 2008, 47, 3242–3244. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Naito, S.; Goto, T.; Sasaki, M. Total synthesis of (+)-neopeltolide. Angew. Chem. Int. Ed. 2008, 47, 4737–4739. [Google Scholar] [CrossRef] [PubMed]
- Vintonyak, V.V.; Maier, M.E. Formal total synthesis of neopeltolide. Org. Lett. 2008, 10, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.; Miller, N.A. Total synthesis of the marine macrolide (+)-neopeltolide. Chem. Commun. 2008, 39, 4708–4710. [Google Scholar] [CrossRef] [PubMed]
- Kartika, R.; Gruffi, T.R.; Taylor, R.E. Concise enantioselective total synthesis of neopeltolide macrolactone highlighted by ether transfer. Org. Lett. 2008, 10, 5047–5050. [Google Scholar] [CrossRef] [PubMed]
- Vintonyak, V.V.; Kunze, B.; Sasse, F.; Maier, M.E. Total synthesis and biological activity of neopeltolide. Chem. Eur. J. 2008, 14, 11132–11140. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Floreancig, P.E. Oxidative carbocation formation in macrocycles: Synthesis of the neopeltolide macrocycle. Angew. Chem. Int. Ed. 2009, 48, 4567–4571. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Park, Y.; Hong, J. Stereoselective synthesis of 2,6-cis-tetrahydropyrans through a tandem allylic oxidation/oxa-Michael reaction promoted by the gem-disubstituent effect: Synthesis of (+)-neopeltolide macrolactone. Angew. Chem. Int. Ed. 2009, 48, 7577–7581. [Google Scholar] [CrossRef] [PubMed]
- Guinchard, X.; Roulland, E. Total synthesis of the antiproliferative macrolide (+)-neopeltolide. Org. Lett. 2009, 11, 4700–4703. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Saito, A.; Naito, S.; Konoki, K.; Yotsu-Yamashita, M.; Sasaki, M. Total synthesis and biological evaluation of (+)-neopeltolide and its analogues. Chem. Eur. J. 2009, 15, 12807–12818. [Google Scholar] [CrossRef] [PubMed]
- Yadav, J.S.; Kumar, G.G.K.S.N. A concise stereoselective formal total synthesis of the cytotoxic macrolide (+)-neopeltolide via Prins cyclization. Tetrahedron 2010, 66, 480–487. [Google Scholar] [CrossRef]
- Fuwa, H.; Saito, A.; Sasaki, M. A concise total synthesis of (+)-neopeltolide. Angew. Chem. Int. Ed. 2010, 49, 3041–3044. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Tu, W.; Floreancig, P.E. Total synthesis of neopeltolide and analogs. Tetrahedron 2010, 66, 4867–4873. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Solorio, D.; Jennings, M.P. Formal synthesis of (−)-neopeltolide featuring a highly stereoselective oxocarbenium formation/reduction sequence. J. Org. Chem. 2010, 75, 4095–4104. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, B.; Zhao, G.; Yang, J.; Xie, X.; She, X. Concise formal synthesis of (+)-neopeltolide. Org. Lett. 2011, 13, 5916–5919. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.V.M.; Reddy, S.V.; Ramakrishna, K.V.S. Synthesis of the macrolactone core of (+)-neopeltolide by transannular cyclization. Org. Biomol. Chem. 2012, 10, 3689–3695. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Samanta, P.K. Stereoselective synthesis of the macrolactone core of (+)-neopeltolide. Org. Lett. 2012, 14, 2346–2349. [Google Scholar] [CrossRef] [PubMed]
- Athe, S.; Chandrasekhar, B.; Roy, S.; Pradhan, T.K.; Ghosh, S. Formal total synthesis of (+)-neopeltolide. J. Org. Chem. 2012, 77, 9840–9845. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Kawakami, M.; Noto, K.; Muto, T.; Suga, Y.; Konoki, K.; Yotsu-Yamashita, M.; Sasaki, M. Concise synthesis and biological assessment of (+)-neopeltolide and a 16-member stereoisomer library of 8,9-dehydroneopeltolide: Identification of pharmacophoric elements. Chem. Eur. J. 2013, 19, 8100–8110. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Shurrush, K.A.; Dawson, Z.L. Enantioselective total synthesis of macrolide (+)-neopeltolide. Org. Biomol. Chem. 2013, 11, 7768–7777. [Google Scholar] [CrossRef] [PubMed]
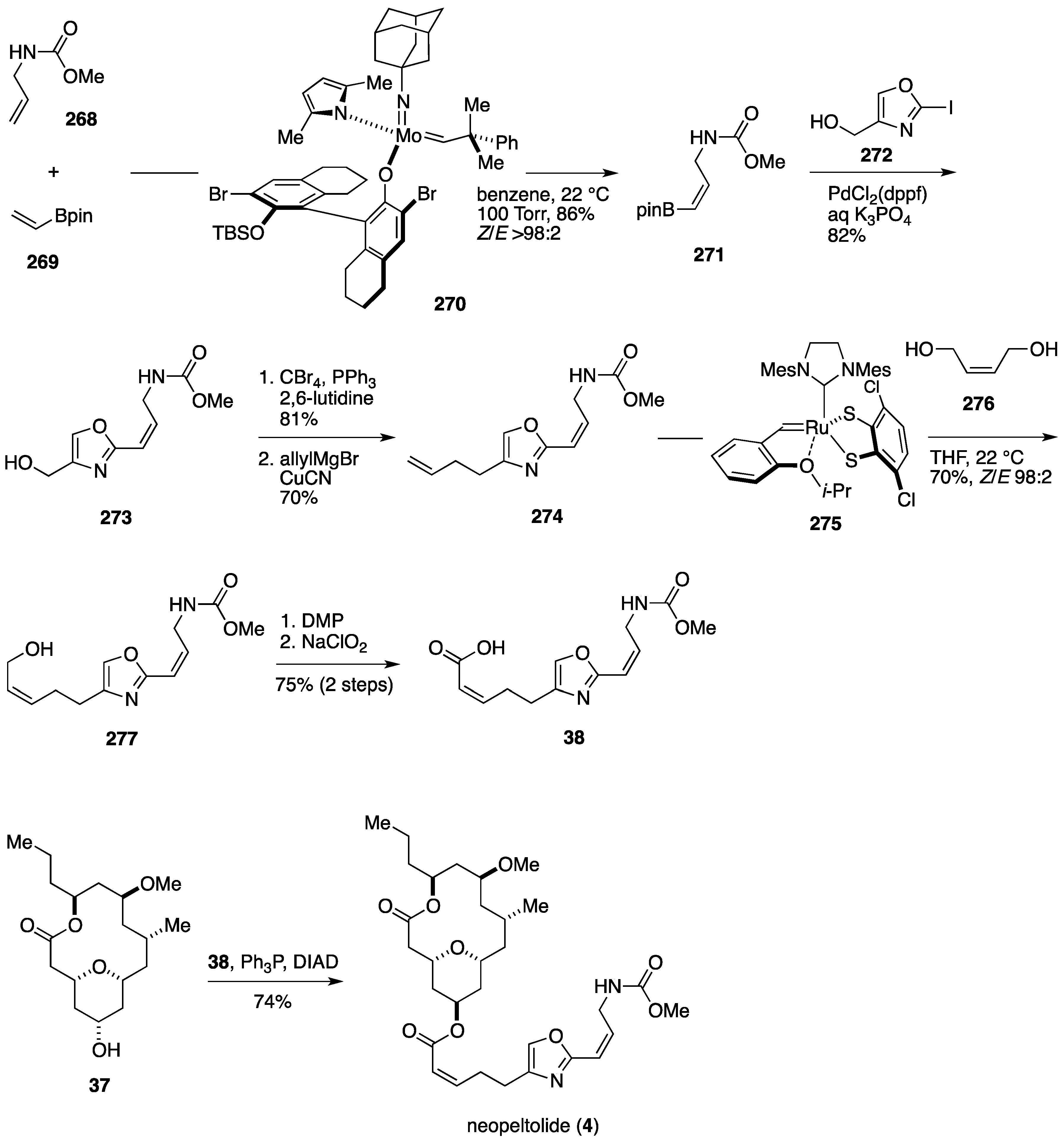
- Yu, M.; Schrock, R.R.; Hoveyda, A.H. Catalyst-controlled stereoselective olefin metathesis as a principal strategy in multistep synthesis design: A concise route to (+)-neopeltolide. Angew. Chem. Int. Ed. 2015, 54, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Balachandran, R.; Day, B.W.; Floreancig, P.E. Synthesis and biological evaluation of neopeltolide and analogs. J. Org. Chem. 2012, 77, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Noguchi, T.; Kawakami, M.; Sasaki, M. Synthesis and biological evaluation of (+)-neopeltolide analogues: Importance of the oxazole-containing side chain. Bioorg. Med. Chem. Lett. 2014, 24, 2415–2419. [Google Scholar] [CrossRef] [PubMed]
- Nasir, N.M.; Ermanis, K.; Clarke, P.A. Strategies for the construction of tetrahydropyran rings in the synthesis of natural products. Org. Biomol. Chem. 2014, 12, 3323–3335. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H. Total synthesis of tetrahydropyran-containing natural products exploiting intramolecular oxa-conjugate cyclization. Heterocycles 2012, 85, 1255–1298. [Google Scholar] [CrossRef]
- Gallon, J.; Reymond, S.; Cossy, J. Neopeltolide, a new promising antitumoral agent. Comptes. Rendus. Chim. 2008, 11, 1463–1476. [Google Scholar] [CrossRef]
- Bai, Y.; Dai, M.J. Strategies and methods for the synthesis of anti-cancer natural product neopeltolide and its analogs. Curr. Org. Chem. 2015, 19, 871–885. [Google Scholar] [CrossRef]
- Huang, H.; Panek, J.S. Stereoselective synthesis of functionalized dihydropyrans via a formal [4+2]-annulation of chiral crotylsilanes. J. Am. Chem. Soc. 2000, 122, 9836–9837. [Google Scholar] [CrossRef]
- Su, Q.; Panek, J.S. Total synthesis of (−)-apicularen A. J. Am. Chem. Soc. 2004, 126, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.T.; Wrona, I.E.; Panek, J.S. Total synthesis of bistramide A. Org. Lett. 2007, 9, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Panek, J.S. Total synthesis of callipeltoside A. Org. Lett. 2004, 6, 4383–4385. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.T.; Panek, J.S. Total synthesis of (−)-kendomycin. Org. Lett. 2008, 10, 3813–3816. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Panek, J.S. Total synthesis of (+)-leucascandrolide A. Angew. Chem. Int. Ed. 2005, 44, 1223–1225. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Klapa, K.; Hansch, M. Zr(Ot-Bu)4-catalyzed Tishchenko reduction of β-hydroxy ketones. Synlett 2005, 91–94. [Google Scholar] [CrossRef]
- Mancuso, A.J.; Huang, S.L.; Swern, D. Oxidation of long-chain and related alcohols to carbonyls by dimethyl sulfoxide “activated” by oxalyl chloride. J. Org. Chem. 1978, 43, 2480–2482. [Google Scholar] [CrossRef]
- Bal, B.S.; Childers, W.E., Jr.; Pinnick, H.W. Oxidation of α,β-unsaturated aldehydes. Tetrahedron 1981, 37, 2091–2096. [Google Scholar] [CrossRef]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef]
- Still, W.C.; Gennari, C. Direct synthesis of Z-unsaturated esters. A useful modification of Horner-Emmons olefination. Tetrahedron Lett. 1983, 24, 4405–4408. [Google Scholar] [CrossRef]
- Hornberger, K.R.; Hamblett, C.L.; Leighton, J.L. Total synthesis of leucascandrolide A. J. Am. Chem. Soc. 2000, 122, 12894–12895. [Google Scholar] [CrossRef]
- Morris, W.J.; Custar, D.W.; Scheidt, K.A. Stereoselective synthesis of tetrahydropyran-4-ones from dioxinones catalyzed by scandium(III) triflate. Org. Lett. 2005, 7, 1113–1115. [Google Scholar] [CrossRef] [PubMed]
- Crane, E.A.; Schiedt, K.A. Prins-type macrocyclizations as an efficient ring-closing strategy in natural product synthesis. Angew. Chem. Int. Ed. 2010, 49, 8316–8326. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R.; Ohkuma, T.; Kitamura, M.; Takaya, H.; Sayo, N.; Kumobayashi, H.; Akutagawa, S. Asymmetric hydrogenation of β-keto carboxylic esters. A practical, purely chemical access to β-hydroxy esters in high enantiomeric purity. J. Am. Chem. Soc. 1987, 109, 5856–5858. [Google Scholar] [CrossRef]
- Basha, A.; Lipton, M.; Weinreb, S.M. A mild, general method for conversion of esters to amides. Tetrahedron Lett. 1977, 18, 4171–4172. [Google Scholar] [CrossRef]
- Nahm, S.; Weinreb, S.M. N-Methoxy-N-methylamides as effective acylating agents. Tetrahedron Lett. 1981, 22, 3815–3818. [Google Scholar] [CrossRef]
- Evans, D.A.; Hoveyda, A.H. Samarium-catalyzed intramolecular Tishchenko reduction of β-hydroxy ketones. A stereoselective approach to the synthesis of differentiated anti 1,3-diol monoesters. J. Am. Chem. Soc. 1990, 112, 6447–6449. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Ishida, A. A convenient method for the preparation of δ-alkoxy-α,β-unsaturated aldehydes by reaction of acetals with 1trimethylsilyloxy-1,3-butadiene. Chem. Lett. 1975, 319–322. [Google Scholar] [CrossRef]
- De Mico, A.; Margarita, R.; Parlanti, L.; Vescovi, A.; Piancatelli, G. A versatile and highly selective hypervalent iodine(III)/2,2,6,6-tetramethyl-1-piperidinyloxyl-mediated oxidation of alcohols to carbonyl compounds. J. Org. Chem. 1997, 62, 6974–6977. [Google Scholar] [CrossRef]
- Mitsunobu, O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981, 1–28. [Google Scholar] [CrossRef]
- Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P. Mitsunobu and related reactions: Advances and applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Janjic, J.; Kozmin, S.A. Synthesis of leucascandrolide A via a spontaneous macrolactolization. J. Am. Chem. Soc. 2002, 124, 13670–13671. [Google Scholar] [CrossRef] [PubMed]
- Crane, E.A.; Zabawa, T.P.; Farmer, R.L.; Scheidt, K.A. Enantioselective synthesis of (−)-exiguolide by iterative stereoselective dioxinone-directed Prins cyclizations. Angew. Chem. Int. Ed. 2011, 50, 9112–9115. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.L.K.; Lee, C.H.A.; Tan, K.T.; Loh, T.P. An unusual approach to the synthesis of enantiomerically cis linear homoallylic alcohols based on the steric interaction mechanism of camphor scaffold. Org. Lett. 2004, 6, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T.; Jung, A. 1,3-Asymmetric induction in addition reactions of chiral β-alkoxy aldehydes: Efficient chelation control via Lewis acidic titanium reagents. J. Am. Chem. Soc. 1983, 105, 4833–4835. [Google Scholar] [CrossRef]
- Breit, B. 1,3-Asymmetric induction in stereoselective rhodium-catalyzed hydroformylation of homomethallylic alcohols. Eur. J. Org. Chem. 1998, 1998, 1123–1134. [Google Scholar] [CrossRef]
- Kubota, K.; Leighton, J.L. A Highly practical and enantioselective reagent for the allylation of aldehydes. Angew. Chem. Int. Ed. 2003, 42, 946–948. [Google Scholar] [CrossRef] [PubMed]
- Des Mazery, R.; Pullez, M.; Lopez, F.; Harutyunyan, S.R.; Minnaard, A.J.; Feringa, B.L. An iterative catalytic route to enantiopure deoxypropionate subunits: Asymmetric conjugate addition of Grignard reagents to α,β-unsaturated thioesters. J. Am. Chem. Soc. 2005, 127, 9966–9977. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, T.; Ling, S.C.; Li, L. Facile reduction of ethyl thiol esters to aldehydes: Application to a total synthesis of (+)-neothramycin A methyl ether. J. Am. Chem. Soc. 1990, 112, 7050–7051. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. A useful 12-I-5 triacetoxyperiodinane (the Dess–Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Kozmin, S.A. Efficient stereochemical relay en route to leucascandrolide A. Org. Lett. 2001, 3, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Narasaka, K.; Pai, F.C. Stereoselective reduction of β-hydroxyketones to 1,3-diols highly selective 1,3-asymmetric induction via boron chelates. Tetrahedron 1984, 40, 2233–2238. [Google Scholar] [CrossRef]
- Laganis, E.D.; Chenard, B.L. Metal silanolates: Organic soluble equivalents for O−2. Tetrahedron Lett. 1984, 25, 5831–5834. [Google Scholar] [CrossRef]
- Tu, W.; Liu, L.; Floreancig, P.E. Diastereoselective tetrahydropyrone synthesis through transition- metal-free oxidative carbon–hydrogen bond activation. Angew. Chem. Int. Ed. 2008, 47, 4184–4187. [Google Scholar] [CrossRef] [PubMed]
- Peh, G.R.; Floreancig, P.E. Cyclopropane compatibility with oxidative carbocation formation: Total synthesis of clavosolide A. Org. Lett. 2012, 14, 5614–5617. [Google Scholar] [CrossRef] [PubMed]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. The Sonogashira reaction: A booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Wang, Z. Highly stereoselective hydrocarbation of terminal alkynes via Pt-catalyzed hydrosilylation/Pd-catalyzed cross-coupling reactions. Org. Lett. 2001, 3, 1073–1076. [Google Scholar] [CrossRef] [PubMed]
- Gooßen, L.J.; Paetzold, J.; Koley, D. Regiocontrolled Ru-catalyzed addition of carboxylic acids to alkynes: Practical protocols for the synthesis of vinyl esters. Chem. Commun. 2003, 706–707. [Google Scholar] [CrossRef]
- Lewis, M.D.; Cha, J.K.; Kishi, Y. Highly stereoselective approaches to α- and β-C-glycopyranosides. J. Am. Chem. Soc. 1982, 104, 4976–4978. [Google Scholar] [CrossRef]
- Brown, H.C.; Jadhav, P.K. Asymmetric carbon–carbon bond formation via β-allyldiisopinocampheylborane. Simple synthesis of secondary homoallylic alcohols with excellent enantiomeric purities. J. Am. Chem. Soc. 1983, 105, 2092–2093. [Google Scholar] [CrossRef]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Connon, S.J.; Blechert, S. Recent developments in olefin cross-metathesis. Angew. Chem. Int. Ed. 2003, 42, 1900–1923. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Gauchet-Prunet, J.A. Diastereoselective synthesis of protected syn 1,3-diols by base-catalyzed intramolecular conjugate addition of hemiacetal-derived alkoxide nucleophiles. J. Org. Chem. 1993, 58, 2446–2453. [Google Scholar] [CrossRef]
- Eschenbrenner-Lux, V.; Kumar, K.; Waldmann, H. The asymmetric hetero-Diels-Alder reaction in the syntheses of biologically relevant compounds. Angew. Chem. Int. Ed. 2014, 53, 11146–11157. [Google Scholar] [CrossRef] [PubMed]
- Dossetter, A.G.; Jamison, T.F.; Jacobsen, E.N. Highly enantio- and diastereoselective hetero-Diels-Alder reactions catalyzed by new chiral tridentate chromium(III) catalysts. Angew. Chem. Int. Ed. 1999, 38, 2398–2400. [Google Scholar] [CrossRef]
- Ouellet, S.G.; Tuttle, J.B.; MacMillan, D.W.C. Enantioselective organocatalytic hydride reduction. J. Am. Chem. Soc. 2005, 127, 32–33. [Google Scholar] [CrossRef] [PubMed]
- Danishefsky, S.; Kitahara, T. Useful diene for the Diels−Alder reaction. J. Am. Chem. Soc. 1974, 96, 7807–7808. [Google Scholar] [CrossRef]
- Schaus, S.E.; Brånalt, J.; Jacobsen, E.N. Asymmetric hetero-Diels-Alder reactions catalyzed by chiral (salen)chromium(III) complexes. J. Org. Chem. 1998, 63, 403–405. [Google Scholar] [CrossRef]
- Gemal, A.L.; Luche, J.L. Lanthanoids in organic synthesis. 6. The reduction of α-enones by sodium borohydride in the presence of lanthanoid chlorides: Synthetic and mechanistic aspects. J. Am. Chem. Soc. 1981, 103, 5454–5459. [Google Scholar]
- Ireland, R.E.; Mueller, R.H.; Willard, A.K. The ester enolate Claisen rearrangement. Stereochemical control through stereoselective enolate formation. J. Am. Chem. Soc. 1976, 98, 2868–2877. [Google Scholar] [CrossRef]
- Ireland, R.E.; Wipf, P.; Armstrong, J.D., III. Stereochemical control in the ester enolate Claisen rearrangement. 1. Stereoselectivity in silyl ketene acetal formation. J. Org. Chem. 1991, 56, 650–657. [Google Scholar] [CrossRef]
- Foubelo, F.; Yus, M. Functionalised organolithium compounds by sulfur–lithium exchange. Chem. Soc. Rev. 2008, 37, 2620–2633. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.A.; Choy, W.; Davis, J.T.; Essenfeld, A.P.; Masamune, S.; Roush, W.R.; Sakai, T. Horner-Wadsworth-Emmons reaction: Use of lithium chloride and an amine for base-sensitive compounds. Tetrahedron Lett. 1984, 25, 2183–2186. [Google Scholar] [CrossRef]
- Hoveyda, A.H.; Zhugralin, A.R. The remarkable metal-catalysed olefin metathesis reaction. Nature 2007, 450, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Metathesis reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4490–4527. [Google Scholar] [CrossRef] [PubMed]
- Gradillas, A.; Pérez-Castells, J. Macrocyclization by ring-closing metathesis in the total synthesis of natural products: Reaction conditions and limitations. Angew. Chem. Int. Ed. 2006, 45, 6086–6101. [Google Scholar] [CrossRef] [PubMed]
- Deiters, A.; Martin, S.F. Synthesis of oxygen- and nitrogen-containing heterocycles by ring-closing metathesis. Chem. Rev. 2004, 104, 2199–2238. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A. Cross-coupling reactions of organoboranes: An easy way to construct C–C bonds (Nobel Lecture). Angew. Chem. Int. Ed. 2011, 50, 6722–6737. [Google Scholar] [CrossRef] [PubMed]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Fuwa, H. Palladium-catalyzed synthesis of N- and O-heterocycles starting from enol phosphates. Synlett 2011, 6–29. [Google Scholar] [CrossRef]
- Fuwa, H.; Sasaki, M. An efficient strategy for the synthesis of endocyclic enol ethers and its application to the synthesis of spiroacetals. Org. Lett. 2008, 10, 2549–2552. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Ishigai, K.; Hashizume, K.; Sasaki, M. Total synthesis and complete stereostructure of gambieric acid A. J. Am. Chem. Soc. 2012, 134, 11984–11987. [Google Scholar] [CrossRef] [PubMed]
- Ishigai, K.; Fuwa, H.; Hashizume, K.; Fukazawa, R.; Cho, Y.; Yotsu-Yamashita, M.; Sasaki, M. Total synthesis and biological evaluation of (+)-gambieric acid A and its analogues. Chem. Eur. J. 2013, 19, 5276–5288. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Sakamoto, K.; Muto, T.; Sasaki, M. Concise synthesis of the C15–C38 fragment of okadaic acid, a specific inhibitor of protein phosphatases 1 and 2A. Tetrahedron 2015, 71, 6369–6383. [Google Scholar] [CrossRef]
- Keck, G.E.; Tarbet, K.H.; Geraci, L.S. Catalytic asymmetric allylation of aldehydes. J. Am. Chem. Soc. 1993, 115, 8467–8468. [Google Scholar] [CrossRef]
- Rai, A.N.; Basu, A. An efficient method for para-methoxybenzyl ether formation with lanthanum triflate. Tetrahedron Lett. 2003, 44, 2267–2269. [Google Scholar] [CrossRef]
- Marshall, J.A.; Johns, B.A. Total synthesis of (+)-discodermolide. J. Org. Chem. 1998, 63, 7885–7892. [Google Scholar] [CrossRef]
- Parikh, J.R.; Doering, W.V.E. Sulfur trioxide in the oxidation of alcohols by dimethyl sulfoxide. J. Am. Chem. Soc. 1967, 89, 5505–5507. [Google Scholar] [CrossRef]
- Giese, B.; Kopping, B.; Göbel, T.; Dickhaut, J.; Thoma, G.; Kulicke, K.; Trach, F. Radical cyclization reactions. Org. React. 1995, 48, 301–856. [Google Scholar]
- Lee, E.; Jin, S.T.; Lee, C.; Min, P.C. β-Alkoxyacrylates in radical cyclizations: Remarkably efficient oxacycle synthesis. Tetrahedron Lett. 1993, 34, 4831–4834. [Google Scholar] [CrossRef]
- Liu, K.; Taylor, R.E.; Kartika, R. Electrophile-induced ether transfer: A new approach to polyketide structural units. Org. Lett. 2006, 8, 5393–5395. [Google Scholar] [CrossRef] [PubMed]
- Burgos, C.H.; Canales, E.; Matos, K.; Soderquist, J.A. Asymmetric allyl- and crotylboration with the robust, versatile, and recyclable 10-TMS-9-borabicyclo[3.3.2]decanes. J. Am. Chem. Soc. 2005, 127, 8044–8049. [Google Scholar] [CrossRef] [PubMed]
- Nising, S.F.; Bräse, S. Recent developments in the field of oxa-Michael reactions. Chem. Soc. Rev. 2012, 41, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Nising, S.F.; Bräse, S. The oxa-Michael reaction: From recent developments to applications in natural product synthesis. Chem. Soc. Rev. 2008, 37, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Ichinokawa, N.; Noto, K.; Sasaki, M. Stereoselective synthesis of 2,6-cis-substituted tetrahydropyrans: Brønsted acid-catalyzed intramolecular oxa-conjugate cyclization of α,β-unsaturated ester surrogates. J. Org. Chem. 2012, 77, 2588–2607. [Google Scholar] [CrossRef] [PubMed]
- Piel, J. A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc. Natl. Acad. Sci. USA 2002, 99, 14002–14007. [Google Scholar] [CrossRef] [PubMed]
- Julien, B.; Tian, Z.Q.; Reid, R.; Reeves, C.D. Analysis of the ambruticin and jerangolid gene clusters of Sorangium cellulosum reveals unusual mechanisms of polyketide biosynthesis. Chem. Biol. 2006, 13, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Sudek, S.; Lopanik, N.B.; Waggoner, L.E.; Hildebrand, M.; Anderson, C.; Liu, H.; Patel, A.; Sherman, D.H.; Haygood, M.G. Identification of the putative bryostatin polyketide synthase gene cluster from “Candidatus Endobugula sertula”, the uncultivated microbial symbiont of the marine bryozoan Bugula neritina. J. Nat. Prod. 2007, 70, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.; Harris, P.W.R.; Brimble, M.A. Synthesis of methyl N-Boc-(2S,4R)-4-methylpipecolate. J. Org. Chem. 2010, 75, 8728–8731. [Google Scholar]
- Kolb, H.C.; VanNieuwenhze, M.S.; Sharpless, K.B. Catalytic asymmetric dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- Hong, C.Y.; Kishi, Y. Total synthesis of onnamide A. J. Am. Chem. Soc. 1991, 113, 9693–9694. [Google Scholar] [CrossRef]
- Maki, B.E.; Scheidt, K.A. N-Heterocyclic carbene-catalyzed oxidation of unactivated aldehydes to esters. Org. Lett. 2008, 10, 4331–4334. [Google Scholar] [CrossRef] [PubMed]
- Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. An effective use of benzoic anhydride and its derivatives for the synthesis of carboxylic esters and lactones: A powerful and convenient mixed anhydride method promoted by basic catalysts. J. Org. Chem. 2004, 69, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Yang, H.; Wuitschik, G. A Ru-catalyzed tandem alkyne−enone coupling/Michael addition: Synthesis of 4-methylene-2,6-cis-tetrahydropyrans. Org. Lett. 2005, 7, 4761–4764. [Google Scholar] [CrossRef] [PubMed]
- de Paule, S.D.; Jeulin, S.; Ratovelomanana-Vidal, V.; Genêt, J.P.; Champion, N.; Dellis, P. Synthesis and molecular modeling studies of SYNPHOS®, a new, efficient diphosphane ligand For ruthenium-catalyzed asymmetric hydrogenation. Eur. J. Org. Chem. 2003, 2003, 1931–1941. [Google Scholar] [CrossRef]
- Lee, K.C.; Lin, M.J.; Loh, T.P. Silicon-assisted propargylic transfer to aldehydes. Chem. Commun. 2004, 21, 2456–2457. [Google Scholar] [CrossRef] [PubMed]
- Nagao, Y.; Hagiwara, Y.; Kumagai, T.; Ochiai, M.; Inoue, T.; Hashimoto, K.; Fujita, E. New C-4-chiral 1,3-thiazolidine-2-thiones: Excellent chiral auxiliaries for highly diastereo-controlled aldol-type reactions of acetic acid and α,β-unsaturated aldehydes. J. Org. Chem. 1986, 51, 2391–2939. [Google Scholar] [CrossRef]
- Hong, S.H.; Sanders, D.P.; Lee, C.W.; Grubbs, R.H. Prevention of undesirable isomerization during olefin metathesis. J. Am. Chem. Soc. 2005, 127, 17160–17161. [Google Scholar] [CrossRef] [PubMed]
- Schaus, S.E.; Brandes, B.D.; Larrow, J.F.; Tokunaga, M.; Hansen, K.B.; Gould, A.E.; Furrow, M.E.; Jacobsen, E.N. Highly selective hydrolytic kinetic resolution of terminal epoxides catalyzed by chiral (salen)CoIII complexes. Practical synthesis of enantioenriched terminal epoxides and 1,2-diols. J. Am. Chem. Soc. 2002, 124, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Reiter, M.; Turner, H.; Gouverneur, V. Intramolecular hetero-Michael addition of β-hydroxyenones for the preparation of highly substituted tetrahydropyranones. Chem. Eur. J. 2006, 12, 7190–7203. [Google Scholar] [CrossRef] [PubMed]
- Florence, G.J.; Cadou, R.F. Studies toward the synthesis of neopeltolide: Synthesis of a ring-closing metathesis macrocyclization precursor. Tetrahedron Lett. 2010, 51, 5761–5763. [Google Scholar] [CrossRef]
- Hari, T.P.A.; Wilke, B.I.; Davey, J.A.; Boddy, C.N. Diastereoselective transannular oxa-conjugate addition generates the 2,6-cis-disubstituted tetrahydropyran of neopeltolide. J. Org. Chem. 2016, 81, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Semmelhack, M.F.; Bodurow, C. Intramolecular alkoxypalladation/carbonylation of alkenes. J. Am. Chem. Soc. 1984, 106, 1496–1498. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Kim, C.; Zhang, N.; Bodurow, C.; Sanner, M.; Dobler, W.; Meier, M. Intramolecular alkoxy-carbonylation of hydroxy alkenes promoted by Pd(II). Pure Appl. Chem. 1990, 62, 2035–2040. [Google Scholar] [CrossRef]
- Kim, I.S.; Ngai, M.Y.; Krische, M.J. Enantioselective iridium-catalyzed carbonyl allylation from the alcohol or aldehyde oxidation level via transfer hydrogenative coupling of allyl acetate: Departure from chirally modified allyl metal reagents in carbonyl addition. J. Am. Chem. Soc. 2008, 130, 14891–14899. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Kim, I.S.; Hassan, A.; del Valle, D.J.; Krische, M.J. 1,n-Glycols as dialdehyde equivalents in iridium-catalyzed enantioselective carbonyl allylation and iterative two-directional assembly of 1,3-polyols. Angew. Chem. Int. Ed. 2009, 48, 5018–5021. [Google Scholar] [CrossRef] [PubMed]
- Hanessian, S.; Giroux, S.; Larsson, A. Efficient allyl to propenyl isomerization in functionally diverse compounds with a thermally modified Grubbs second-generation catalyst. Org. Lett. 2006, 8, 5481–5484. [Google Scholar]
- Garber, S.B.; Kingsbury, J.S.; Gray, B.L.; Hoveyda, A.H. Efficient and recyclable monomeric and dendritic Ru-based metathesis catalysts. J. Am. Chem. Soc. 2000, 122, 8168–8179. [Google Scholar] [CrossRef]
- Bai, Y.; Davis, D.C.; Dai, M. Synthesis of tetrahydropyran/tetrahydrofuran-containing macrolides by palladium-catalyzed alkoxycarbonylative macrolactonizations. Angew. Chem. Int. Ed. 2014, 53, 6519–6522. [Google Scholar] [CrossRef] [PubMed]
- Tanner, D.; Somfai, P. Enantioselective total synthesis of (+)-negamycin. Tetrahedron Lett. 1988, 29, 2373–2376. [Google Scholar] [CrossRef]
- Finan, J.M.; Kishi, Y. Reductive ring openings of allyl-alcohol epoxides. Tetrahedron Lett. 1982, 23, 2719–2722. [Google Scholar] [CrossRef]
- Saravanan, P.; Chandrasekhar, M.; Anand, R.V.; Singh, V.K. An efficient method for deprotection of acetals. Tetrahedron Lett. 1998, 39, 3091–3092. [Google Scholar] [CrossRef]
- Martinelli, M.J.; Nayyar, N.K.; Moher, E.D.; Dhokte, U.P.; Pawlak, J.M.; Vaidyanathan, R. Dibutyltin oxide catalyzed selective sulfonylation of α-chelatable primary alcohols. Org. Lett. 1999, 1, 448–450. [Google Scholar] [CrossRef]
- Hanessian, S.; Ugolini, A.; Dubé, D.; Glamyan, A. Facile access to (S)-1,2,4-butanetriol and its derivatives. Can. J. Chem. 1984, 62, 2146–2147. [Google Scholar] [CrossRef]
- Mori, Y.; Takeuchi, A.; Kageyama, H.; Suzuki, M. A convergent general protocol for syn-1,3-polyols. Tetrahedron Lett. 1988, 29, 5423–5426. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Hirao, I. An efficient method for the alkynylation of oxiranes using alkynyl boranes. Tetrahedron Lett. 1983, 24, 391–394. [Google Scholar] [CrossRef]
- Roush, W.R.; Adam, M.A.; Peseckis, S.M. Regioselectivity of the reactions of trialkylaluminum reagents with 2,3-epoxyalcohols: Application to the synthesis of α-chiral aldehydes. Tetrahedron Lett. 1983, 24, 1377–1380. [Google Scholar] [CrossRef]
- Garegg, P.J.; Samuelsson, B. Conversion of vicinal diols into olefins using triphenylphosphine and triiodoimidazole. Synthesis 1979, 1979, 813–814. [Google Scholar] [CrossRef]
- Malcolmson, S.J.; Meek, S.J.; Sattely, E.S.; Schrock, R.R.; Hoveyda, A.H. Highly efficient molybdenum-based catalysts for enantioselective alkene metathesis. Nature 2008, 456, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Hoveyda, A.H.; Malcolmson, S.J.; Meek, S.J.; Zhugralin, A.R. Catalytic enantioselective olefin metathesis in natural product synthesis. Chiral metal-based complexes that deliver high enantioselectivity and more. Angew. Chem. Int. Ed. 2010, 49, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Ibrahem, I.; Hasegawa, M.; Schrock, R.R.; Hoveyda, A.H. Enol ethers as substrates for efficient Z- and enantioselective ring-opening/cross-metathesis reactions promoted by stereogenic-at-Mo complexes: Utility in chemical synthesis and mechanistic attributes. J. Am. Chem. Soc. 2012, 134, 2788–2799. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Radomkit, S.; O’Brien, J.M.; Hoveyda, A.H. Metal-free catalytic enantioselective C–B bond formation: (Pinacolato)boron conjugate additions to α,β-unsaturated ketones, esters, Weinreb amides, and aldehydes promoted by chiral N-heterocyclic carbenes. J. Am. Chem. Soc. 2012, 134, 8277–8285. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Haeffner, F.; Schrock, R.R.; Hoveyda, A.H. Molybdenum-based complexes with two aryloxides and a pentafluoroimido ligand: Catalysts for efficient Z-selective synthesis of a macrocyclic trisubstituted alkene by ring-closing metathesis. Angew. Chem. Int. Ed. 2013, 52, 1939–1943. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.J.; Khan, R.K.M.; Torker, S.; Hoveyda, A.H. Broadly applicable Z- and diastereoselective ring-opening/cross-metathesis catalyzed by a dithiolate Ru complex. Angew. Chem. Int. Ed. 2014, 53, 1968–1972. [Google Scholar] [CrossRef] [PubMed]





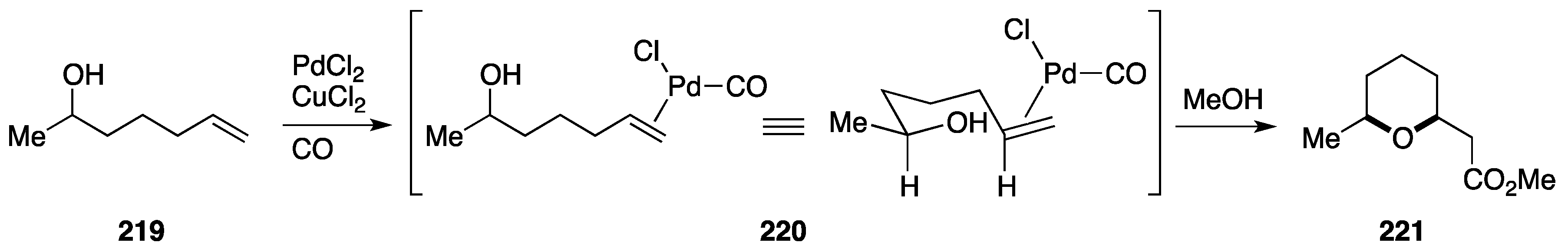



© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuwa, H. Contemporary Strategies for the Synthesis of Tetrahydropyran Derivatives: Application to Total Synthesis of Neopeltolide, a Marine Macrolide Natural Product. Mar. Drugs 2016, 14, 65. https://doi.org/10.3390/md14040065
Fuwa H. Contemporary Strategies for the Synthesis of Tetrahydropyran Derivatives: Application to Total Synthesis of Neopeltolide, a Marine Macrolide Natural Product. Marine Drugs. 2016; 14(4):65. https://doi.org/10.3390/md14040065
Chicago/Turabian StyleFuwa, Haruhiko. 2016. "Contemporary Strategies for the Synthesis of Tetrahydropyran Derivatives: Application to Total Synthesis of Neopeltolide, a Marine Macrolide Natural Product" Marine Drugs 14, no. 4: 65. https://doi.org/10.3390/md14040065
APA StyleFuwa, H. (2016). Contemporary Strategies for the Synthesis of Tetrahydropyran Derivatives: Application to Total Synthesis of Neopeltolide, a Marine Macrolide Natural Product. Marine Drugs, 14(4), 65. https://doi.org/10.3390/md14040065