
Synthesis of the Oligosaccharides Related to Branching Sites of Fucosylated Chondroitin Sulfates from Sea Cucumbers

Abstract
: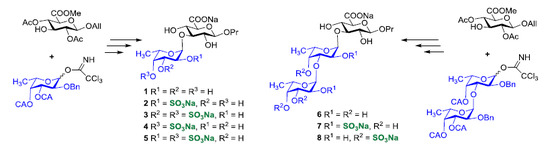
1. Introduction
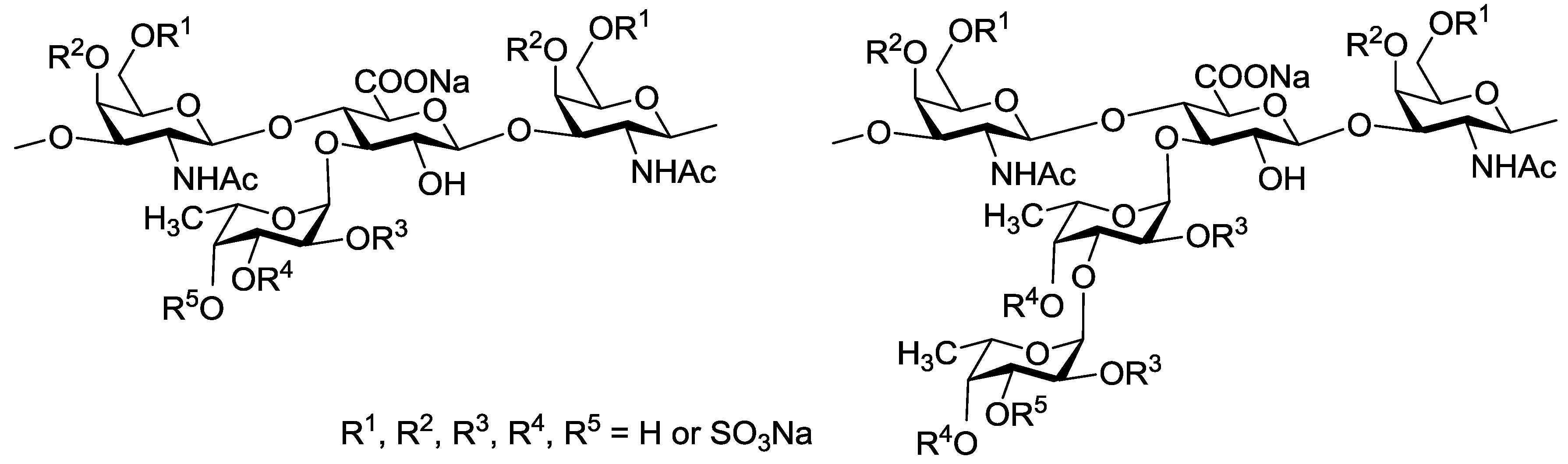
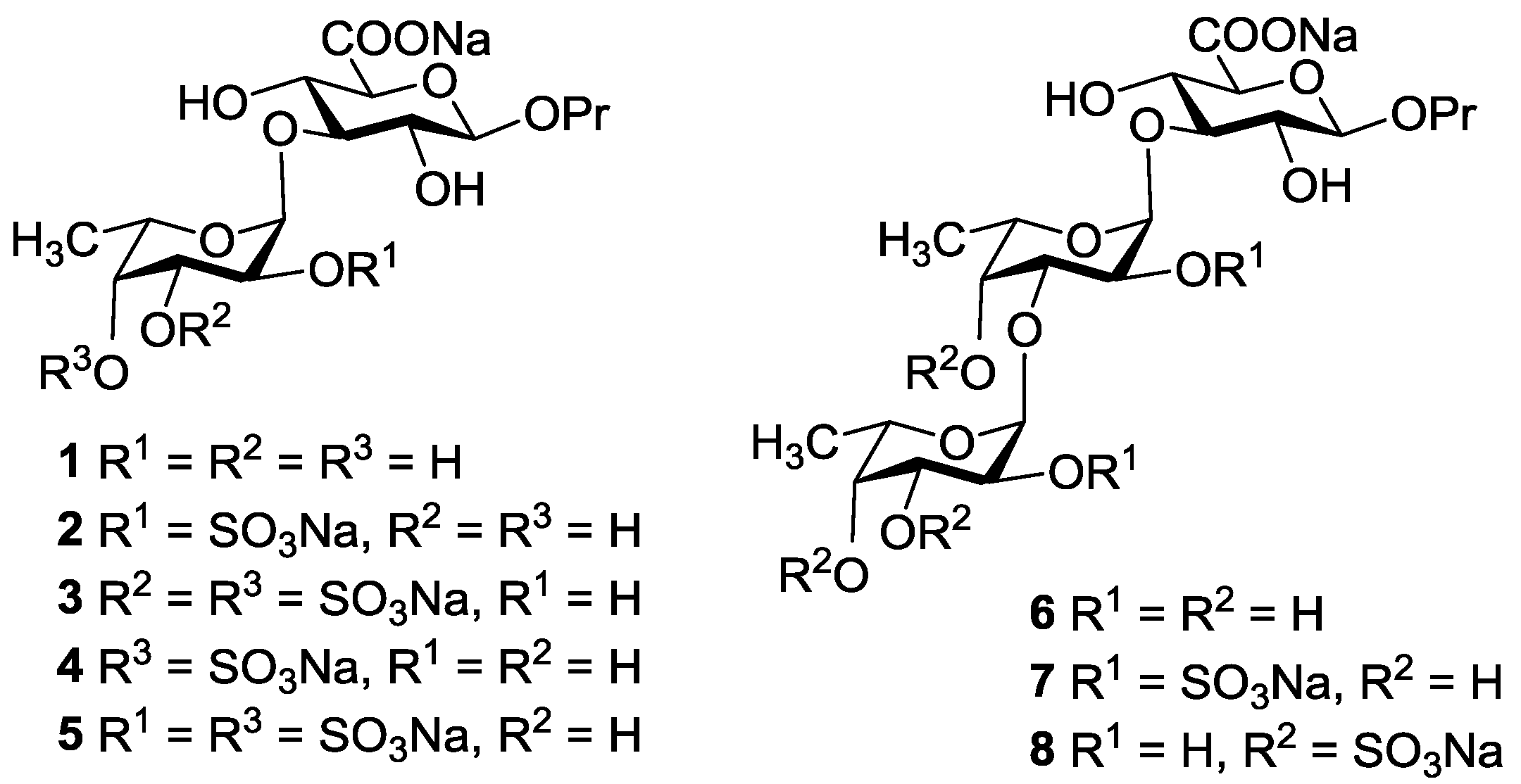
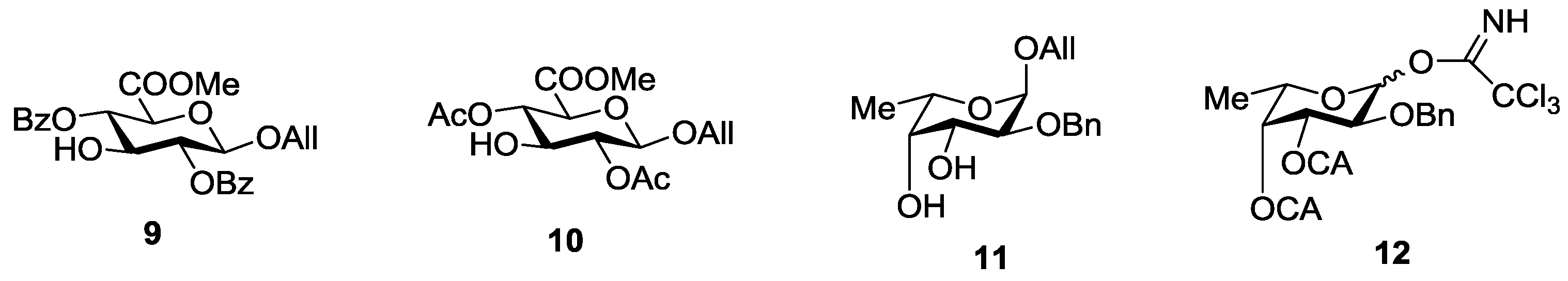
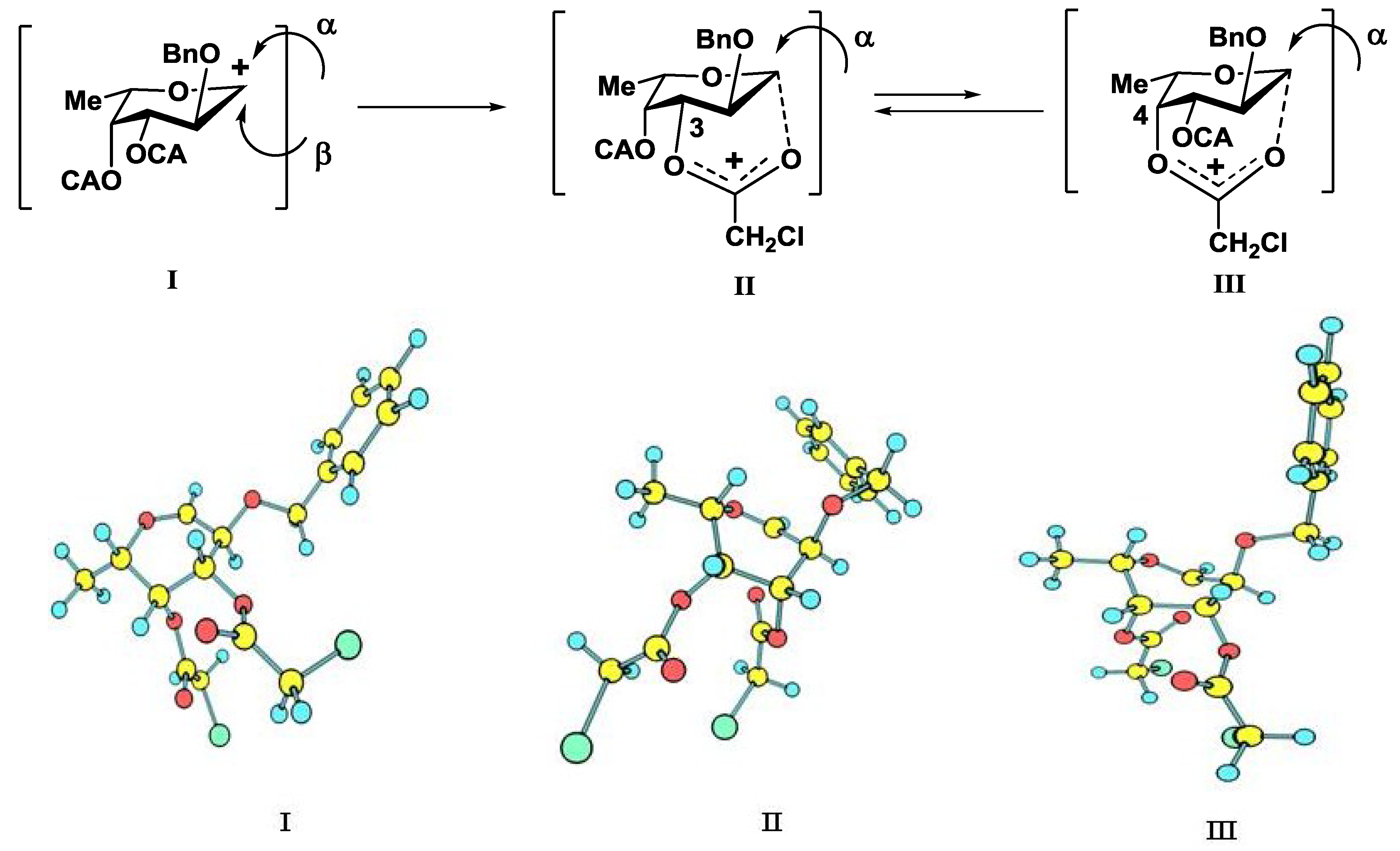
2. Results and Discussion
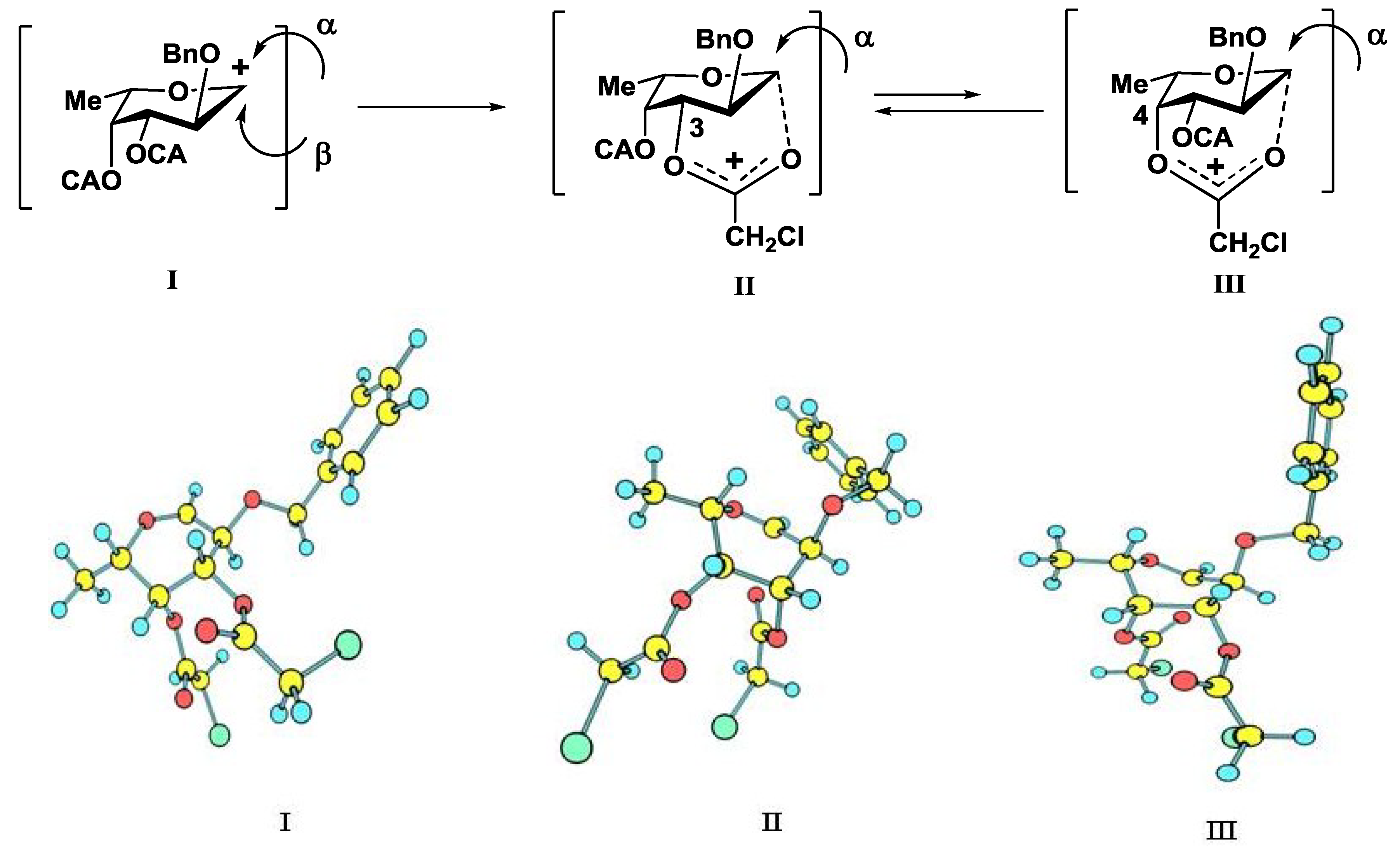

{kind=link}
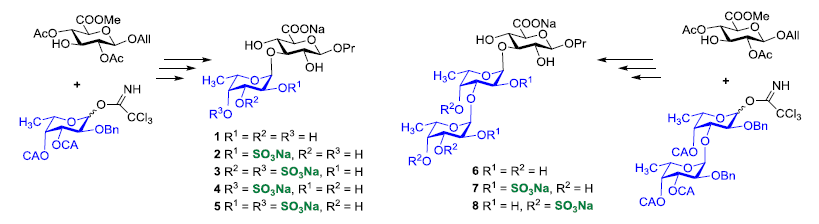
{kind=link}
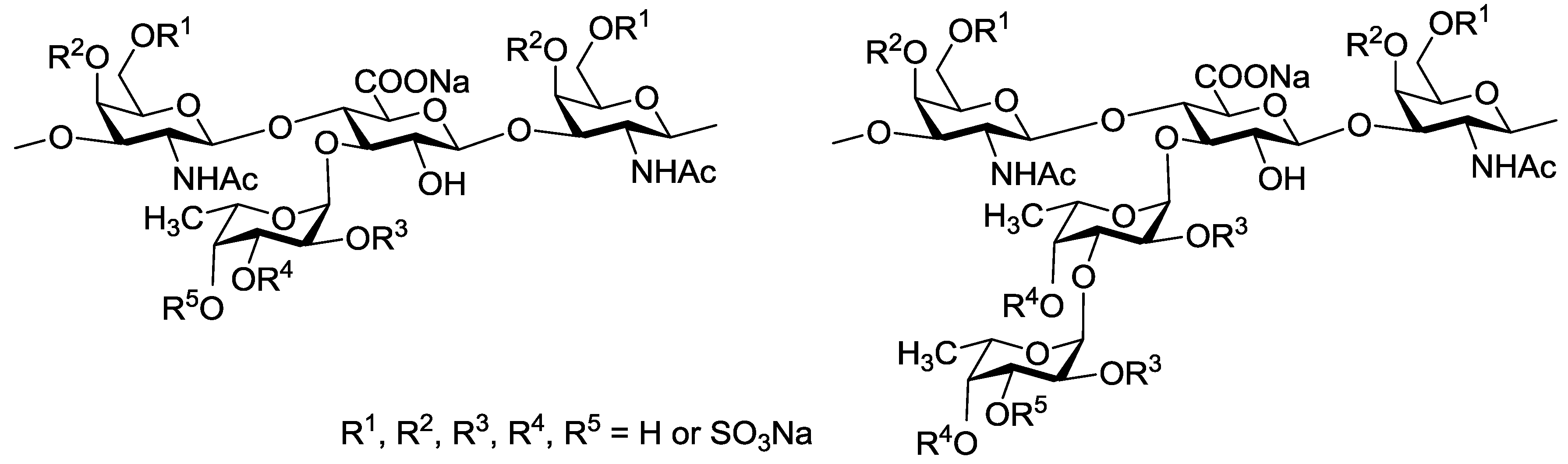
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | The Stabilizing Group | Stabilization Energy Calculated at MM+ Level, Kcal/Mole | Stabilization Energy Calculated at SCF/MP2 Level, Kcal/Mole |
|---|---|---|---|
 | 3-O-Ac | 11.6 | 15.5 |
| 4-O-Bz | 9.7 | 15.5 | |
 | 3-O-C(O)CH2Cl | 11.1 | 10.4 |
| 4-O-C(O)CH2Cl | 6.2 | 8.6 |

| Compound | Residue | C1 | C2 | C3 | C4 | C5 | C6 |
|---|---|---|---|---|---|---|---|
| 1 | →3)-β-d-GlcA | 103.2 | 74.8 | 83.5 | 71.7 | 78.0 | 176.9 |
| α-l-Fuc-(1→3 | 100.7 | 69.6 | 70.8 | 73.2 | 68.1 | 16.5 | |
| 2 | →3)-β-d-GlcA | 103.0 | 74.7 | 83.2 | 71.6 | 77.9 | 176.8 |
| α-l-Fuc-(1→3 | 98.4 | 76.8 | 68.7 | 73.3 | 68.0 | 16.5 | |
| 3 | →3)-β-d-GlcA | 103.5 | 74.6 | 83.5 | 71.6 | 78.1 | 176.8 |
| α-l-Fuc-(1→3 | 100.5 | 67.8 | 76.6 | 80.4 | 67.8 | 17.0 | |
| 4 | →3)-β-d-GlcA | 102.1 | 73.4 | 82.2 | 70.5 | 76.8 | 176.8 |
| α-l-Fuc-(1→3 | 99.1 | 68.6 | 68.6 | 81.0 | 66.4 | 17.0 | |
| 5 | →3)-β-d-GlcA | 103.0 | 74.7 | 83.2 | 71.5 | 77.9 | 176.8 |
| α-l-Fuc-(1→3 | 98.3 | 76.6 | 67.8 | 82.1 | 67.4 | 16.9 | |
| 6 | →3)-β-d-GlcA | 103.1 | 74.6 | 83.6 | 71.7 | 78.0 | 176.8 |
| →3)-α-l-Fuc-(1→3 | 100.7 | 70.7 | 73.2 | 69.5 | 68.1 | 16.5 | |
| α-l-Fuc-(1→3 | 96.5 | 69.3 | 68.2 | 75.9 | 67.9 | 16.5 | |
| 7 | →3)-β-d-GlcA | 103.1 | 75.2 | 83.2 | 71.6 | 78.0 | 176.9 |
| →3)-α-l-Fuc-(1→3 | 98.5 | 74.7 | 74.3 | 70.0 | 68.2 | 16.5 | |
| α-l-Fuc-(1→3 | 95.5 | 76.5 | 68.6 | 73.4 | 67.6 | 16.5 | |
| 8 | →3)-β-d-GlcA | 102.1 | 73.4 | 82.2 | 70.5 | 76.8 | 176.8 |
| →3)-α-l-Fuc-(1→3 | 99.1 | 68.6 | 74.0 | 81.2 | 66.4 | 16.9 | |
| α-l-Fuc-(1→3 | 100.5 | 67.8 | 76.5 | 80.4 | 67.8 | 17.0 |
| Compound | Residue | H1 | H2 | H3 | H4 | H5 | H6 |
|---|---|---|---|---|---|---|---|
| 1 | →3)-β-d-GlcA | 4.48 | 3.51 | 3.60 | 3.63 | 3.74 | - |
| α-l-Fuc-(1→3 | 5.27 | 3.79 | 3.92 | 3.82 | 4.37 | 1.20 | |
| 2 | →3)-β-d-GlcA | 4.49 | 3.55 | 3.65 | 3.65 | 3.74 | - |
| α-l-Fuc-(1→3 | 5.57 | 4.48 | 4.05 | 3.90 | 4.44 | 1.21 | |
| 3 | →3)-β-d-GlcA | 4.48 | 3.55 | 3.67 | 3.63 | 3.74 | - |
| α-l-Fuc-(1→3 | 5.36 | 4.00 | 4.65 | 4.90 | 4.52 | 1.27 | |
| 4 | →3)-β-d-GlcA | 4.47 | 3.50 | 3.62 | 3.62 | 3.73 | - |
| α-l-Fuc-(1→3 | 5.30 | 3.83 | 4.02 | 4.62 | 4.50 | 1.23 | |
| 5 | →3)-β-d-GlcA | 4.47 | 3.55 | 3.63 | 3.66 | 3.73 | - |
| α-l-Fuc-(1→3 | 5.60 | 4.47 | 4.17 | 4.69 | 4.55 | 1.27 | |
| 6 | →3)-β-d-GlcA | 4.49 | 3.55 | 3.63 | 3.65 | 3.74 | - |
| →3)-α-l-Fuc-(1→3 | 5.31 | 3.98 | 3.84 | 4.04 | 4.31 | 1.21 | |
| α-l-Fuc-(1→3 | 5.09 | 3.82 | 3.95 | 3.95 | 4.37 | 1.21 | |
| 7 | →3)-β-d-GlcA | 4.49 | 3.63 | 3.65 | 3.67 | 3.74 | - |
| →3)-α-l-Fuc-(1→3 | 5.60 | 4.57 | 4.07 | 4.10 | 4.42 | 1.24 | |
| α-l-Fuc-(1→3 | 5.34 | 4.45 | 4.12 | 3.91 | 4.49 | 1.26 | |
| 8 | →3)-β-d-GlcA | 4.46 | 3.55 | 3.63 | 3.65 | 3.73 | - |
| →3)-α-l-Fuc-(1→3 | 5.32 | 4.00 | 4.08 | 4.76 | 4.46 | 1.24 | |
| α-l-Fuc-(1→3 | 5.20 | 3.92 | 4.62 | 4.90 | 4.46 | 1.27 |
3. Experimental Section
3.1. Materials
3.2. Characterization of Compounds
3.3. Chemical Synthesis
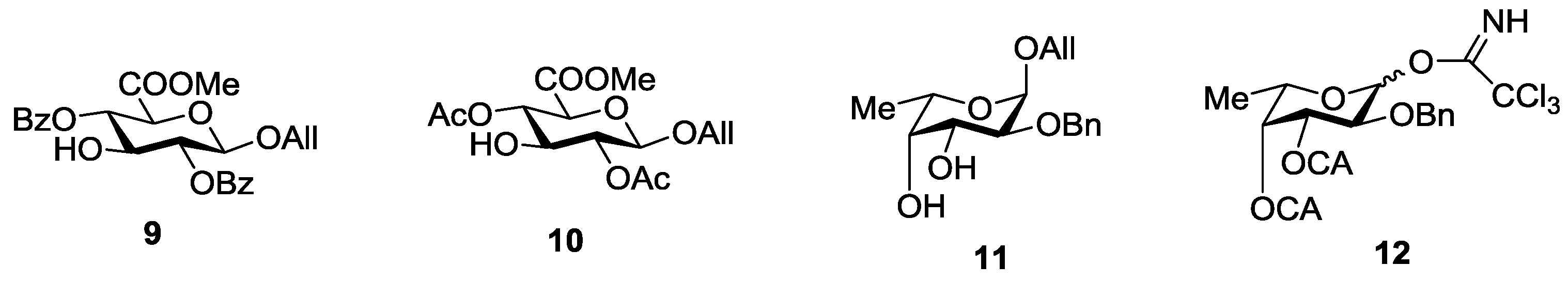
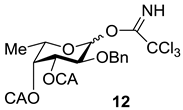
3.3.1. 2-O-Benzyl-3,4-di-O-chloroacetyl-l-fucopyranosyl Trichloroacetimidate (12)
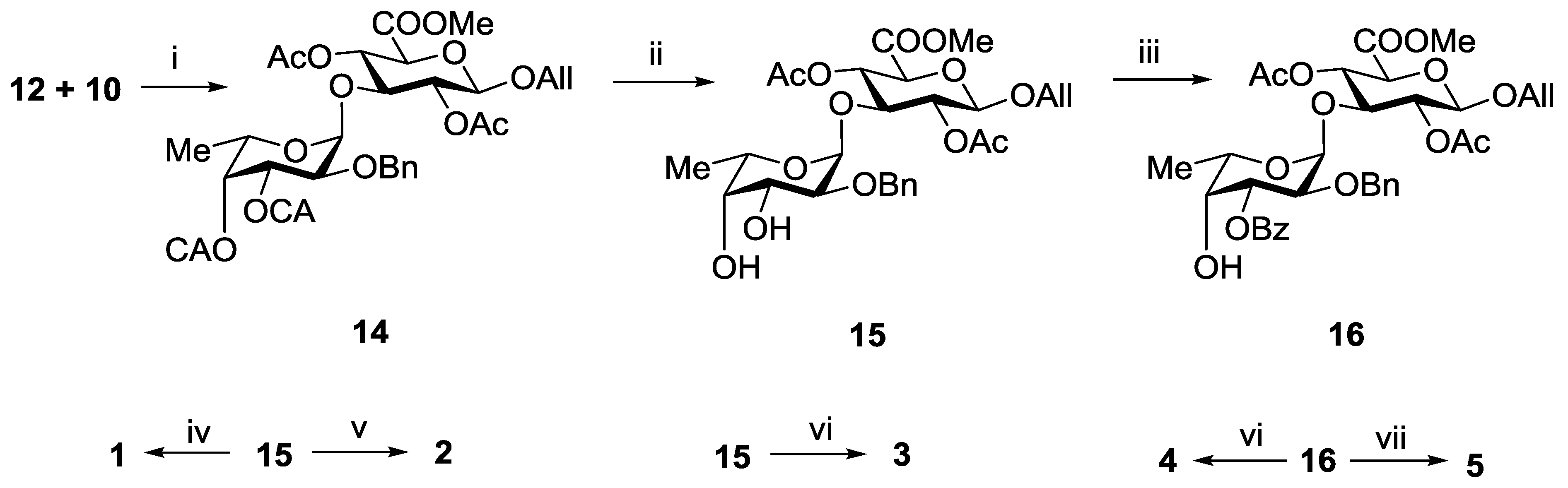
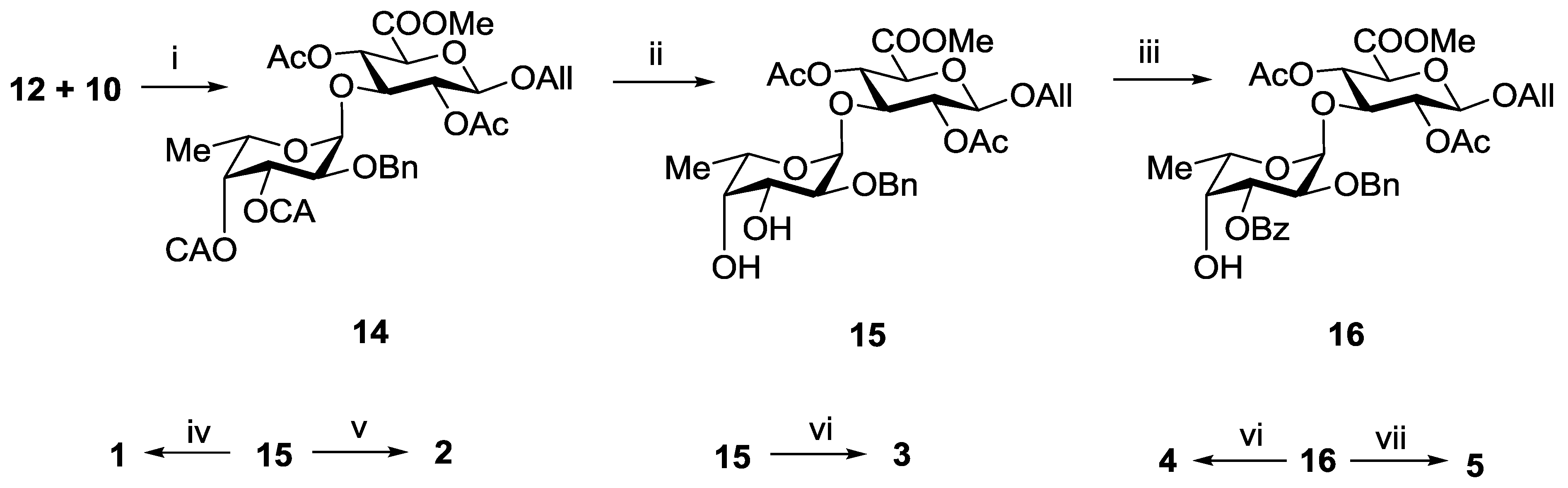
3.3.2. Allyl 2-O-benzyl-3,4-di-O-chloroacetyl-α-l-fucopyranosyl-(1→3)-(methyl 2,4-di-O-acetyl-β-d-glucopyranosyl Uronate) (14)
3.3.3. Allyl 2-O-benzyl-α-l-fucopyranosyl-(1→3)-(methyl 2,4-di-O-acetyl-β-d-glucopyranosyl Uronate) (15)
3.3.4. Allyl 2-O-benzyl-3-O-benzoyl-α-l-fucopyranosyl-(1→3)-(methyl 2,4-di-O-acetyl-β-d-glucopyranosyl Uronate) (16)
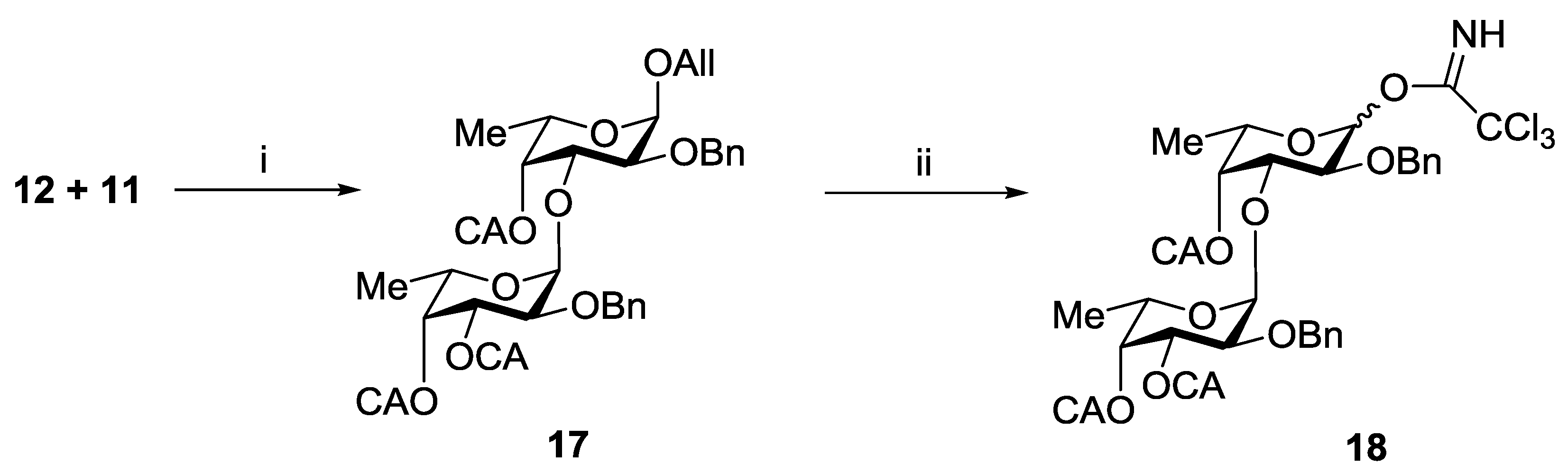
3.3.5. Allyl 2-O-benzyl-3,4-di-O-chloroacetyl-α-l-fucopyranosyl-(1→3)-2-O-benzyl-4-O-cloroacetyl-α-l-fucopyranoside (17)
3.3.6. 2-O-Benzyl-3,4-di-O-chloroacetyl-α-l-fucopyranosyl-(1→3)-2-O-benzyl-4-O-cloroacetyl-l-fucopyranosyl Trichloroacetimidate (18)
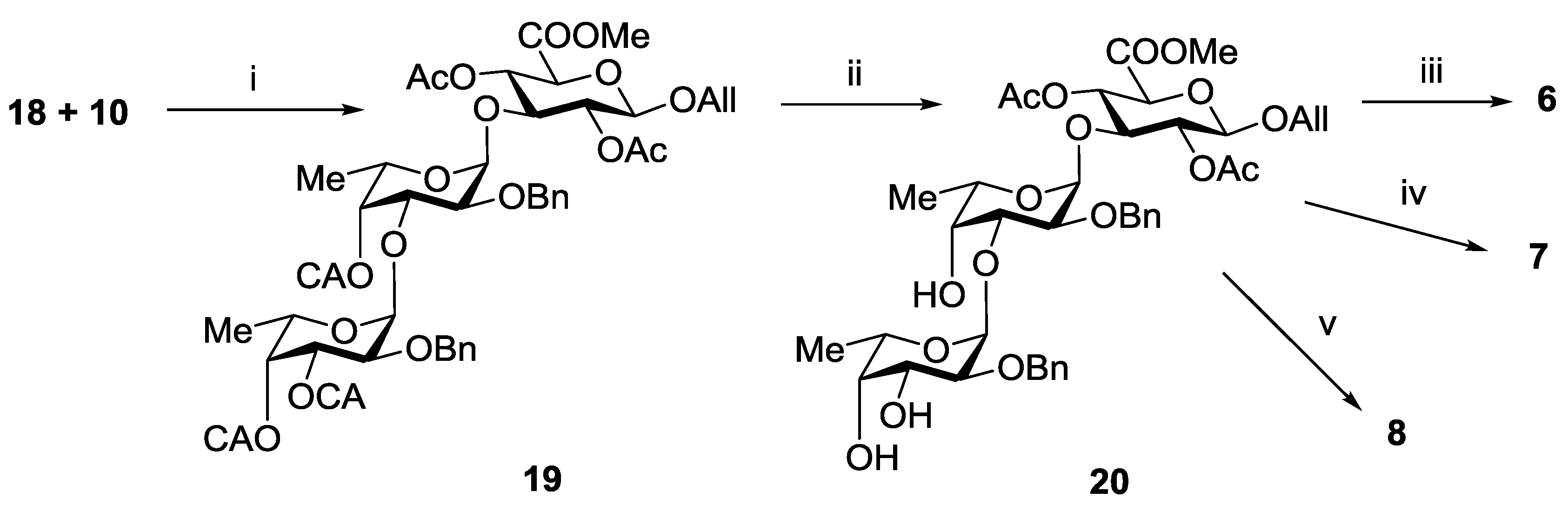
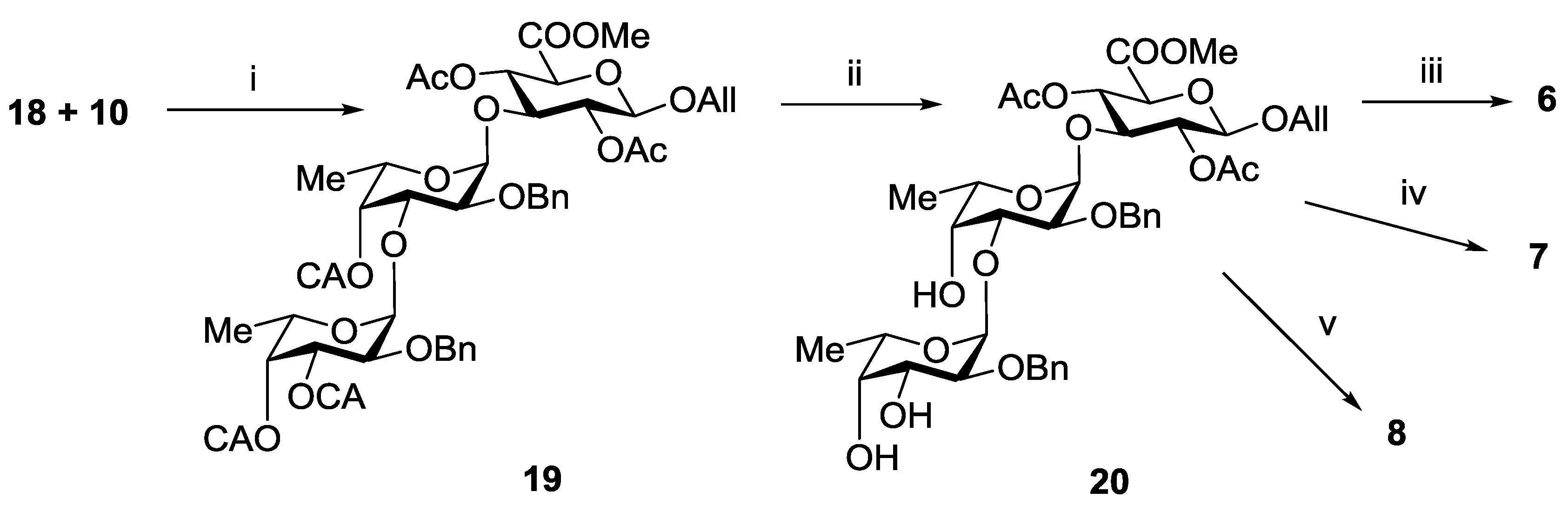
3.3.7. Allyl 2-O-benzyl-3,4-di-O-chloroacetyl-α-l-fucopyranosyl-(1→3)-2-O-benzyl-4-O-chloroacetyl-α-l-fucopyranosyl-(1→3)-(methyl 2,4-di-O-acetyl-β-d-glucopyranosyl Uronate) (19)
3.3.8. Allyl 2-O-benzyl-α-l-fucopyranosyl-(1→3)-2-O-benzyl-α-l-fucopyranosyl-(1→3)-(methyl 2,4-di-O-acetyl-β-d-glucopyranosyl Uronate) (20)
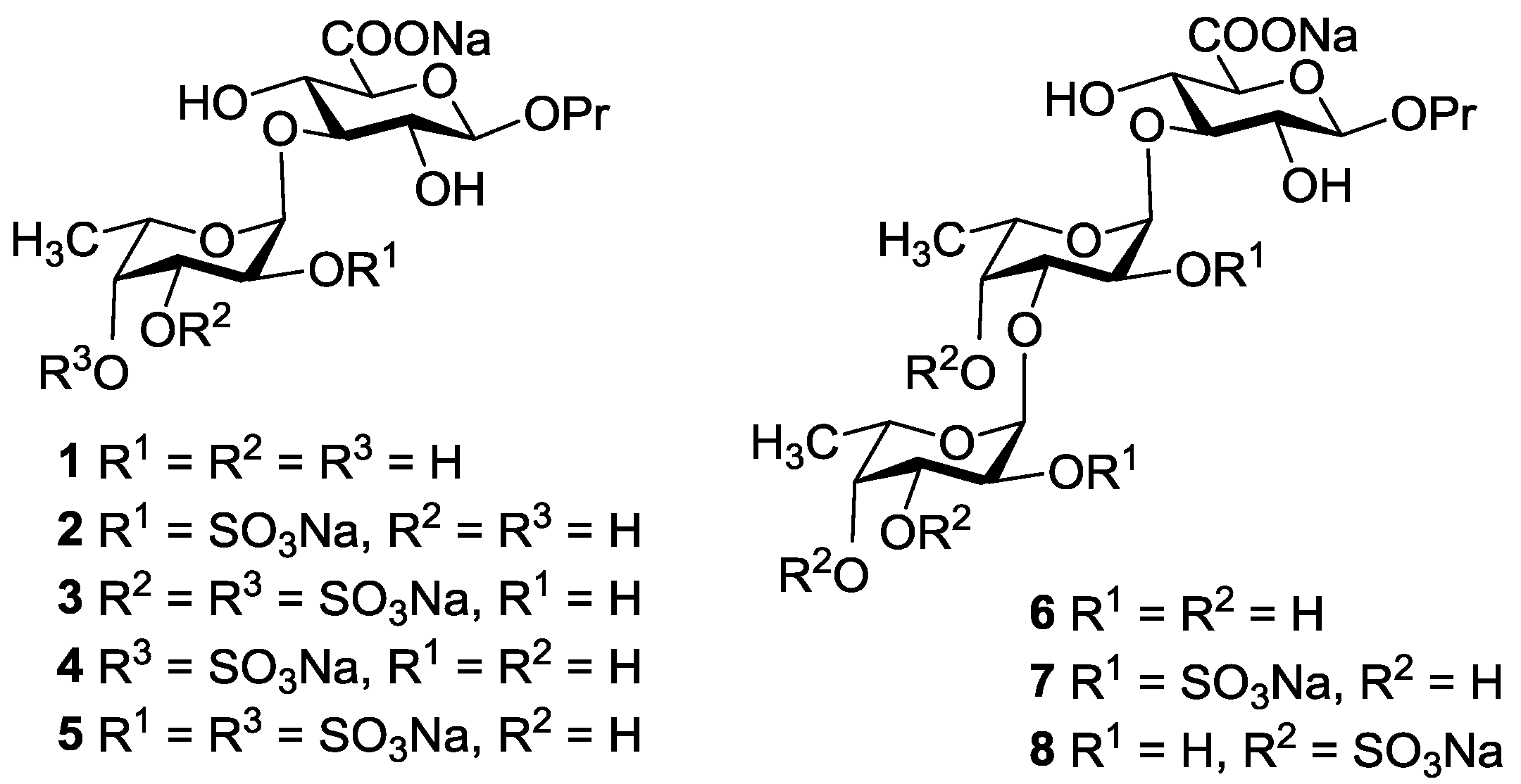
3.3.9. Propyl α-l-fucopyranosyl-(1→3)-β-d-glucopyranosyl Uronic Acid Sodium Salt (1)
3.3.10. Propyl 2-O-sulfonato-α-l-fucopyranosyl-(1→3)-β-d-glucopyranosyl Uronic Acid Disodium Salt (2)
3.3.11. Propyl 3,4-di-O-sulfonato-α-l-fucopyranosyl-(1→3)-β-d-glucopyranosyl Uronic Acid Trisodium Salt (3)
3.3.12. Propyl 4-O-sulfonato-α-l-fucopyranosyl-(1→3)-β-d-glucopyranosyl Uronic Acid Disodium Salt (4)
3.3.13. Propyl 2,4-di-O-sulfonato-α-l-fucopyranosyl-(1→3)-β-d-glucopyranosyl Uronic Acid Trisodium Salt (5)
3.3.14. Propyl α-l-fucopyranosyl-(1→3)-α-l-fucopyranosyl-(1→3)-β-d-glucopyranosyl Uronic Acid Sodium Salt (6)
3.3.15. Propyl 2-O-sulfonato-α-l-fucopyranosyl-(1→3)-2-O-sulfonato-α-l-fucopyranosyl-(1→3)-β-d-glucopyranosyl Uronic Acid Trisodium Salt (7)
3.3.16. Propyl 3,4-di-O-sulfonato-α-l-fucopyranosyl-(1→3)-4-O-sulfonato-α-l-fucopyranosyl-(1→3)-β-d-glucopyranosyl Uronic Acid Tetrasodium Salt (8)
3.4. Calculations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Galanaud, J.P.; Laroche, J.P.; Righini, M. The history and historical treatments of deep vein thrombosis. J. Thomb. Haemost. 2013, 3, 402–411. [Google Scholar]
- Petitou, M.; van Boeckel, C.A. A synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew. Chem. Int. Ed. Engl. 2004, 43, 3118–3133. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.L.; Varki, A.; Borsig, L. Heparin attenuates metastasis mainly due to inhibition of P- and L-selectin, but non-anticoagulant heparins can have additional effects. Thromb. Res. 2007, 120, S107–S111. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brown, J.R.; Varki, A.; Esko, J.D. Heparin’s anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J. Clin. Invest. 2002, 110, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H. Holothurian Fucosylated Chondroitin Sulfate. Mar. Drugs 2014, 12, 232–254. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L.; Wang, L.; Cavalcante, M.C.; Cardilo-Reis, L.; Ferreira, P.L.; Mourão, P.A.; Esko, J.D.; Pavão, M.S. Selectin blocking activity of a fucosylated chondroitin sulfate glycosaminoglycan from sea cucumber. Effect on tumor metastasis and neutrophil recruitment. J. Biol. Chem. 2007, 282, 14984–14991. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, R.J.; Oliveira, S.N.; Pomin, V.H.; Mecawi, A.S.; Araujo, I.G.; Mourão, P.A. Effects of oversulfated and fucosylated chondroitin sulfates on coagulation. Challenges for the study of anticoagulant polysaccharides. Thromb. Haemost. 2010, 103, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, D.; Wang, S.; Tao, L.; Wang, A.; Chen, W.; Zhu, Z.; Zheng, S.; Gao, X.; Lu, Y. Holothurian glycosaminoglycan inhibits metastasis and thrombosis via targeting of nuclear factor-κB/tissue factor/Factor Xa pathway in melanoma B16F10 cells. PLoS One 2013, 8, e56557. [Google Scholar] [CrossRef] [PubMed]
- Panagos, C.G.; Thomson, D.S.; Moss, C.; Hughes, A.D.; Kelly, M.S.; Liu, Y.; Chai, W.; Venkatasamy, R.; Spina, D.; Page, C.P.; et al. Fucosylated chondroitin sulfates from the body wall of the sea cucumber Holothuria. forskali: Conformation, selectin binding, and biological activity. J. Biol. Chem. 2014, 289, 28284–28298. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Wu, M.; Xu, L.; Lian, W.; Xiang, J.; Lu, F.; Gao, N.; Xiao, C.; Wang, S.; Zhao, J. Comparison of physicochemical characteristics and anticoagulant activities of polysaccharides from three sea cucumbers. Mar. Drugs 2013, 11, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Myron, P.; Siddiquee, S.; Al Azad, S. Fucosylated chondroitin sulfate diversity in sea cucumbers: A review. Carbohydr. Polym. 2014, 112, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Mourão, P.A.; Giumarães, B.; Mulloy, B.; Thomas, S.; Gray, E. Antithrombotic activity of a fucosylated chondroitin sulphate from echinoderm: Sulphated fucose branches on the polysaccharide account for its antithrombotic action. Br. J. Haematol. 1998, 101, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kang, H.; Wu, M.; Zeng, W.; Li, Z.; Gao, Y.; Cui, J.; Wang, J.; Feng, H.; Yu, L. Depolymerized Glycosaminoglycan from Thelenota. ananas and Preparation Method Thereof. US Patent 20120270834 A1, 25 October 2012. [Google Scholar]
- Chen, S.; Xue, C.; Yin, L.; Tang, Q.; Yu, G.; Chai, W. Comparison of structures and anticoagulant activities of fucosylated chondroitin sulfates from different sea cucumbers. Carbohydr. Polym. 2011, 83, 688–695. [Google Scholar] [CrossRef]
- Kariya, Y.; Watabe, S.; Kyogashima, M.; Ishihara, M.; Ishii, T. Structure of fucose branches in the glycosaminoglycan from the body wall of the sea cucumber Stichopus. japonicus. Carbohydr. Res. 1997, 297, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Tamura, J.; Tanaka, H.; Nakamura, A.; Takeda, N. Synthesis of β-d-GalNAc(4,6-diS)(1→4) [α-l-Fuc(2,4-diS)(1→3)]-β-d-GlcA, a novel trisaccharide unit of chondroitin sulfate with a fucose branch. Tetrahedr. Lett. 2013, 54, 3940–3943. [Google Scholar] [CrossRef]
- Kornilov, A.V.; Sukhova, E.V.; Nifantiev, N.E. Preparative route to glucuronyl donors bearing temporary protecting group at O-3 via 6,3-lactonisation by Bz2O or Piv2O. Carbohydr. Res. 2001, 336, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Gerbst, A.G.; Ustuzhanina, N.E.; Grachev, A.A.; Tsvetkov, D.E.; Khatuntseva, E.A.; Whitefield, D.M.; Berces, A.; Nifantiev, N.E. Synthesis, NMR and conformational studies of fucoidan fragments. Part 3. Effect of benzoyl group at O-3 on stereoselectivity of glycosylation by 3-O- and 3,4-di-O-benzoylated 2-O-benzylfucosyl bromides. J. Carbohydr. Chem. 2001, 20, 821–831. [Google Scholar] [CrossRef]
- Komarova, B.S.; Ustyuzhanina, N.E.; Tsvetkov, Y.E.; Nifantiev, N.E. Stereocontrol of 1,2-cis-Glycosylation by remote O-Acyl protecting groups. In Modern Synthetic Methods in Carbohydrate Chemistry—From Monosaccharides to Complex Glycoconjugates; Vidal, S., Wertz, D., Eds.; Wiley-VCH: Weinheim, Germany, 2013; pp. 125–159. [Google Scholar]
- Ustyuzhanina, N.; Krylov, V.; Grachev, A.; Gerbst, A.; Nifantiev, N. Synthesis, NMR and conformational studies of fucoidan fragments. part 8: Convergent block-wise synthesis of long chain linear and 2,3-branched oligosaccharides. Synthesis 2006, 23, 4017–4031. [Google Scholar]
- Gerbst, A.G.; Ustuzhanina, N.E.; Grachev, A.A.; Tsvetkov, D.E.; Khatuntseva, E.A.; Nifant’ev, N.E. Effect of the nature of protecting group at O-4 on stereoselectivity of glycosylation by 4-O-substituted 2,3-di-O-benzylfucosyl bromides. Mend. Commun. 1999, 9, 114–116. [Google Scholar] [CrossRef]
- Hypercube, Inc. Available online: http://www.hyper.com (accessed on 27 January 2015).
- Allinger, N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comp. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods II. Applications. J. Comp. Chem. 1989, 10, 221–264. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef]
- Khatuntseva, E.A.; Ustuzhanina, N.E.; Zatonskii, G.V.; Shashkov, A.S.; Usov, A.I.; Nifant’ev, N.E. Synthesis, NMR and conformational studies of fucoidan fragments. Part 1: Desulfated 2,3- and 3,4-branched trisaccharide fragments and constituting disaccharides. J. Carbohydr. Chem. 2000, 19, 1151–1173. [Google Scholar] [CrossRef]
- Belyakov, P.A.; Kadentsev, V.I.; Chizhov, A.O.; Kolotyrkina, N.G.; Shashkov, A.S.; Ananikov, V.P. Mechanistic insight into organic and catalytic reactions by joint studies using mass spectrometry and NMR spectroscopy. Mendel. Commun. 2010, 20, 125–131. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ustyuzhanina, N.E.; Fomitskaya, P.A.; Gerbst, A.G.; Dmitrenok, A.S.; Nifantiev, N.E. Synthesis of the Oligosaccharides Related to Branching Sites of Fucosylated Chondroitin Sulfates from Sea Cucumbers. Mar. Drugs 2015, 13, 770-787. https://doi.org/10.3390/md13020770
Ustyuzhanina NE, Fomitskaya PA, Gerbst AG, Dmitrenok AS, Nifantiev NE. Synthesis of the Oligosaccharides Related to Branching Sites of Fucosylated Chondroitin Sulfates from Sea Cucumbers. Marine Drugs. 2015; 13(2):770-787. https://doi.org/10.3390/md13020770
Chicago/Turabian StyleUstyuzhanina, Nadezhda E., Polina A. Fomitskaya, Alexey G. Gerbst, Andrey S. Dmitrenok, and Nikolay E. Nifantiev. 2015. "Synthesis of the Oligosaccharides Related to Branching Sites of Fucosylated Chondroitin Sulfates from Sea Cucumbers" Marine Drugs 13, no. 2: 770-787. https://doi.org/10.3390/md13020770
APA StyleUstyuzhanina, N. E., Fomitskaya, P. A., Gerbst, A. G., Dmitrenok, A. S., & Nifantiev, N. E. (2015). Synthesis of the Oligosaccharides Related to Branching Sites of Fucosylated Chondroitin Sulfates from Sea Cucumbers. Marine Drugs, 13(2), 770-787. https://doi.org/10.3390/md13020770