
Biological Potential of Chitinolytic Marine Bacteria


Abstract
:1. Introduction
2. Results
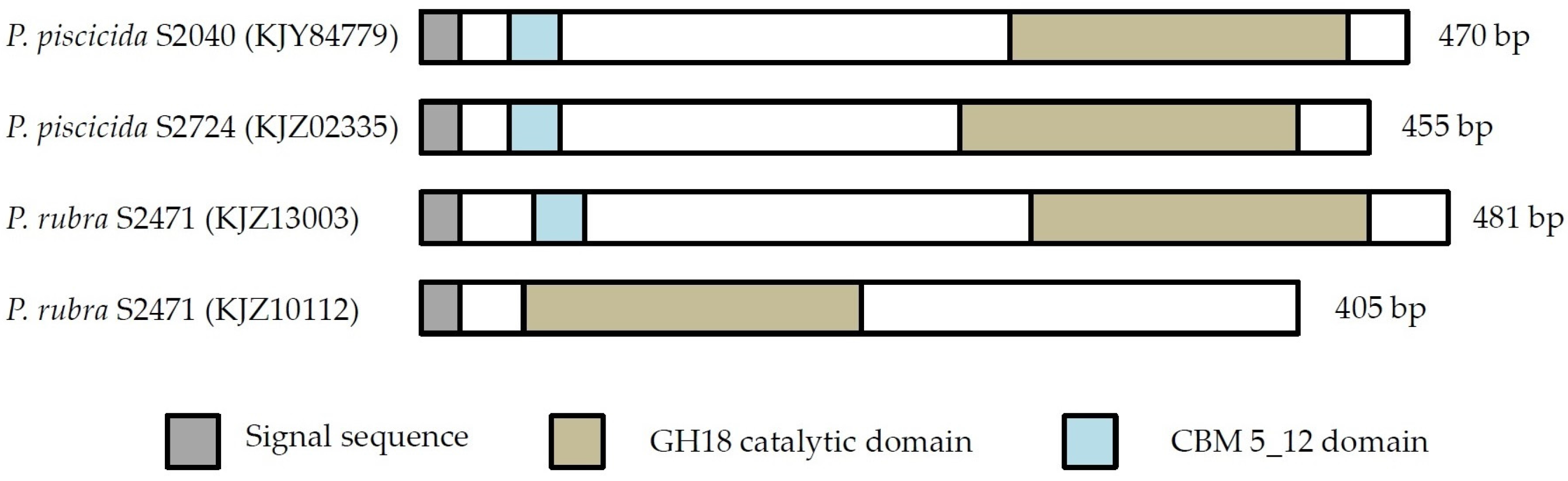
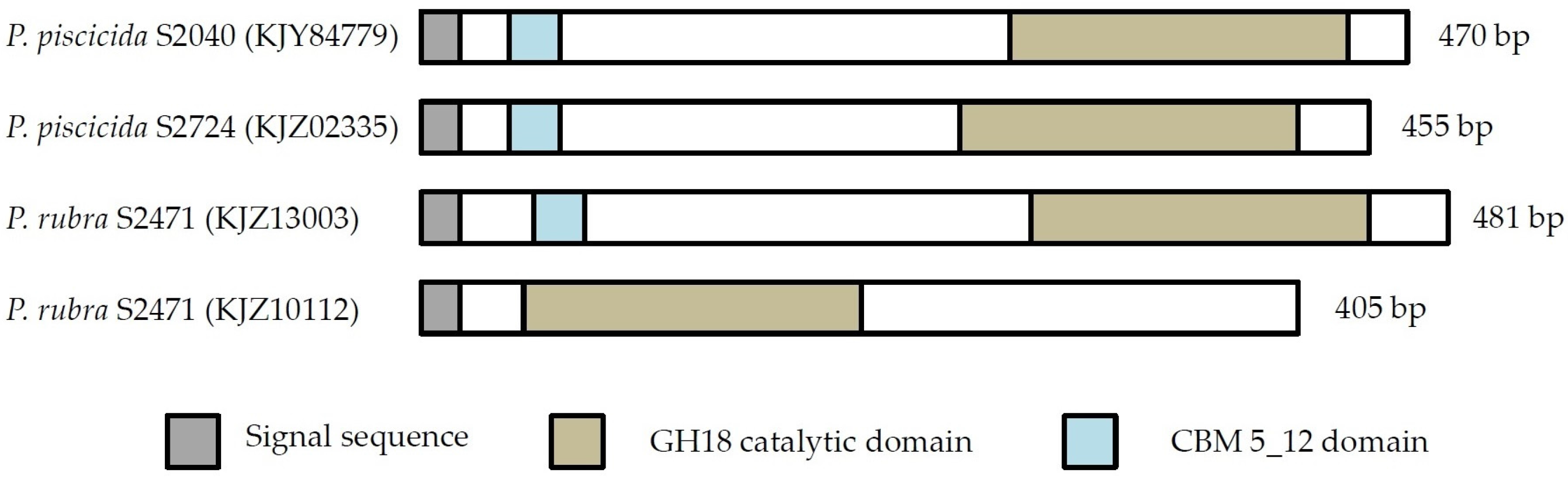
2.1. Chitin Degrading Activity and in Silico Analysis
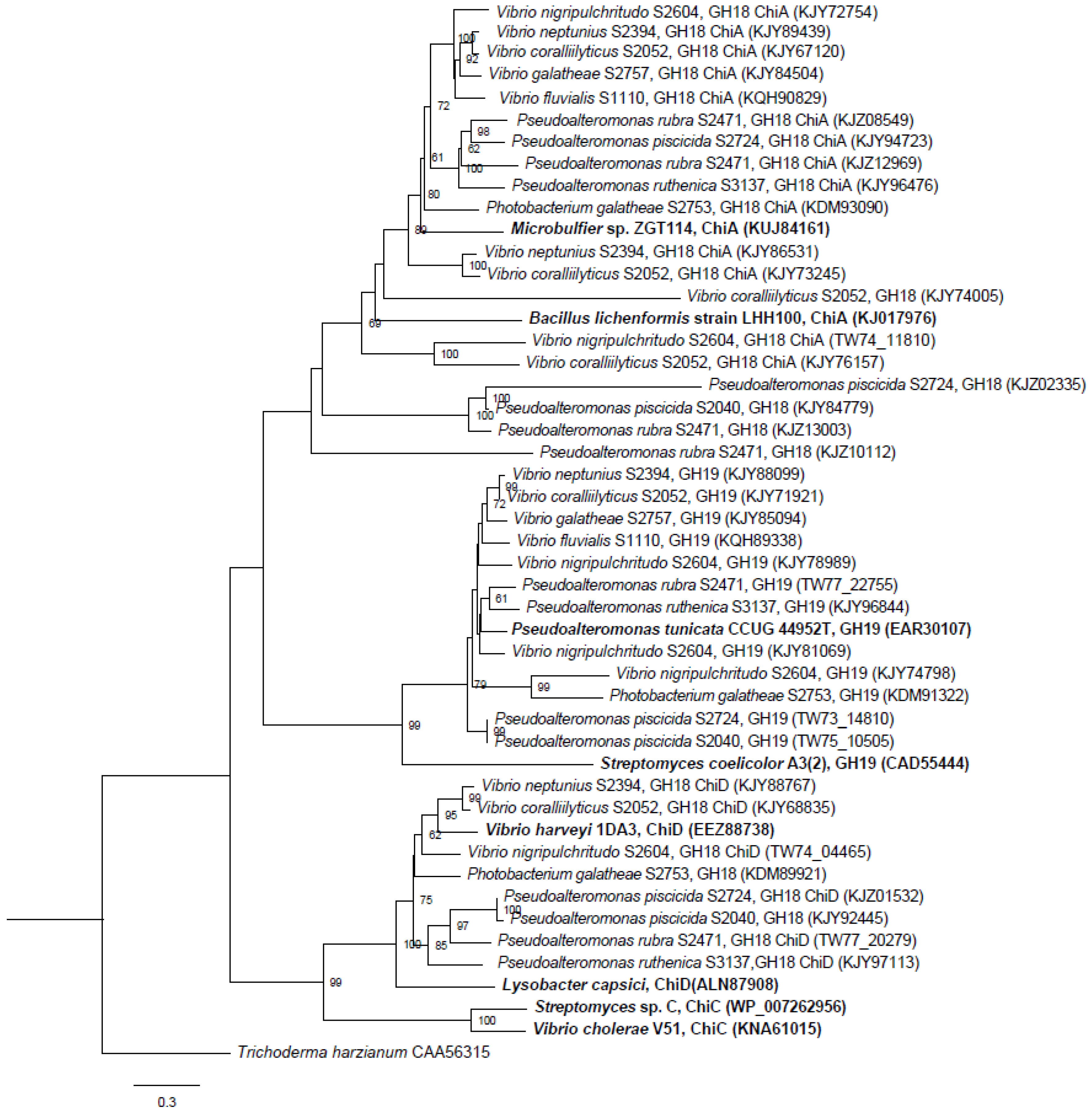
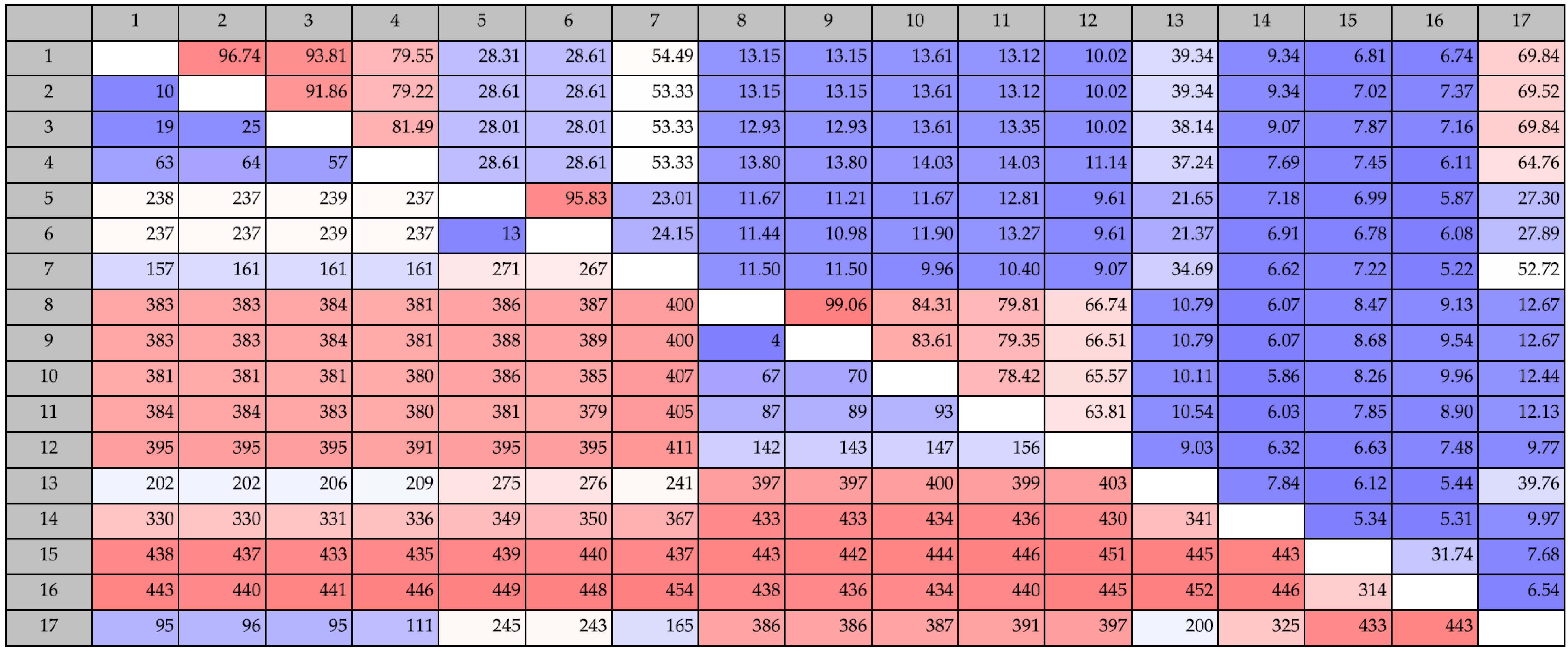
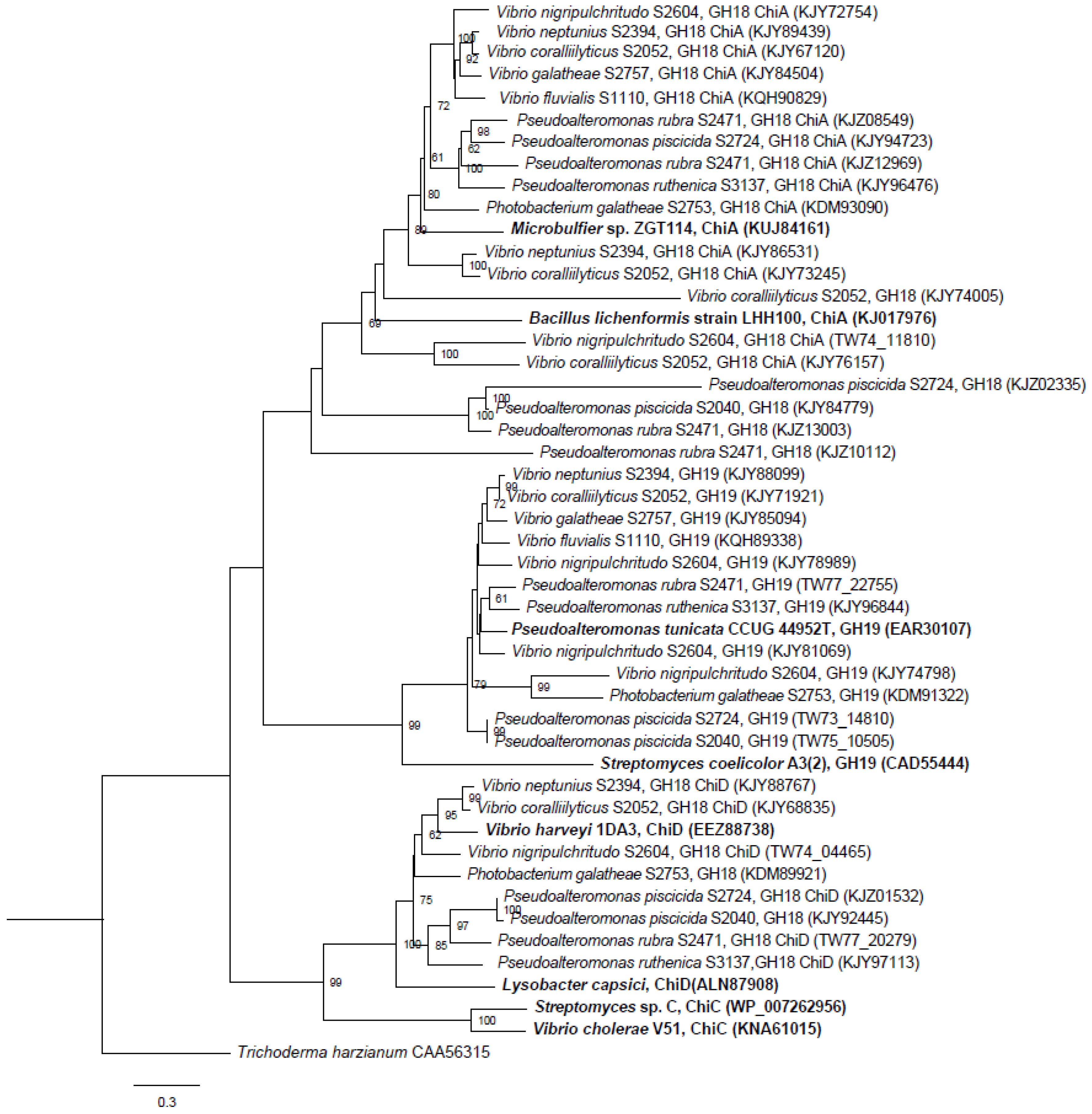
2.1.1. Phylogenetic Analysis of Chitinases
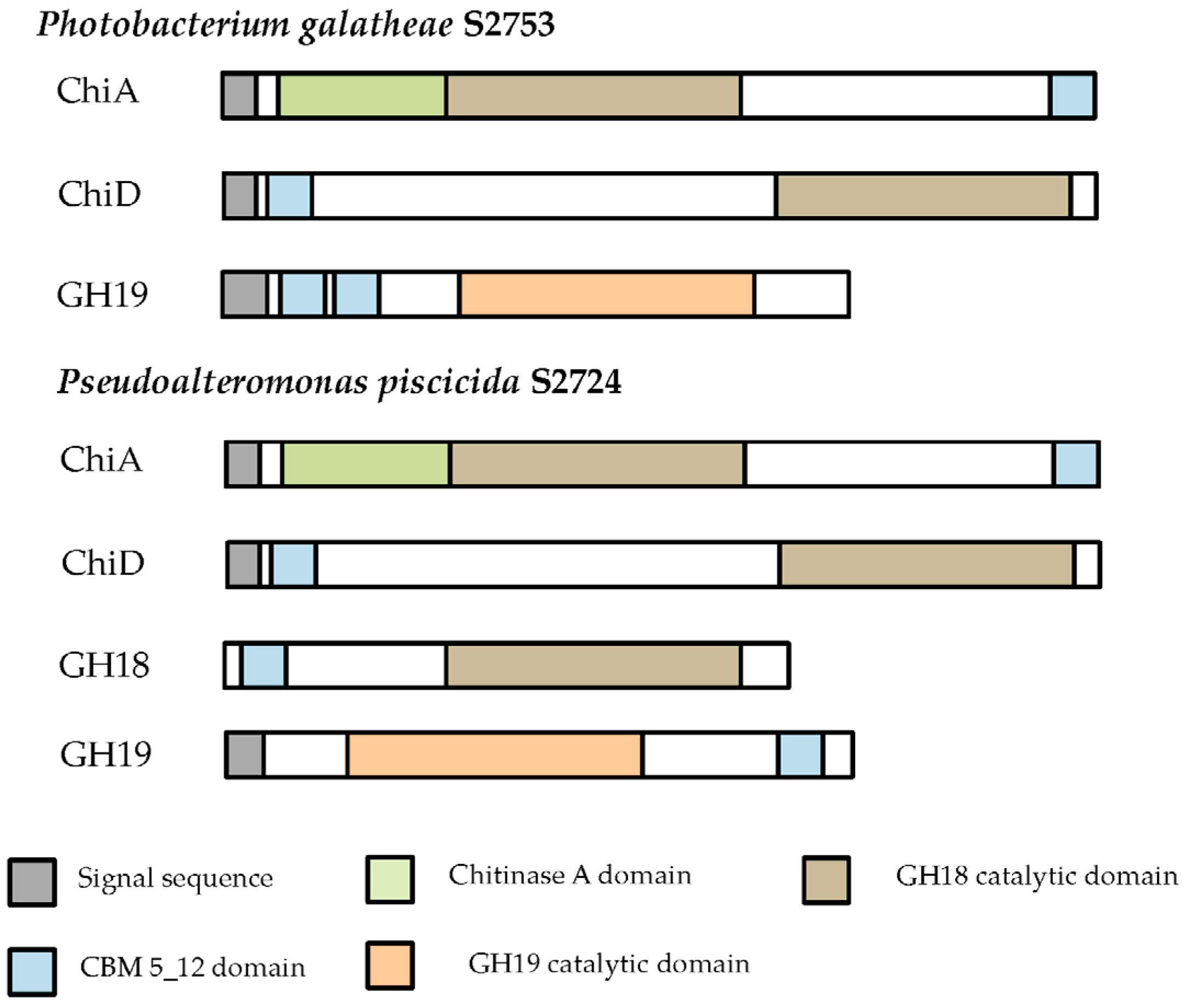
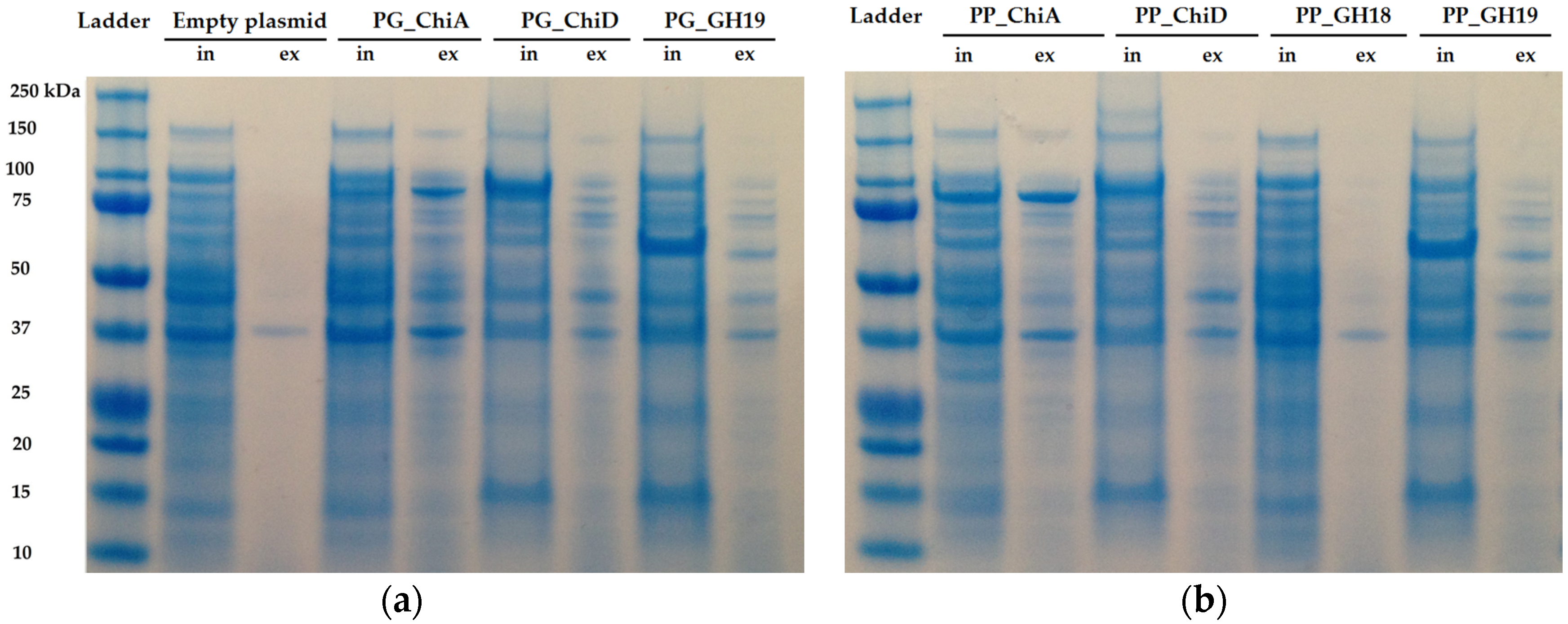
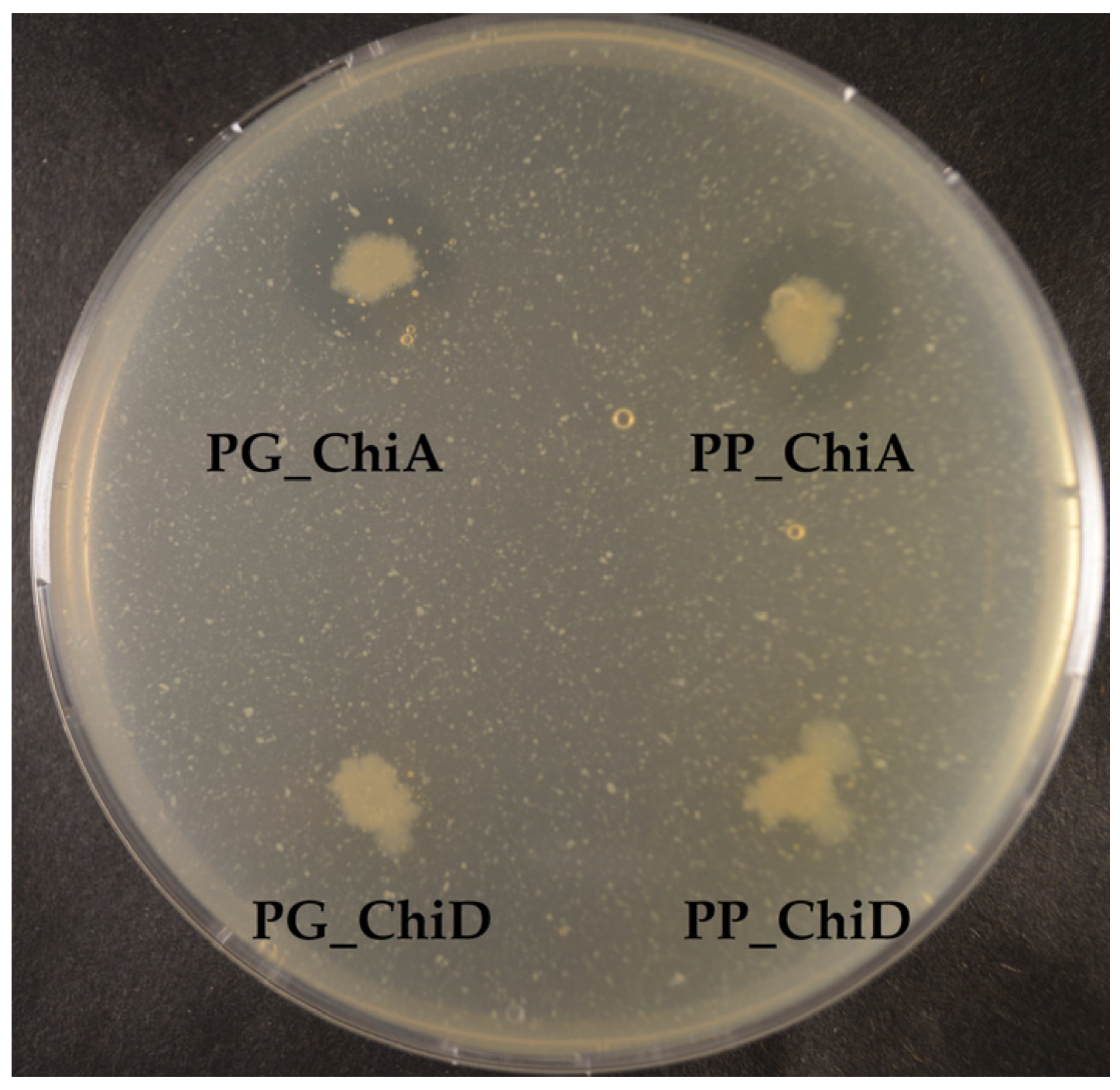
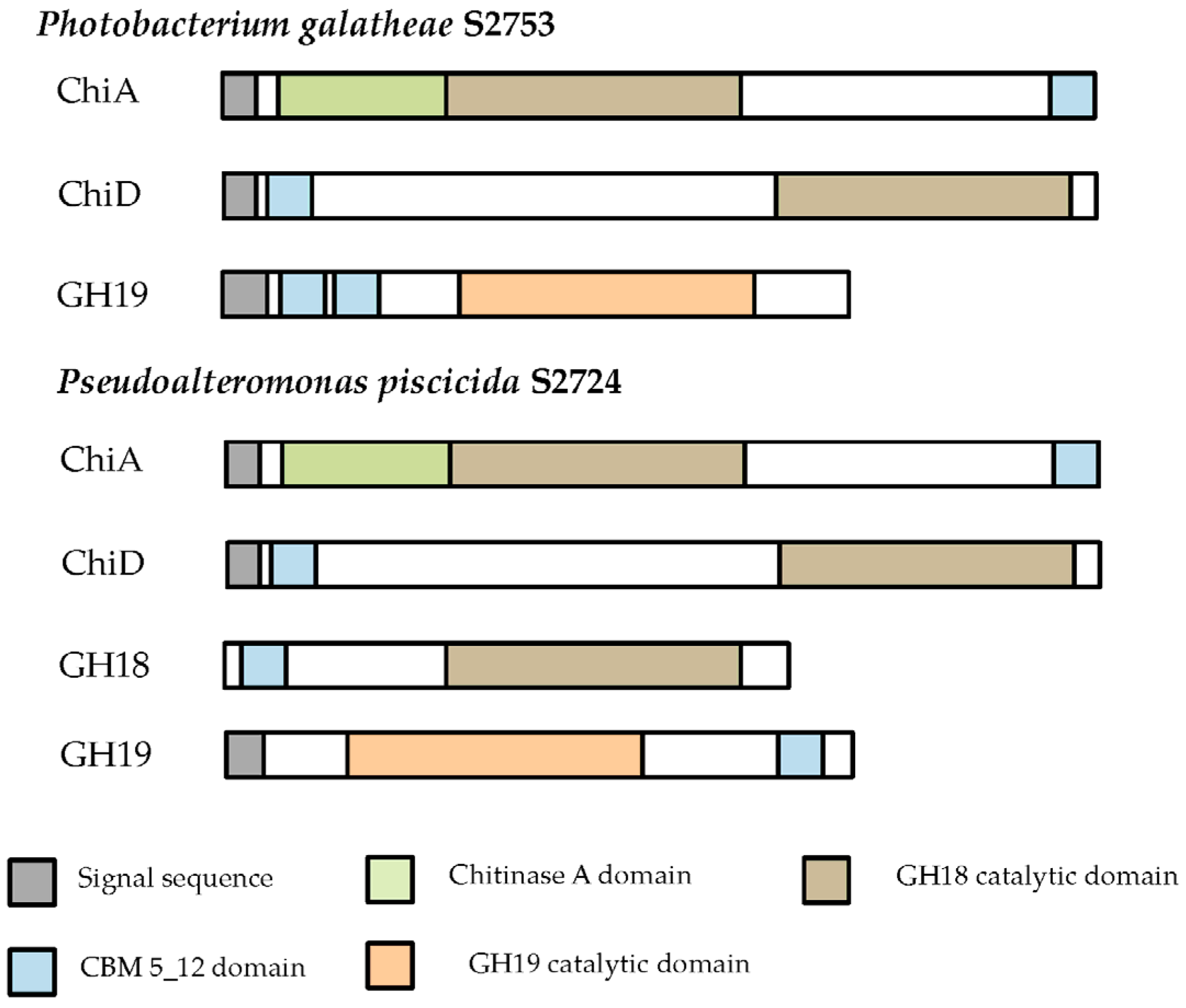
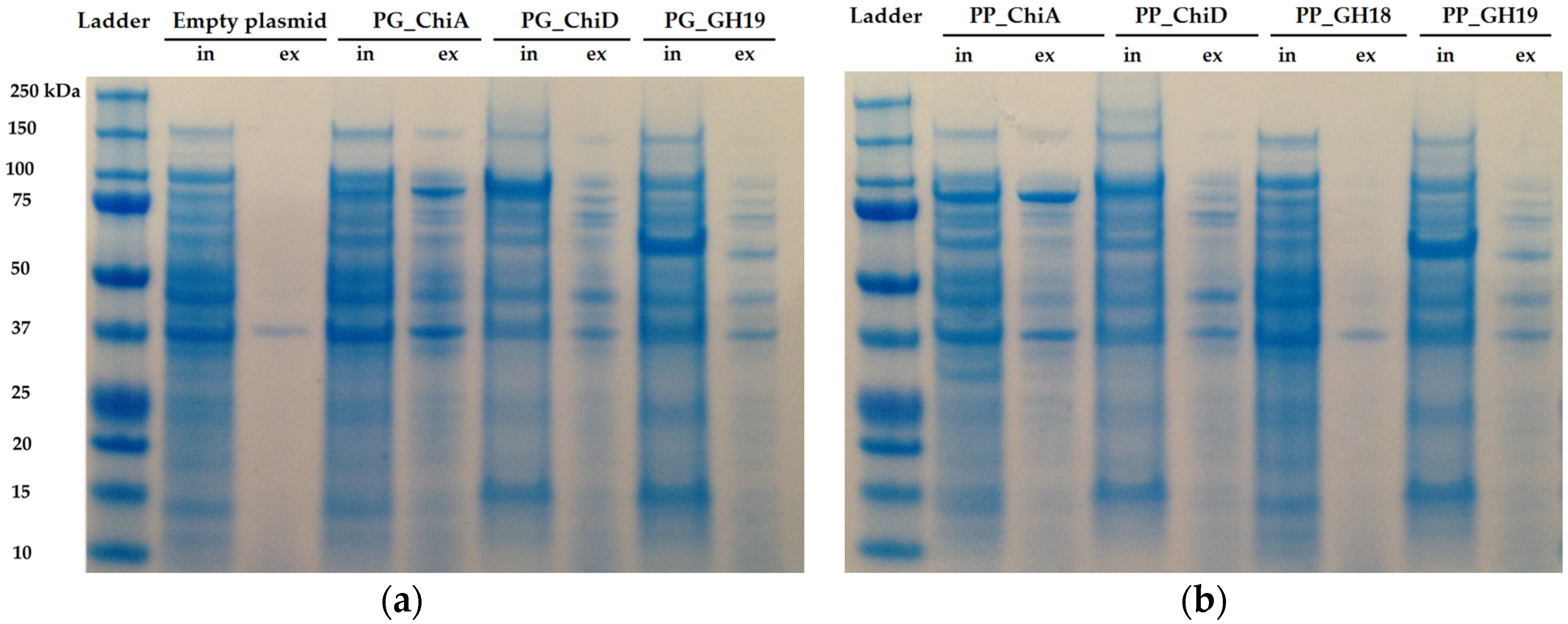
2.1.2. Cloning of Chitinases from S2753 and S2724
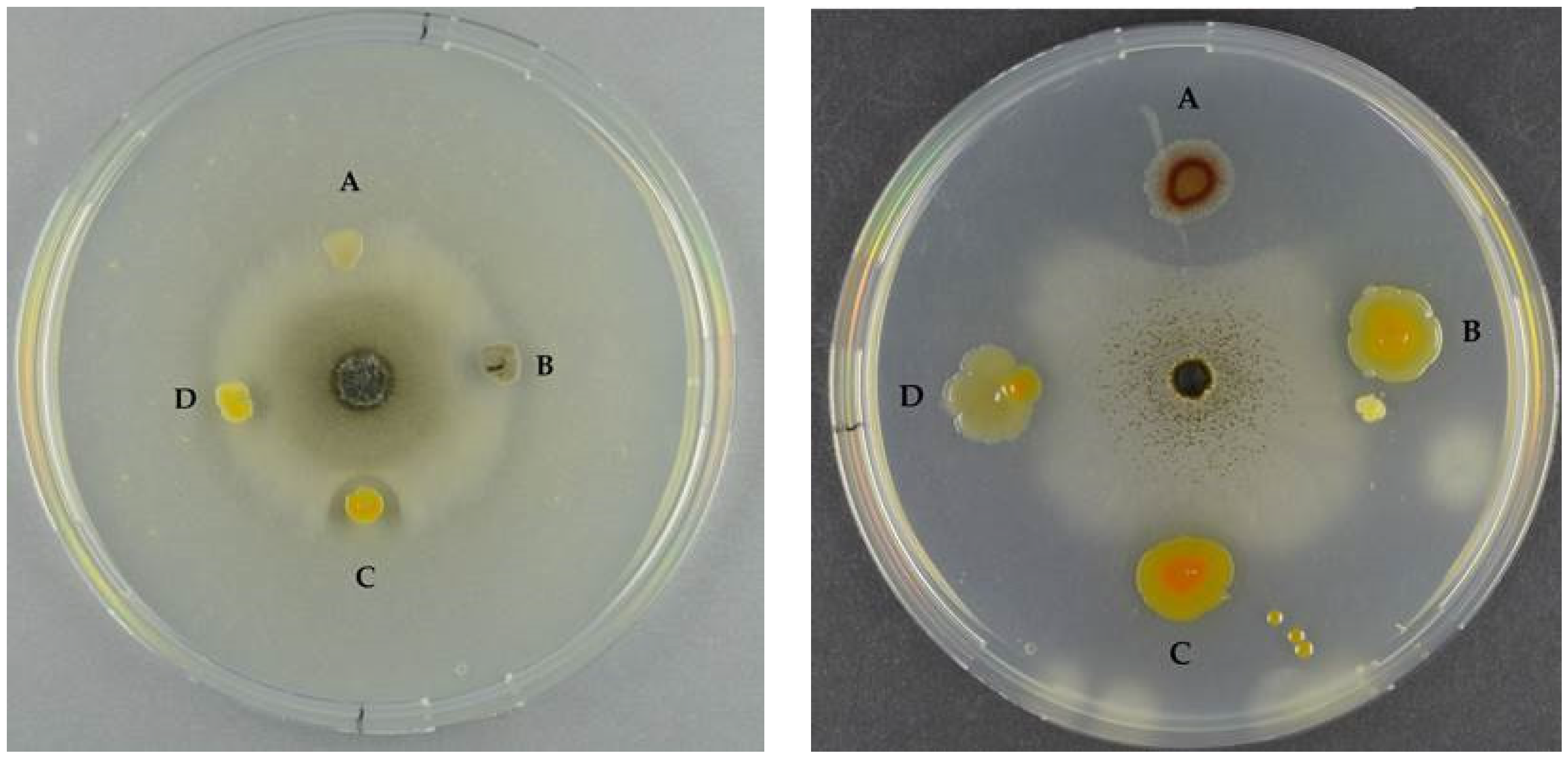
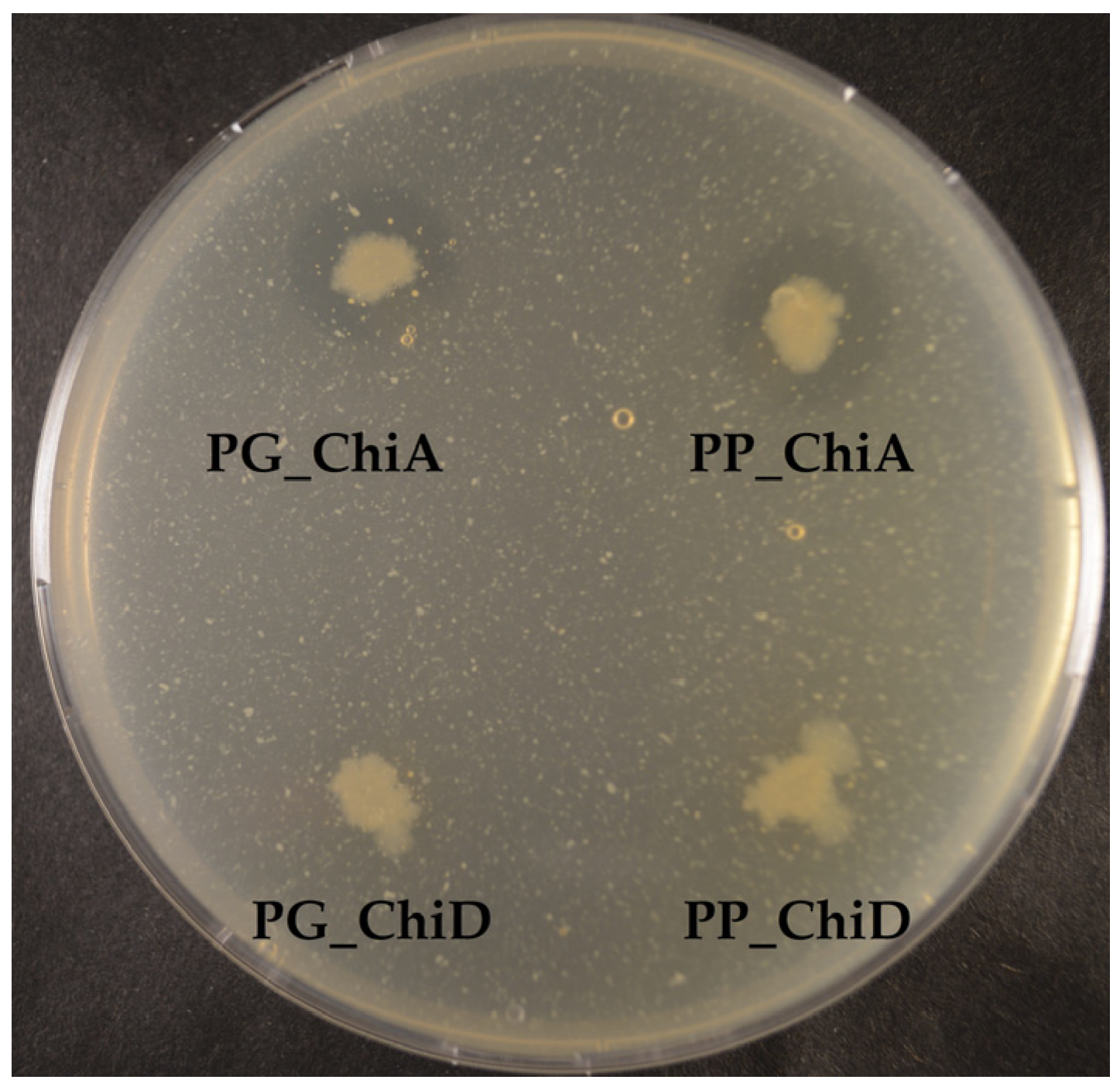
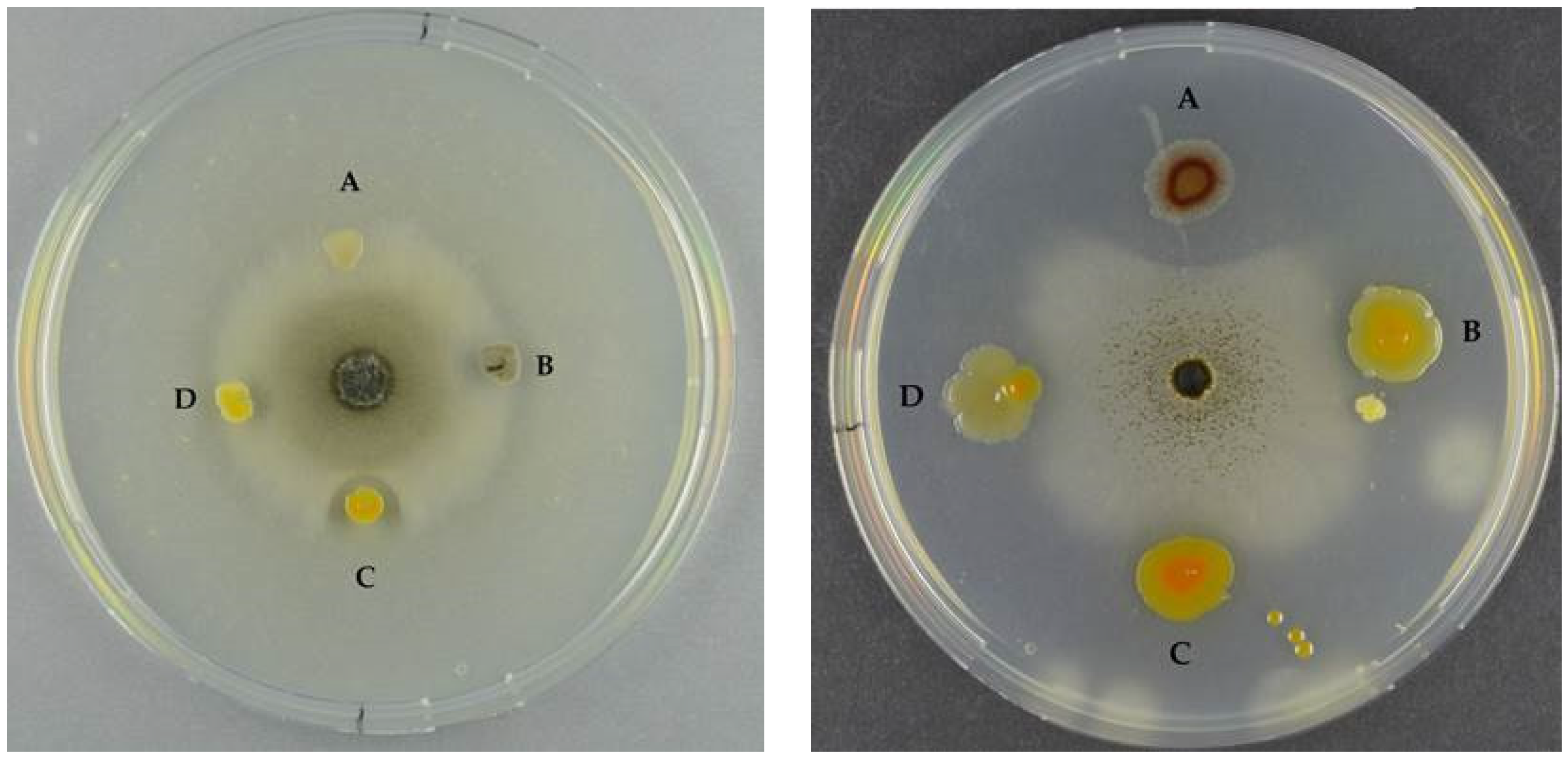
2.1.3. Antifungal Activity
3. Discussion
4. Materials and Methods
4.1. Strains and Plasmids
4.2. Preparation of Colloidal Chitin
4.3. Chitinase and Chitosanase Activity Screening
4.4. In Silico Analysis
4.5. Phylogenetic Analysis of Chitinases
4.6. Construction of Plasmids for Chitinase Expression
4.7. Protein Expression and SDS-Page Analysis
4.8. Antifungal Activity
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zobell, C.E.; Rittenberg, S.C. The Occurrence and Characteristics of Chitinoclastic Bacteria in the Sea. J Bacteriol. 1938, 35, 275–287. [Google Scholar] [PubMed]
- Gooday, G.W. Physiology of microbial degradation of chitin and chitosan. Biodegradation 1990, 1, 177–190. [Google Scholar] [CrossRef]
- Hayes, M.; Carney, B.; Slater, J.; Brück, W. Mining marine shellfish wastes for bioactive molecules: Chitin and chitosan—Part B: Applications. Biotechnol. J. 2008, 3, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Xi, C. Don’t waste seafood waste. Nature 2015, 524, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.; Sotelo, C.G.; Chapela, M.J.; Pérez-Martin, R.I. Towards sustainable and efficient use of fishery resources: Present and future trends. Trends Food Sci. Technol. 2007, 18, 29–36. [Google Scholar] [CrossRef]
- Kim, S.; Mendis, E. Bioactive compounds from marine processing byproducts—A review. J. Food Res. 2006, 39, 383–393. [Google Scholar] [CrossRef]
- Kawase, T.; Yokokawa, S.; Saito, A.; Fujii, T.; Miyashita, K.; Watanabe, T. Comparison of Enzymatic and Antifungal Properties between Family 18 and 19 Chitinases from S. coelicolor A3(2). Biosci. Biotechnol. Biochem. 2014, 8451, 988–998. [Google Scholar]
- García-Fraga, B.; Da Silva, A.F.; López-Seijas, J.; Sieiro, C. A novel family 19 chitinase from the marine-derived Pseudoalteromonas tunicata CCUG 44952T: Heterologous expression, characterization and antifungal activity. Biochem. Eng. J. 2015, 93, 84–93. [Google Scholar] [CrossRef]
- Hoster, F.; Schmitz, J.E.; Daniel, R. Enrichment of chitinolytic microorganisms: Isolation and characterization of a chitinase exhibiting antifungal activity against phytopathogenic fungi from a novel Streptomyces strain. Appl. Microbiol. Biotechnol. 2005, 66, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.I.; Hocking, A.D. Introduction. In Fungi and Food Spoilage; Springer Science: New York, NY, USA, 2009; p. 1. [Google Scholar]
- Andersen, B.; Dosen, I.; Lewinska, A.M.; Nielsen, K.F. Pre-contamination of new gypsum wallboard with potentially harmful fungal species. Indoor Air 2016. [Google Scholar] [CrossRef] [PubMed]
- Brzezinska, M.S.; Jankiewicz, U.; Burkowska, A.; Walczak, M. Chitinolytic microorganisms and their possible application in environmental protection. Curr. Microbiol. 2014, 68, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cai, J.; Xie, C.; Liu, C.; Chen, Y. Purification and partial characterization of a 36-kDa chitinase from Bacillus thuringiensis subsp. colmeri, and its biocontrol potential. Enzyme Microb. Technol. 2010, 46, 252–256. [Google Scholar] [CrossRef]
- Beier, S.; Bertilsson, S. Bacterial chitin degradation-mechanisms and ecophysiological strategies. Front. Microbiol. 2013, 4, 149. [Google Scholar] [CrossRef] [PubMed]
- Svitil, A.; Chadhain, S.; Moore, J.; Kirchman, D. Chitin Degradation Proteins Produced by the Marine Bacterium Vibrio harveyi Growing on Different Forms of Chitin. Appl. Environ. Microbiol. 1997, 63, 408–413. [Google Scholar] [PubMed]
- Orikoshi, H.; Nakayama, S.; Miyamoto, K.; Hanato, C.; Yasuda, M.; Inamori, Y.; Tsujibo, H. Roles of four chitinases (ChiA, ChiB, ChiC, and ChiD) in the chitin degradation system of marine bacterium Alteromonas sp. strain O-7. Appl. Environ. Microbiol. 2005, 71, 1811–1815. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kanai, R.; Kawase, T.; Tanabe, T.; Mitsutomi, M.; Sakuda, S.; Miyashita, K. Family 19 chitinases of Streptomyces species: Characterization and distribution. Microbiology 1999, 145, 3353–3363. [Google Scholar] [CrossRef] [PubMed]
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.; Liu, Z.; Zhai, H.; Sørlie, M.; Eisjink, V. An Oxidative Enzyme Boosting the enzymatic conversion of recalcitrant polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Fushinobu, S. Metalloproteins: A new face for biomass breakdown. Nat. Chem. Biol. 2013, 10, 88–89. [Google Scholar] [CrossRef] [PubMed]
- Levasseur, A.; Drula, E.; Lombard, V.; Coutinho, P.M.; Henrissat, B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 2013, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Hamre, A.G.; Eide, K.B.; Wold, H.H.; Sørlie, M. Activation of enzymatic chitin degradation by a lytic polysaccharide monooxygenase. Carbohydr. Res. 2015, 407, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Kadokura, K.; Rokutani, A.; Yamamoto, M.; Ikegami, T.; Sugita, H.; Itoi, S.; Hakamata, W.; Oku, T.; Nishio, T. Purification and characterization of Vibrio parahaemolyticus extracellular chitinase and chitin oligosaccharide deacetylase involved in the production of heterodisaccharide from chitin. Appl. Microbiol. Biotechnol. 2007, 75, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Kadokura, K.; Ikegami, T.; Shigeta, Y.; Kumaki, Y.; Hakamata, W.; Oku, T.; Nishio, T. Heterodisaccharide 4-O-(N-acetyl-beta-d-glucosaminyl)-d-glucosamine is a specific inducer of chitinolytic enzyme production in Vibrios harboring chitin oligosaccharide deacetylase genes. Glycobiology 2009, 19, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Hamer, S.N.; Cord-Landwehr, S.; Planas, A.; Waegeman, H.; Moerschbacher, B.M.; Kolkenbrock, S. Enzymatic production of defined chitosan oligomers with a specific pattern of acetylation using a combination of chitin oligosaccharide deacetylases. Sci. Rep. 2015, 5, 8716. [Google Scholar] [CrossRef] [PubMed]
- Gram, L.; Melchiorsen, J.; Bruhn, J.B. Antibacterial activity of marine culturable bacteria collected from a global sampling of ocean surface waters and surface swabs of marine organisms. Mar. Biotechnol. 2010, 12, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Wietz, M.; Månsson, M.; Gram, L. Chitin stimulates production of the antibiotic andrimid in a Vibrio coralliilyticus strain. Environ. Microbiol. Rep. 2011, 3, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Giubergia, S.; Phippen, C.; Gotfredsen, C.H.; Nielsen, K.F.; Gram, L. Influence of niche-specific nutrients on secondary metabolism in Vibrionaceae. Appl. Environ. Microbiol. 2016, 82, 4035–4044. [Google Scholar] [CrossRef] [PubMed]
- Machado, H.; Sonnenschein, E.C.; Melchiorsen, J.; Gram, L. Genome mining reveals unlocked bioactive potential of marine Gram-negative bacteria. BMC Genom. 2015, 16, 158. [Google Scholar] [CrossRef] [PubMed]
- Ramazzina, I.; Cendron, L.; Folli, C.; Berni, R.; Monteverdi, D.; Zanotti, G.; Percudani, R. Logical Identification of an Allantoinase Analog (puuE) Recruited from Polysaccharide Deacetylases. J. Biol. Chem. 2008, 283, 23295–23304. [Google Scholar] [CrossRef] [PubMed]
- Boer, H.; Munck, N.; Natunen, J.; Wohlfahrt, G.; Söderlund, H.; Renkonen, O.; Koivula, A. Differential recognition of animal type β4-galactosylated and -fucosylated chito-oligosaccharides by two family 18 chitinases from Trichoderma harzianum. Glycobiology 2004, 14, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Vaaje-Kolstad, G.; Bunæs, A.C.; Mathiesen, G.; Eijsink, V.G.H. The chitinolytic system of Lactococcus lactis ssp. lactis comprises a nonprocessive chitinase and a chitin-binding protein that promotes the degradation of α- And β-chitin. FEBS J. 2009, 276, 2402–2415. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sugawara, N.; Suzuki, M.; Uchiyama, T.; Katouno, F.; Nikaidou, N.; Watanabe, T. Chitinases A, B, and C1 of Serratia marcescens 2170 produced by recombinant Escherichia coli: Enzymatic properties and synergism on chitin degradation. Biosci. Biotechnol. Biochem. 2002, 66, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Katouno, F.; Nikaidou, N.; Nonaka, T.; Sugiyama, J.; Watanabe, T. Roles of the Exposed Aromatic Residues in Crystalline Chitin Hydrolysis by Chitinase A from Serratia marcescens 2170. J. Biol. Chem. 2001, 276, 41343–41349. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Yokokawa, S.; Saito, A.; Fujii, T.; Nikaidou, N.; Miyashita, K.; Watanabe, T. Comparison of enzymatic and antifungal properties between family 18 and 19 chitinases from S. coelicolor A3(2). Biosci. Biotechnol. Biochem. 2006, 70, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Jones, C.M.; Mohit, V.; Hallin, S.; Bertilsson, S. Global phylogeography of chitinase genes in aquatic metagenomes. Appl. Environ. Microbiol. 2011, 77, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Ito, Y.; Yamada, T.; Hashimoto, M.; Sekine, S.; Tanaka, H. The Roles of the C-Terminal Domain and Type and Type III Domains of Chitinase A1 from Bacillus circulans WL-12 in chitin degradation. J. Bacteriol. 1994, 176, 4465–4472. [Google Scholar] [CrossRef] [PubMed]
- Katouno, F.; Taguchi, M.; Sakurai, K.; Uchiyama, T.; Nikaidou, N.; Nonaka, T.; Sugiyama, J.; Watanabe, T. Importance of Exposed Aromatic Residues in Chitinase B from Serratia marcescens 2170 for Crystalline Chitin Hydrolysis. J. Biochem. 2004, 168, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Limón, M.C.; Lora, J.M.; García, I.; De La Cruz, J.; Llobell, A.; Benítez, T.; Pintor-Toro, J.A. Primary structure and expression pattern of the 33-kDa chitinase gene from the mycoparasitic fungus Trichoderma harzianum. Curr. Genet. 1995, 28, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Armand, S.; Hata, T.; Nikaidou, N.; Henrissat, B.; Mitsutomi, M.; Watanabe, T. A modular family 19 chitinase found in the prokaryotic organism Streptomyces griseus HUT 6037. J. Bacteriol. 1996, 178, 5065–5070. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Taniguchi, H.; Kitaoka, M. A reducing-end-acting chitinase from Vibrio proteolyticus belonging to glycoside hydrolase family 19. Appl. Microbiol. Biotechnol. 2008, 78, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Kojima, M.; Yoshikawa, T.; Mitsuda, N.; Araki, K.; Kawaguchi, T.; Miyatake, K.; Arai, M.; Fukamizo, T. A novel type of family 19 chitinase from Aeromonas sp. No. 10S-24. Eur. J. Biochem. 2003, 270, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Garbulewska, E.; Sato, K.; Kato, Y.; Nogawa, M.; Taguchi, G.; Shimosaka, M. Isolation of genes coding for chitin-degrading enzymes in the novel chitinolytic bacterium, Chitiniphilus shinanonensis, and characterization of a gene coding for a family 19 chitinase. JBIOSC 2012, 113, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Tsujibo, H.; Orikoshi, H.; Baba, N.; Miyamoto, K.; Yasuda, M.; Miyahara, M. Identification and Characterization of the Gene Cluster Involved in Chitin Degradation in a Marine Bacterium, Alteromonas sp. Appl. Environ. Microbiol. 2002, 68, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Shimosaka, M.; Ando, Y.; Nishiyama, K. Species-specific distribution of a modular family 19 chitinase gene in Burkholderia gladioli. FEMS Microbiol. Ecol. 2001, 37, 135–141. [Google Scholar] [CrossRef]
- Nakagawa, Y.S.; Kudo, M.; Loose, J.S.M.; Ishikawa, T.; Totani, K. A small lytic polysaccharide monooxygenase from Streptomyces griseus targeting alpha and beta chitin. FEBS J. 2015, 282, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, L.X.; Wang, X.; Roseman, S. The chitin catabolic cascade in the marine bacterium Vibrio cholerae: Characterization of a unique chitin oligosaccharide deacetylase. Glycobiology 2007, 17, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Uehera, R.; Shiraishu, H.; Hakamata, W.; Nishio, T. Chitin Oligosaccharide Deacetylase from Shewanella woodyi ATCC51908. J. Appl. Glycosci. 2015. [Google Scholar] [CrossRef]
- Liu, J.; Jia, Z.; Li, S.; Li, Y.; You, Q.; Zhang, C.; Zheng, X.; Xiong, G.; Zhao, J.; Qi, C.; et al. Identification and characterization of a chitin deacetylase from a metagenomic library of deep-sea sediments of the Arctic Ocean. Gene 2016, 590, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Park, R.D.; Muzzarelli, R.A.A. Chitin deacetylases: Properties and applications. Mar. Drugs 2010, 8, 24–46. [Google Scholar] [CrossRef] [PubMed]
- De Fuente-Salcido, N.M.; Casados-Vázquez, L.E.; García-Pérez, A.P.; Barboza-Pérez, U.E.; Bideshi, D.K.; Salcedo-hernández, R.; Almendarez, B.E.G.; Barboza-Corona, J.E. The endochitinase ChiA Btt of Bacillus thuringiensis 2803 and its potential use to control the phytopathogen Colletotrichum gloeosporioides. Microbiol. Open 2016, 5, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.D.P.; Silva, F.A.D.; de Castro Landim, P.G.; da Cruz, P.R.; de Brito, T.L.; de Medeiros, S.C.; Oliveira, J.T.A.; Vasconcelos, I.M.; Pereira, H.D.; Grangeiro, T.B. Expression and efficient secretion of a functional chitinase from Chromobacterium violaceum in Escherichia coli. BMC Biotechnol. 2013, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Suma, K.; Podile, A.R. Chitinase A from Stenotrophomonas maltophilia shows transglycosylation and antifungal activities. Bioresour. Technol. 2013, 133, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Rapid Annotation Using Subsystem Technology Server. Available online: http://rast.nmpdr.org/ (accessed on 14 December 2016).
- The National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 14 December 2016).
- Pfam. Available online: http://pfam.xfam.org/ (accessed on 14 December 2016).
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, 279–285. [Google Scholar] [CrossRef] [PubMed]
- BLAST. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 13 December 2016).
- SignalP. Available online: http://www.cbs.dtu.dk/services/SignalP/ (accessed on 13 December 2016).
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 13 December 2016).
- Nour-Eldin, H.H.; Hansen, B.G.; Nørholm, M.H.H.; Jensen, J.K.; Halkier, B.A. Advancing uracil-excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Res. 2006, 34, e122. [Google Scholar] [CrossRef] [PubMed]
- Nørholm, M.H.H. A mutant Pfu DNA polymerase designed for advanced uracil-excision DNA engineering. BMC Biotechnol. 2010, 10, 21. [Google Scholar]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Chem. 1976, 254, 248–254. [Google Scholar] [CrossRef]
- Compute PI. Available online: http://web.expasy.org/compute_pi/ (accessed on 13 December 2016).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Species | Chitinase Activity | # Of Chitinolytic Enzymes | CSS Type | |||||
|---|---|---|---|---|---|---|---|---|---|
| GH18 (ChiA) | GH18 (ChiD) | GH18 (U) | GH19 | COD | LPMO | ||||
| S2753 | P. galatheae | ++ | 1 | 1 * | 0 | 1 | 0 | 1 | ChiS |
| S2052 | V. coralliilyticus | + | 4 ** | 1 | 0 | 1 | 1 | 2 | ChiS |
| S2604 | V. nigripulchritudo | + | 2 | 1 | 0 | 3 | 1 | 1 | ChiS |
| S2394 | V. neptunius | + | 2 | 1 | 0 | 1 | 1 | 2 | ChiS |
| S2757 | V. galatheae | + | 1 | 0 | 0 | 1 | 1 | 0 | ChiS |
| S1110 | V. fluvialis | + | 1 | 0 | 0 | 1 | 0 | 1 | ChiS |
| S2040 | P. piscicida | ++ | 0 | 1 * | 1 | 1 | 1 | 1 | CdsS |
| S2724 | P. piscicida | ++ | 1 | 1 | 1 | 1 | 1 | 2 | CdsS |
| S3137 | P. ruthenica | + | 1 | 1 | 0 | 1 | 0 | 1 | CdsS |
| S2471 | P. rubra | + | 2 | 1 | 2 | 1 | 0 | 2 | CdsS |
| S3431 | P. fuliginea | − | 0 | 0 | 0 | 0 | 0 | 0 | CdsS |
| Protein | Name | Length (Amino Acids) | Expected Size (kDa) |
|---|---|---|---|
| KJY84504 | PG_ChiA | 834 | 87.7 |
| KDM89921 | PG_ChiD | 846 | 87.9 |
| KJY85094 | PG_GH19 | 539 | 59.4 |
| KJY94723 | PP_ChiA | 822 | 87.5 |
| KJZ01532 | PP_ChiD | 850 | 90.4 |
| KJZ02335 | PP_GH18 | 455 | 49.3 |
| TW73_14810 * | PP_GH19 | 479 | 53.1 |
| Strain | Activity against Fungal Strains | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Penicillium chrysogenum | Stachybotrys chartarum | Chaetomium globosum | Neosartorya hiratsukae | Aspergillus niger | Fusarium oxysporum | Botrytis cinerea | ||||||||
| A | P | A | P | A | P | A | P | A | P | A | P | A | P | |
| S2753 | + | nt | + | sg | − | nt | + | nt | + | nt | + | nt | + | sg |
| S2052 | + | + | + | sg | − | − | − | + | + | + | − | − | + | sg |
| S2604 | − | + | − | sg | − | + | + | + | + | + | − | − | + | sg |
| S2394 | − | nt | − | sg | − | nt | − | nt | + | nt | − | nt | + | sg |
| S2757 | − | − | − | sg | − | + | − | − | + | − | − | − | + | sg |
| S1110 | + | + | − | sg | − | − | − | − | + | + | − | − | + | sg |
| S2040 | ++ | ++ | ++ | sg | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | sg |
| S2724 | + | + | + | sg | − | − | − | − | + | + | − | − | + | sg |
| S3137 | + | ++ | − | sg | − | + | + | + | + | + | − | + | + | sg |
| S2471 | + | − | − | sg | − | + | − | − | + | ++ | − | − | + | sg |
| S3431 | + | nt | + | nt | − | nt | − | nt | + | nt | − | nt | + | sg |
| Strain | Species | Accession Number |
|---|---|---|
| S2753 | Photobacterium galatheae | JMIB01 |
| S2052 | Vibrio coralliilyticus | JXXR01 |
| S2604 | Vibrio nigripulchritudo | JXXT01 |
| S2394 | Vibrio neptunius | JXXU01 |
| S2757 | Vibrio galatheae | JXXV01 |
| S1110 | Vibrio fluvialis | LKHR01 |
| S2040 | Pseudoalteromonas piscicida | JXXW01 |
| S2724 | Pseudoalteromonas piscicida | JXXX01 |
| S3137 | Pseudoalteromonas ruthenica | JXXZ01 |
| S2471 | Pseudoalteromonas rubra | JXYA01 |
| S3431 | Pseudoalteromonas fuliginea | JJNY01 |
| Strain/Plasmid | Details | Reference |
|---|---|---|
| Escherichia coli Top10 | Cloning host | Invitrogen, C404010, Paisley, United Kingdom |
| Escherichia coli BL21 (DE3) | Expression host | Novagen, Madison, WI, USA |
| pBAD_Myc_HisA | Cloning and expression vector | Thermo Scientific, V44001, Waltham, MA, USA |
| PG_ChiA | pBAD_Myc_HisA vector containing the EA58_02560 gene | This study |
| PG_ChiD | pBAD_Myc_HisA vector containing the EA58_19900 gene | This study |
| PG_GH19 | pBAD_Myc_HisA vector containing the EA58_12180 gene | This study |
| PP_ChiA | pBAD_Myc_HisA vector containing the TW73_17595 gene | This study |
| PP_ChiD | pBAD_Myc_HisA vector containing the TW73_14030 gene | This study |
| PP_GH18 | pBAD_Myc_HisA vector containing the TW73_13265 gene | This study |
| PP_GH19 | pBAD_Myc_HisA vector containing the TW73_14810 gene | This study |
| Primer | Sequence 5′–3′ |
|---|---|
| pBAD_Myc_HisA_fw | AATTCGAAGCUTGGGCCCGAA |
| pBAD_Myc_HisA_rv | ATGGTTAATUCCTCCTGTTAGCC |
| PG_ChiA_fw | AATTAACCAUGTCTTTCAATAAGTTGAGTCCTATTGC |
| PG_ChiA_rv | AGCTTCGAATUCTGGCAGTTTGCTGCACCCA |
| PG_ChiD_fw | AATTAACCAUGCGTAAAACTCTGATTCAGACAGCTGT |
| PG_ChiD_rv | AGCTTCGAATUCTGAGCGTTCATAGCATCCAGCTTC |
| PG_GH19_fw | AATTAACCAUGAAACAAAAACTGTCCCCTCAATGGG |
| PG_GH19_rv | AGCTTCGAATUCTCAACGGTGACACCATAATATTTCTGG |
| PP_ChiA_fw | AATTAACCAUGAAACTTAATAAAATAACCAGCTATATAGGACTTG |
| PP_ChiA_rv | AGCTTCGAATUGTTAGTTACTGCCTTCCATACATCAGC |
| PP_ChiD_fw | AATTAACCAUGAAACCAACTTCTATATTACGATTGGCTTGG |
| PP_ChiD_rv | AGCTTCGAATUATTTCCTTGATTCATCTGCGTTAATTTATCGC |
| PP_GH18_fw | AATTAACCAUGGAAGTTGCACTGGCGGTTGACT |
| PP_GH18_rv | AGCTTCGAATUCTGACATTGATAGCTTGGTGTTACACCA |
| PP_GH19_fw | AATTAACCAUGAACAGTCTAAAATTAGCGACCGCAGTT |
| PP_GH19_rv | AGCTTCGAATUGTTAACCGCTAACCAAGGACCCG |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulsen, S.S.; Andersen, B.; Gram, L.; Machado, H. Biological Potential of Chitinolytic Marine Bacteria. Mar. Drugs 2016, 14, 230. https://doi.org/10.3390/md14120230
Paulsen SS, Andersen B, Gram L, Machado H. Biological Potential of Chitinolytic Marine Bacteria. Marine Drugs. 2016; 14(12):230. https://doi.org/10.3390/md14120230
Chicago/Turabian StylePaulsen, Sara Skøtt, Birgitte Andersen, Lone Gram, and Henrique Machado. 2016. "Biological Potential of Chitinolytic Marine Bacteria" Marine Drugs 14, no. 12: 230. https://doi.org/10.3390/md14120230
APA StylePaulsen, S. S., Andersen, B., Gram, L., & Machado, H. (2016). Biological Potential of Chitinolytic Marine Bacteria. Marine Drugs, 14(12), 230. https://doi.org/10.3390/md14120230