Synthesis and Biological Evaluation of Novel 3-Alkylpyridine Marine Alkaloid Analogs with Promising Anticancer Activity

Abstract
:
1. Introduction
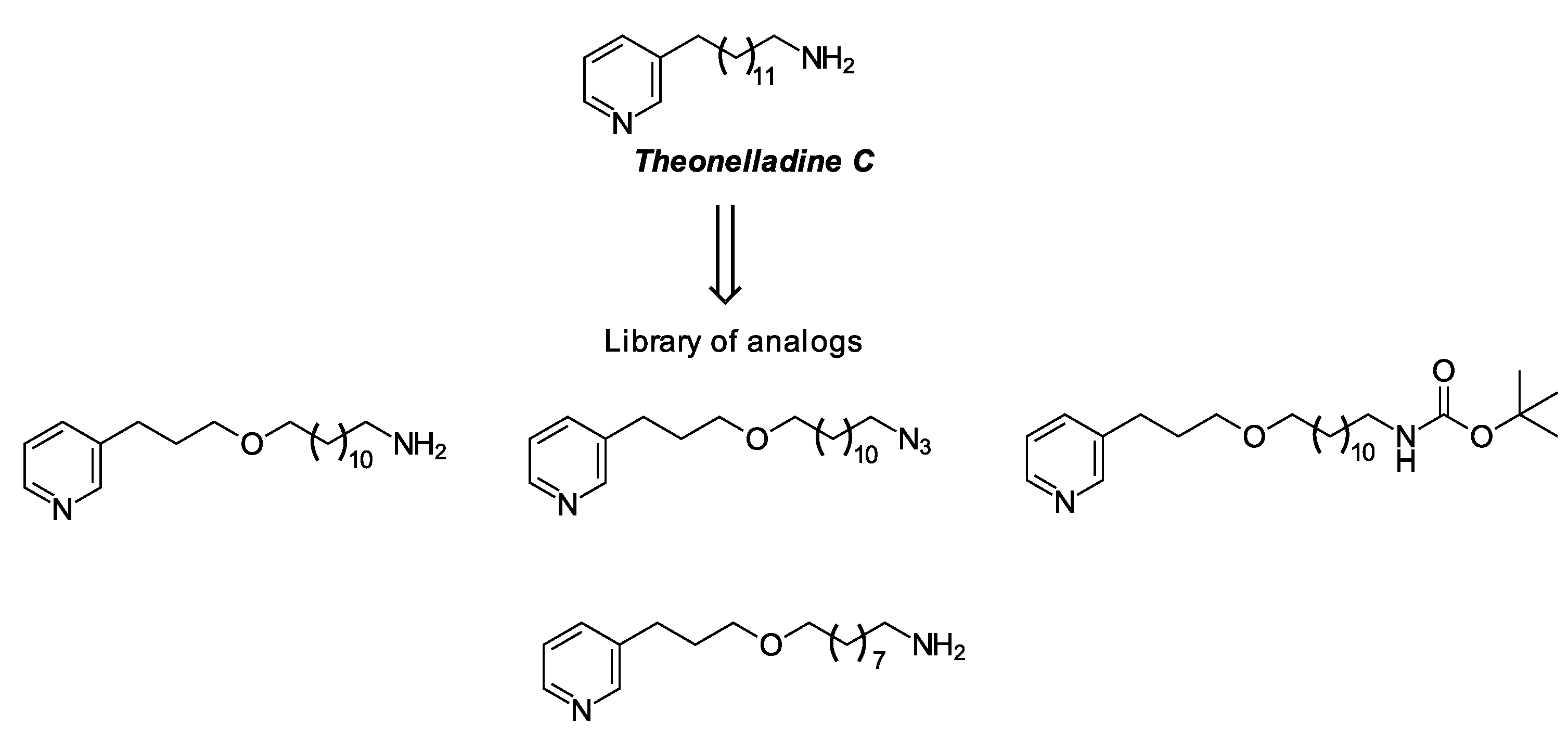
2. Results and Discussion
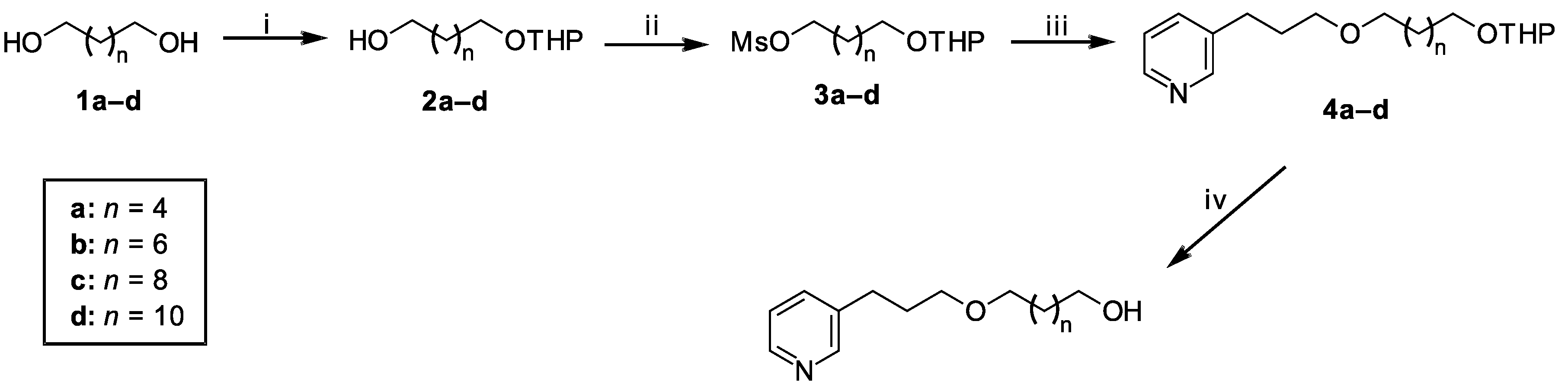
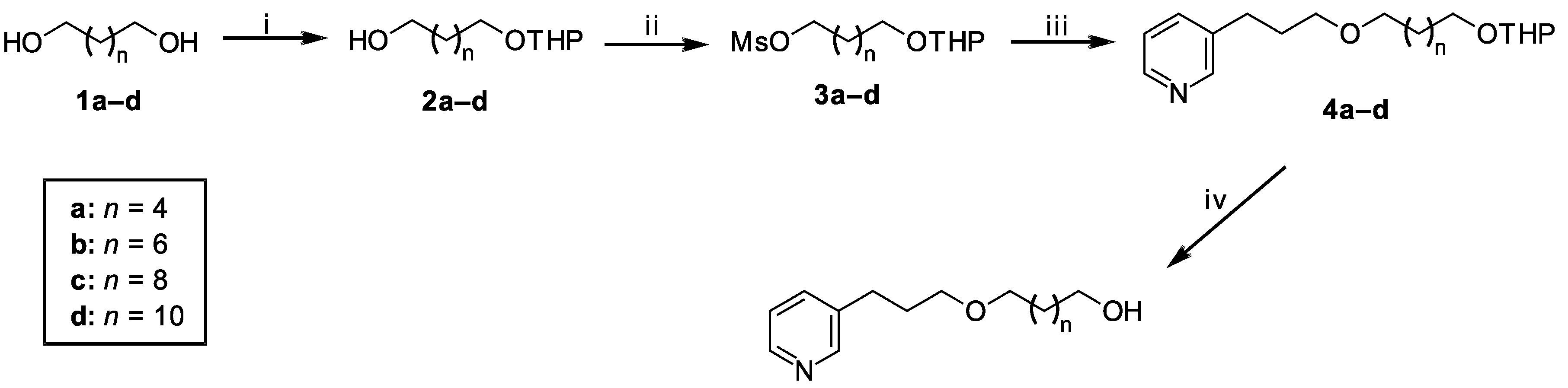
2.1. Chemistry
Synthesis
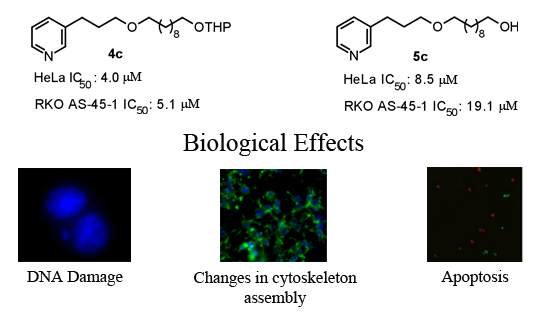
2.2. Biological Evaluation
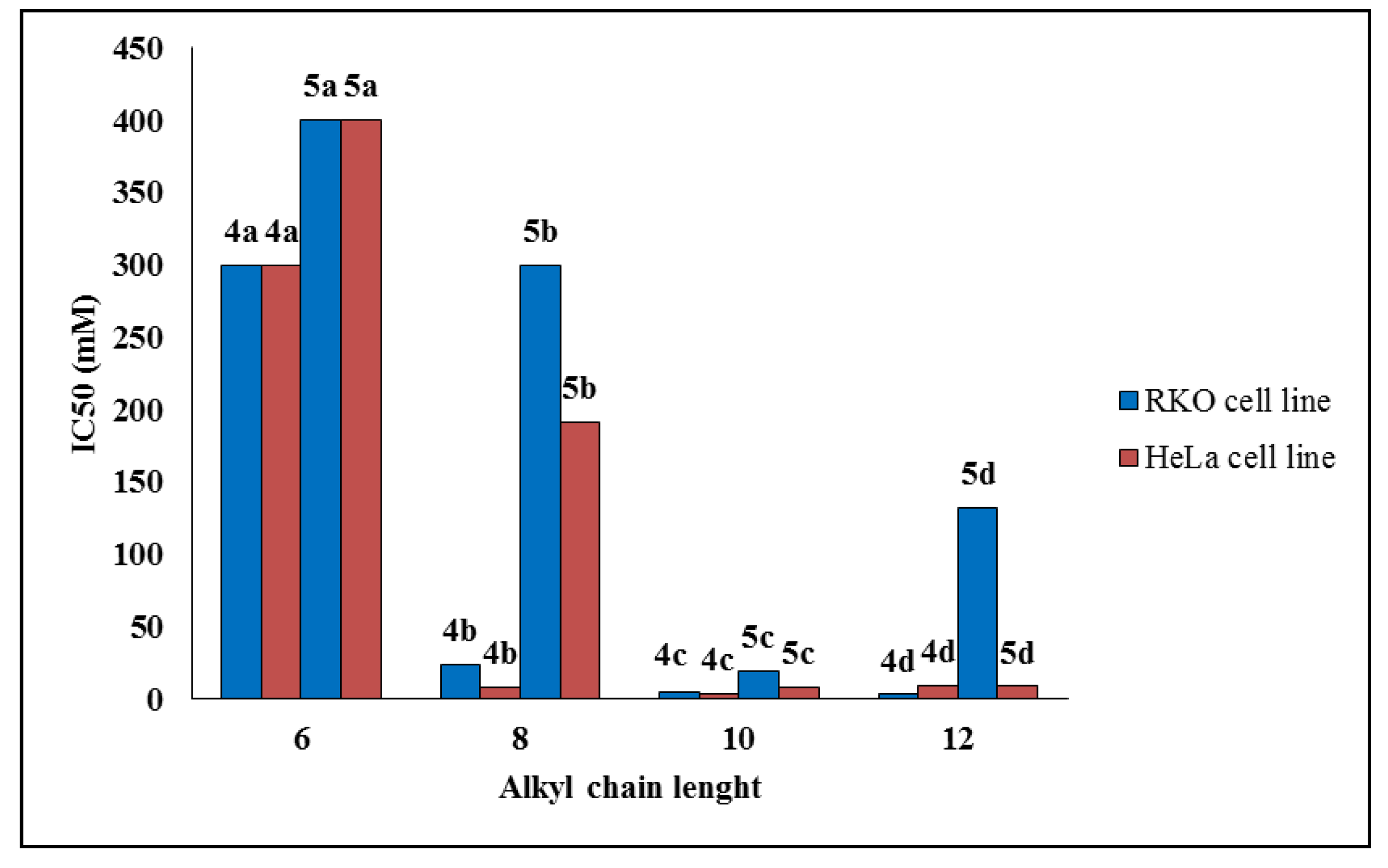
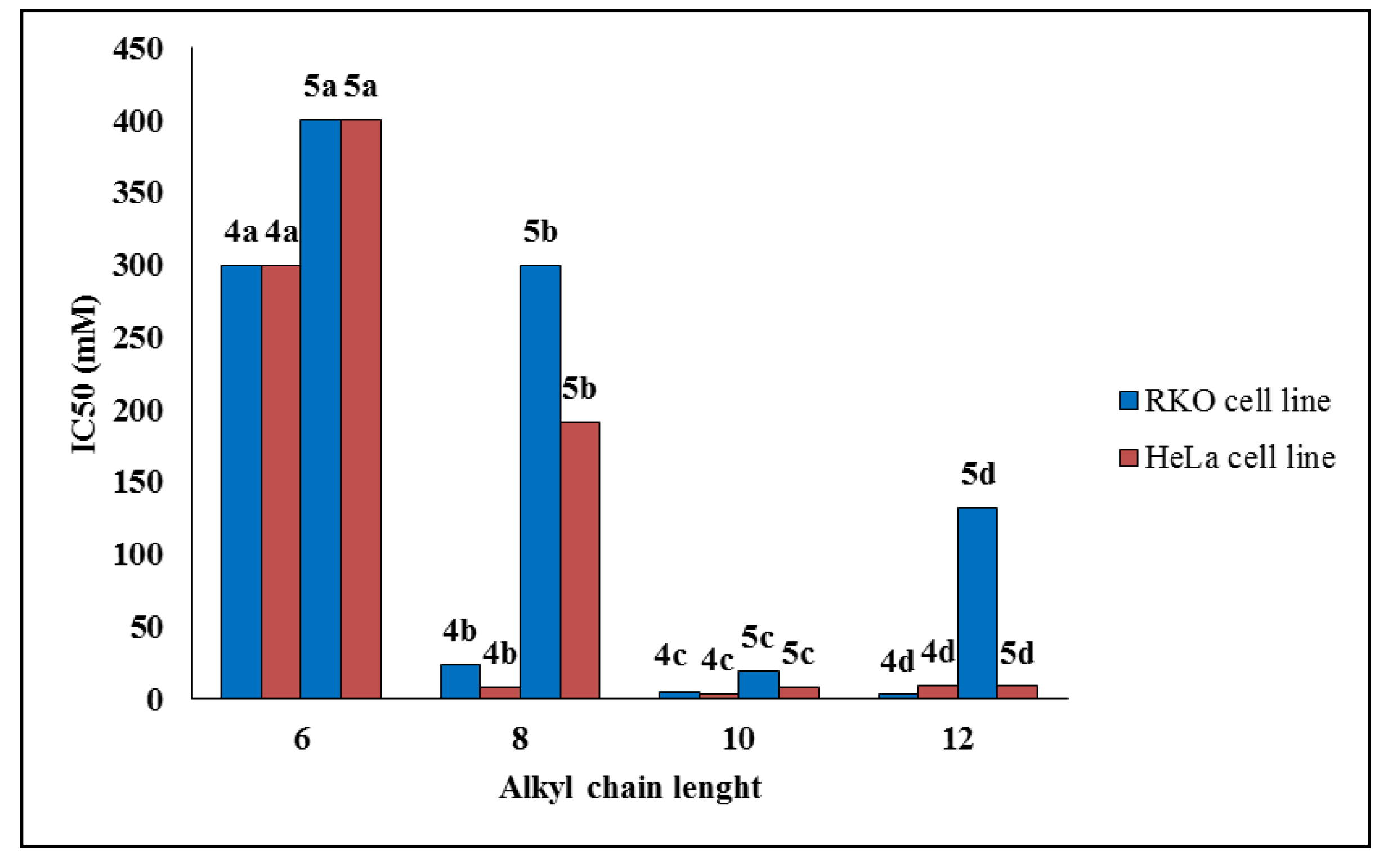
2.2.1. Cell Viability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) ± SD * | Selectivity Index (SI) | |||
|---|---|---|---|---|---|
| RKO AS-45-1 | HeLa | WI-26VA4 | RKO AS-45-1 | HeLa | |
| 4a | >300 | >300 | >300 | ND ** | ND |
| 4b | 23.7 ± 1.7 | 8.1 ± 2.7 | 37.8 ± 5.0 | 1.59 | 4.66 |
| 4c | 5.1 ± 1.1 | 4.0 ± 0.8 | 6.4 ± 0.7 | 1.25 | 1.60 |
| 4d | 3.2 ± 1.7 | 9.4 ± 0.7 | 11.3 ± 1.4 | 3.53 | 1.20 |
| 5a | >400 | >400 | >400 | ND | ND |
| 5b | >300 | 191.8 ± 10.1 | 167.0 ± 10.5 | ND | 0.87 |
| 5c | 19.1 ± 4.4 | 8.5 ± 2.4 | 99.1 ± 11.2 | 5.18 | 11.65 |
| 5d | 131.9 ± 16.8 | 8.8 ± 1.9 | 34.1 ± 6.5 | 0.25 | 3.87 |
| Etoposide | 1.4 ± 0.6 | 2.7 ± 0.4 | ND | - | - |
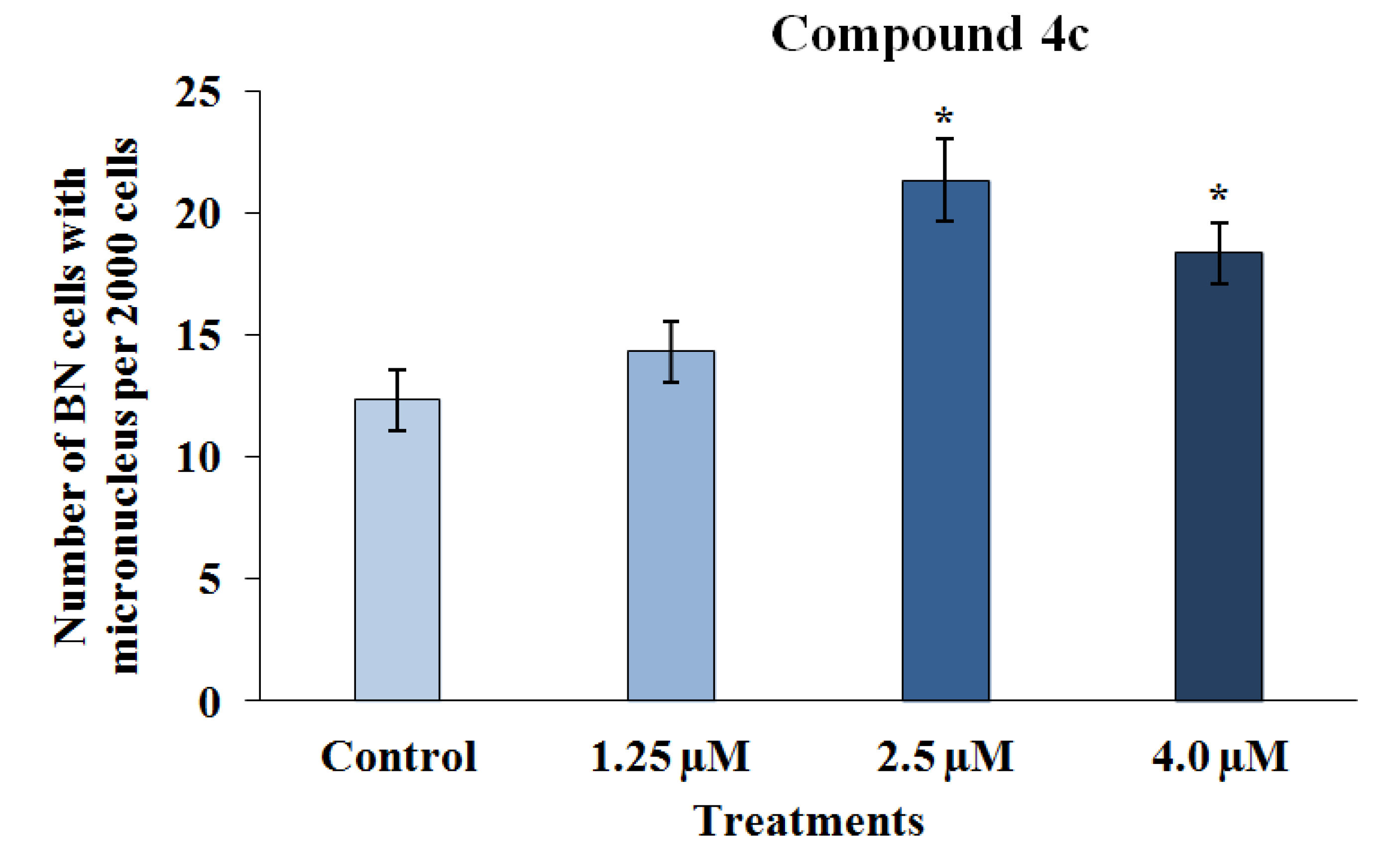
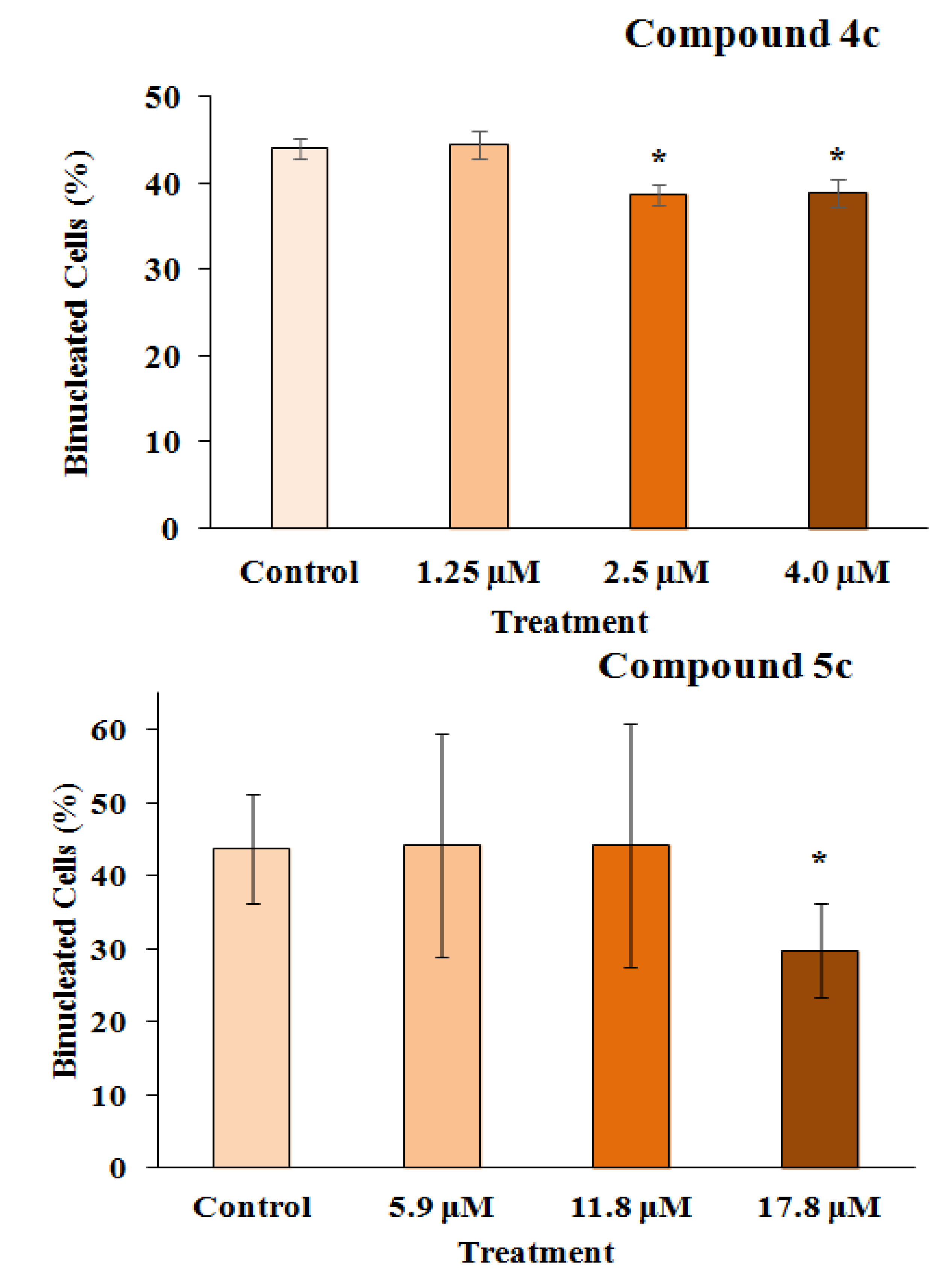
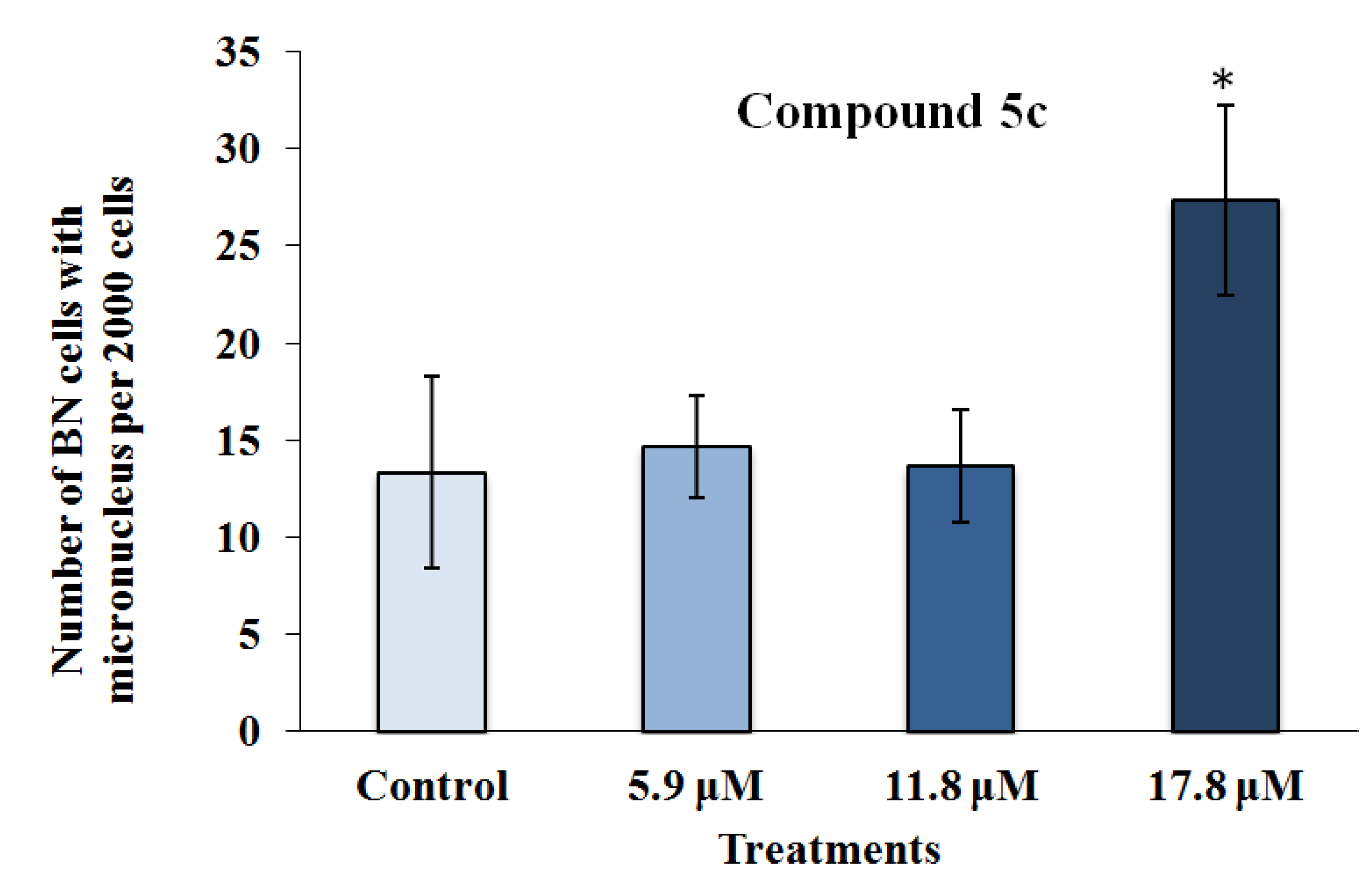
2.2.2. Mutagenicity Assay
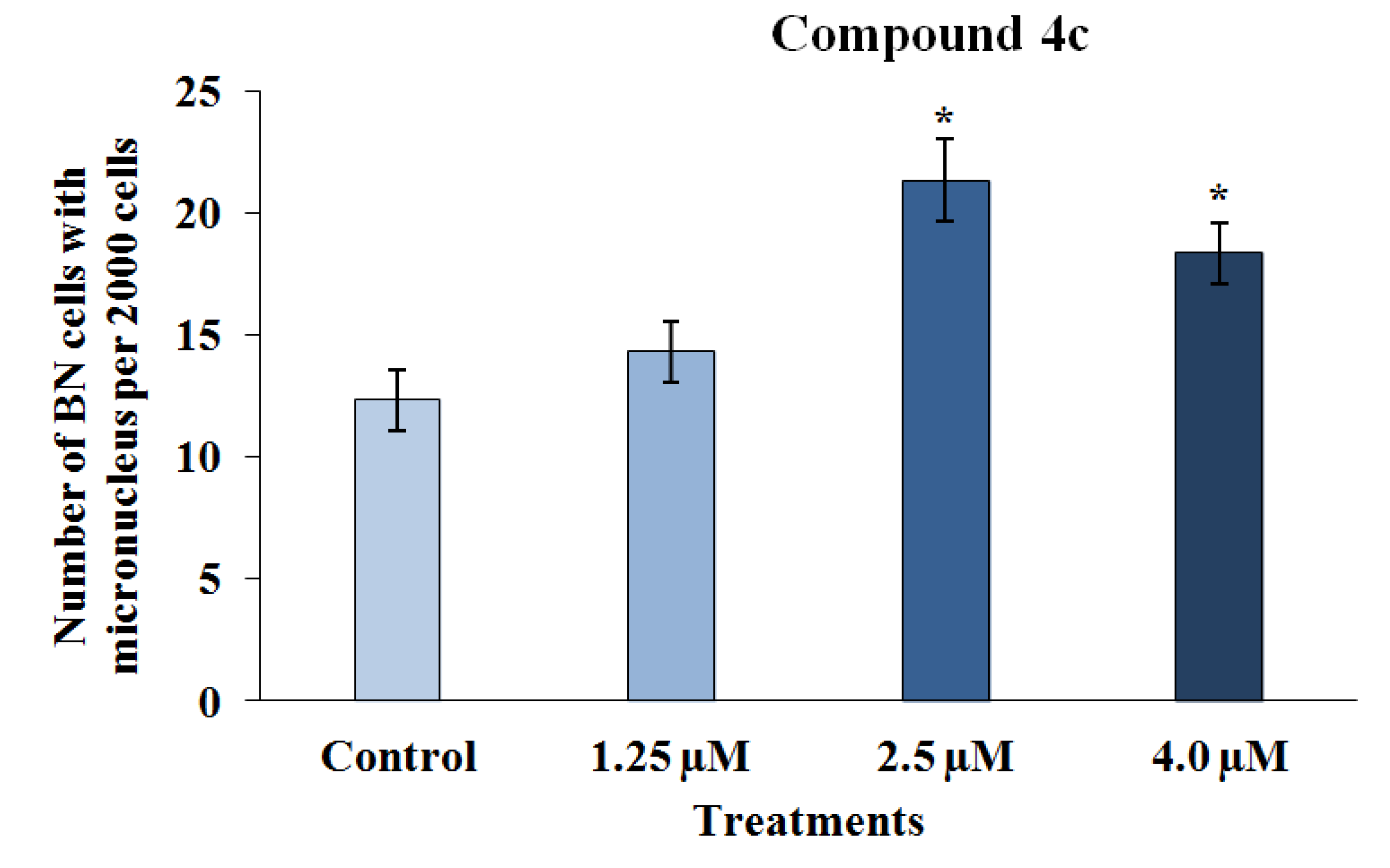
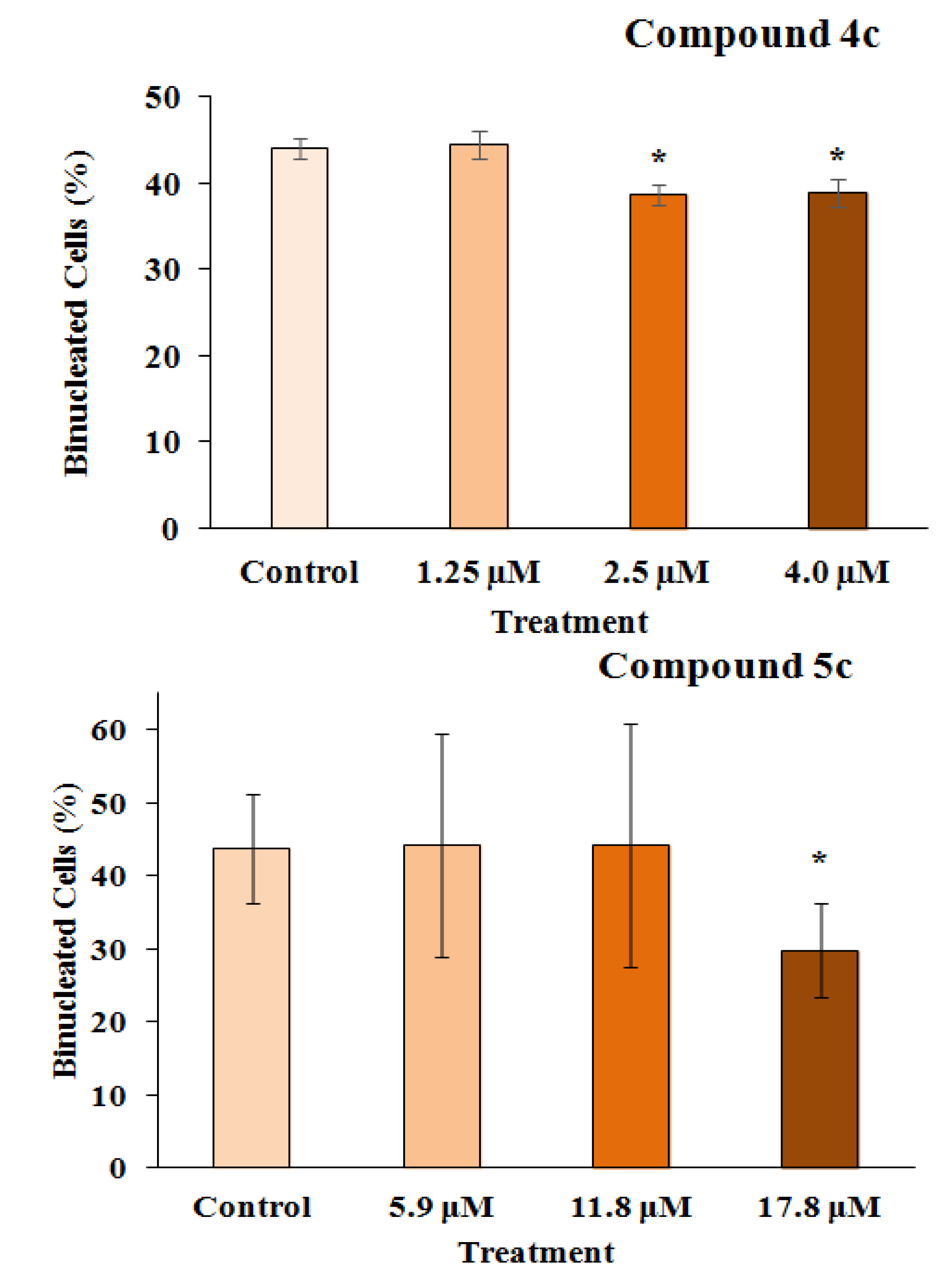
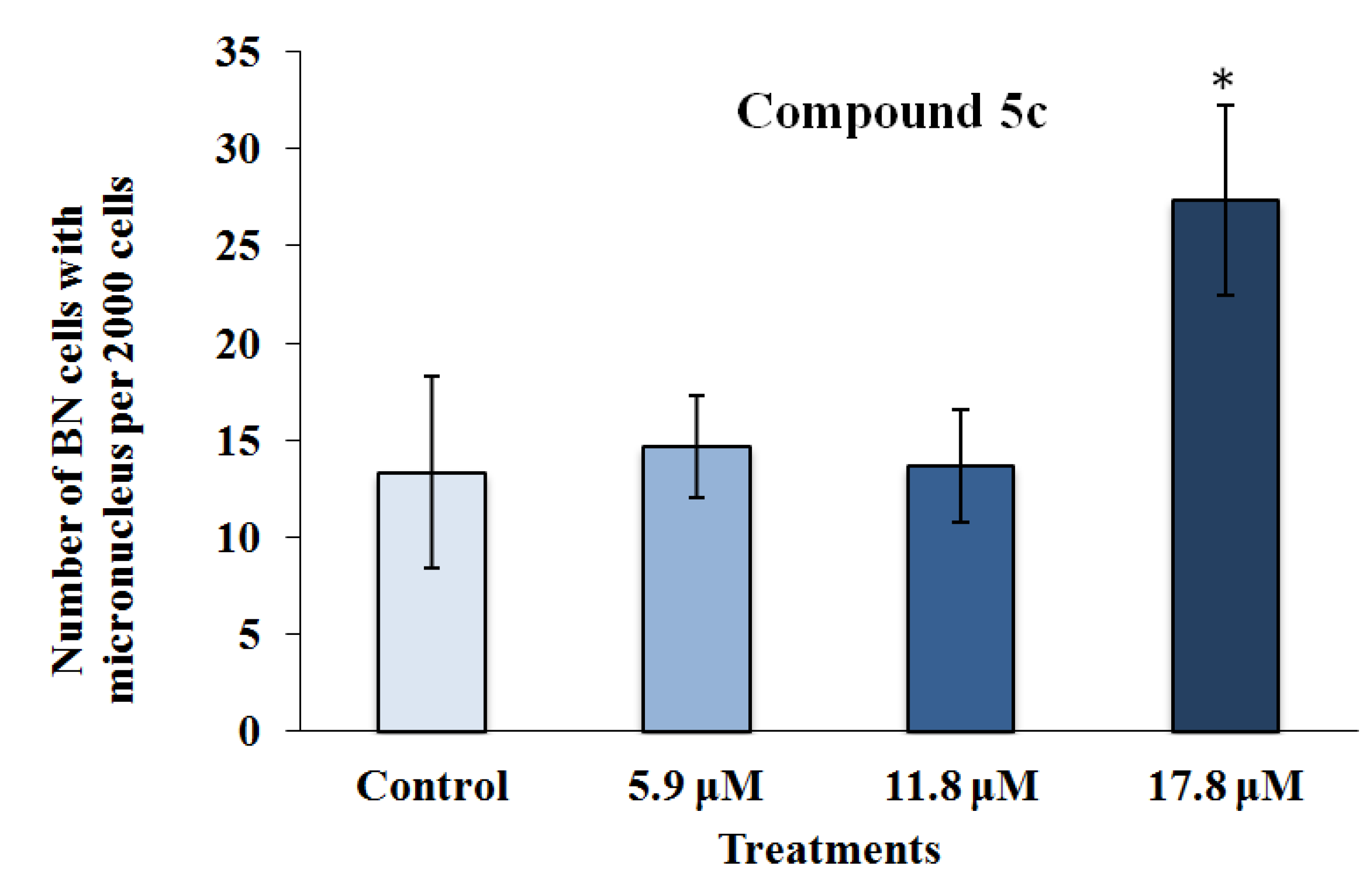
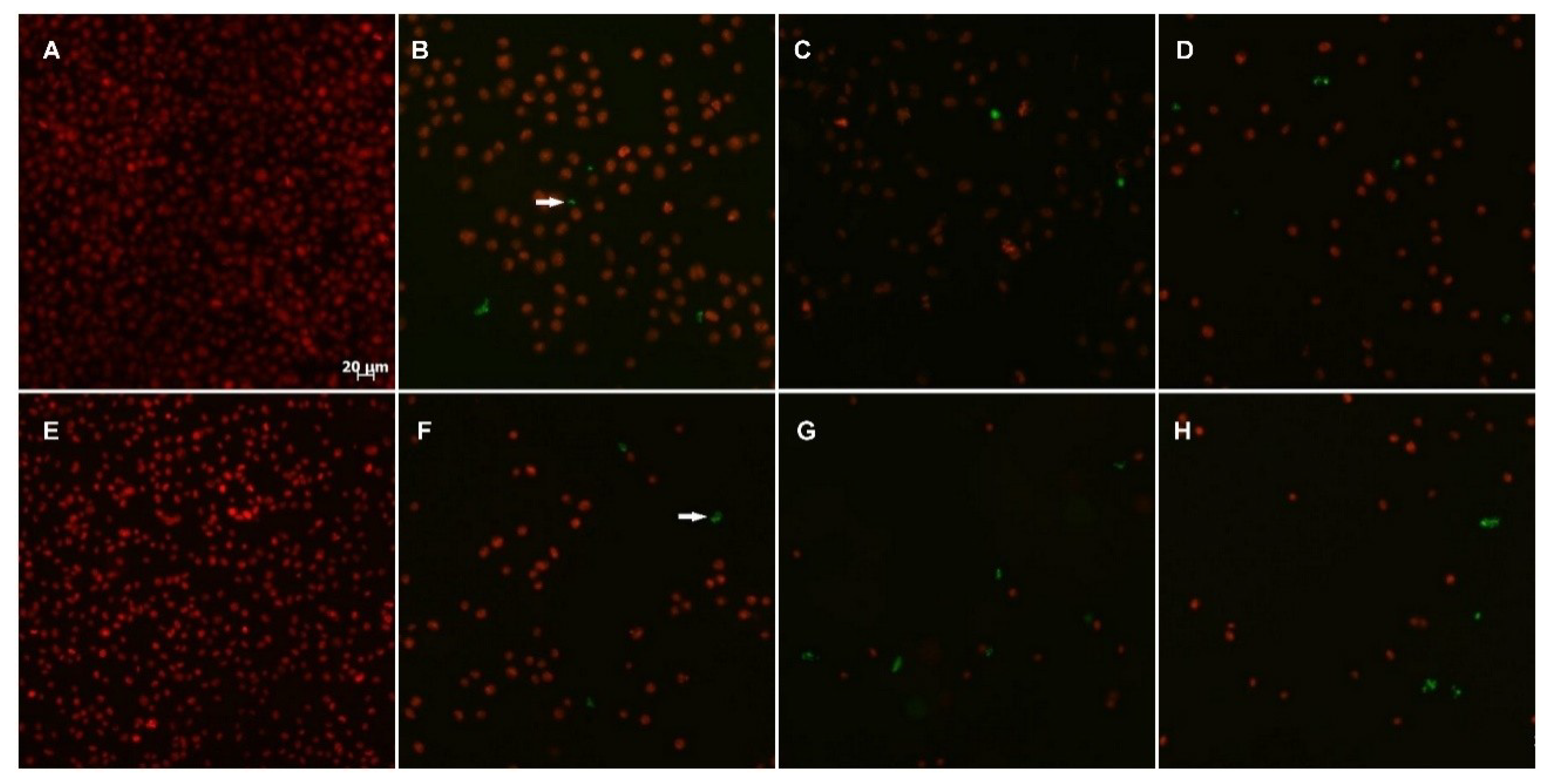
2.2.3. TUNEL Assay
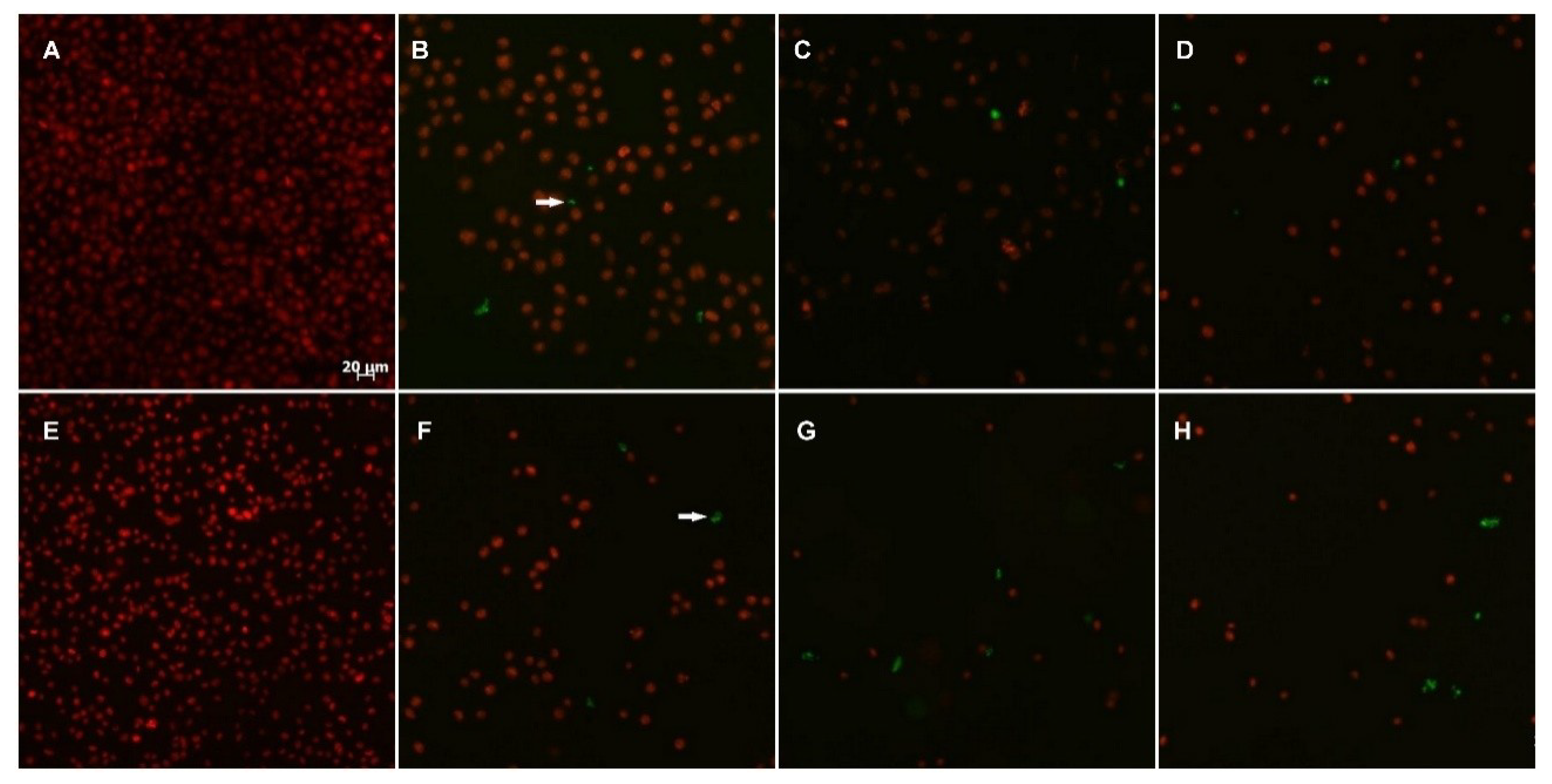
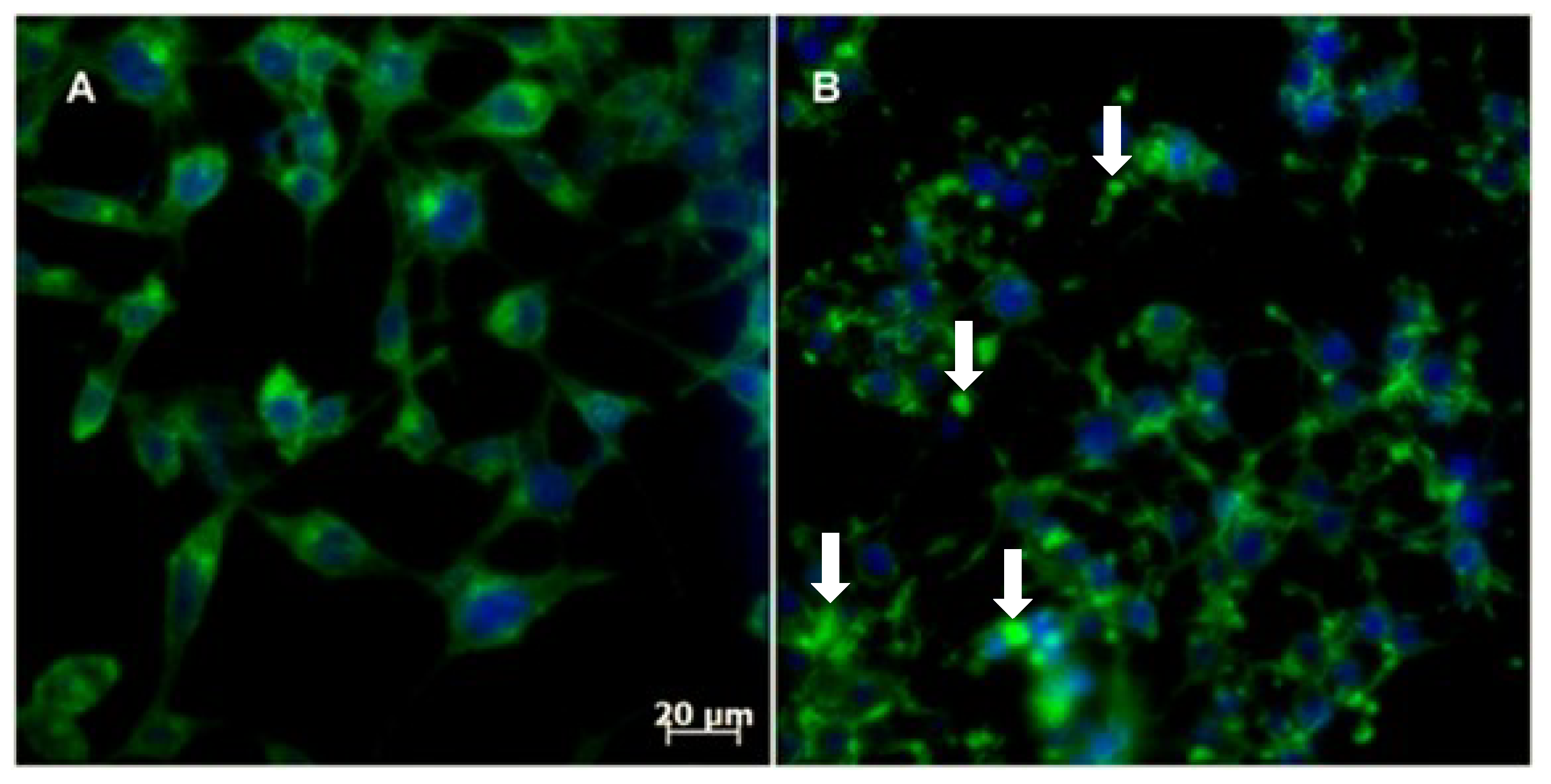
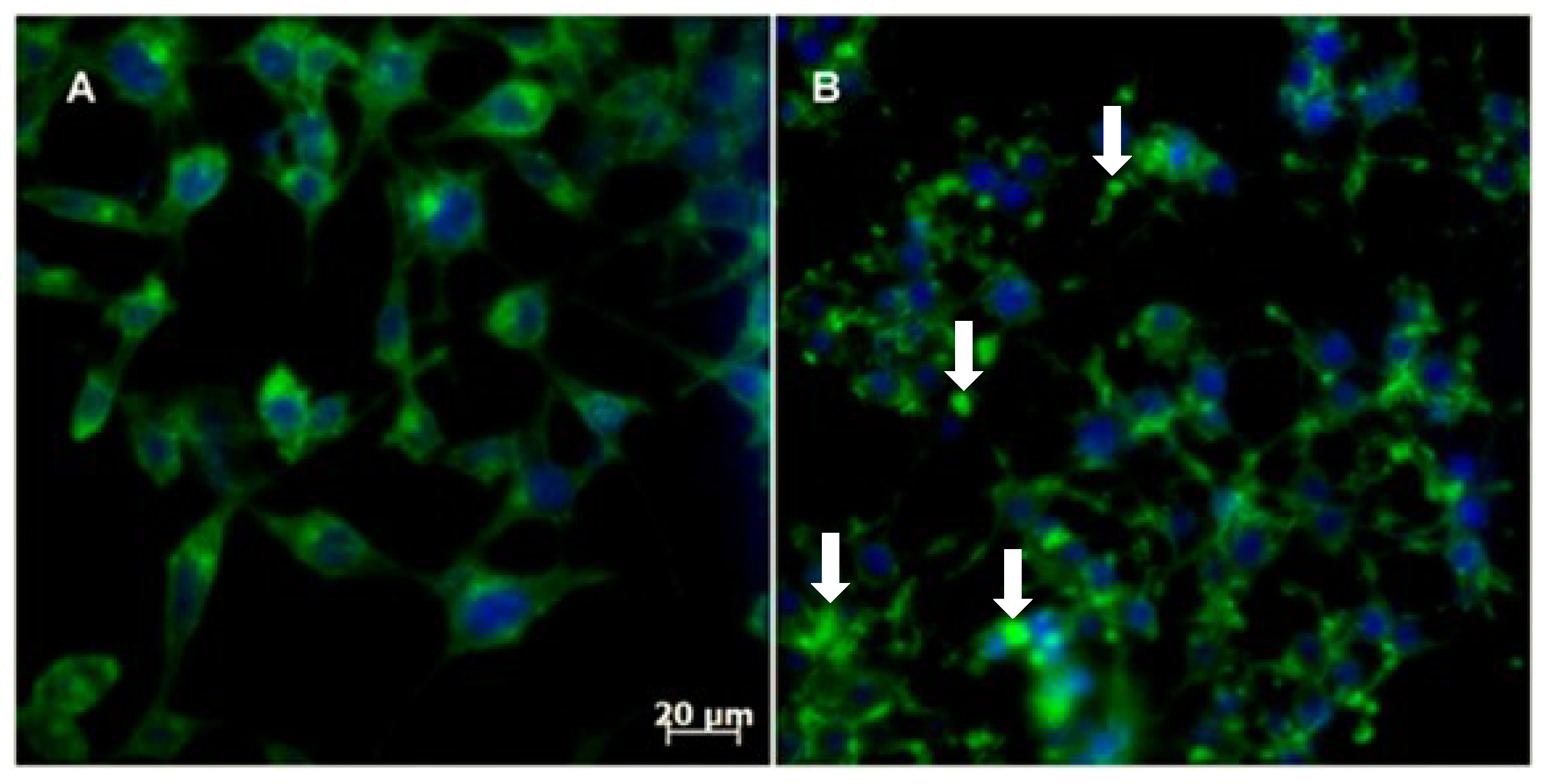
2.2.4. Cytoskeleton Assembly
3. Experimental Section
3.1. Chemistry
3.2. Synthesis
3.3. Drug Samples
3.3.1. Cytotoxicity Assay
3.3.2. DNA Nick-End Labeling by the TUNEL Method and Immunofluorescence
3.3.3. Actin Polymerization Detected by Fluorescence Optical Microscopy
3.3.4. Cytokinesis-Block Micronucleus Assay
3.3.5. Frequency of Binucleation (Mitotic Index)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vineis, P.; Wild, C.P. Global cancer patterns: Causes and prevention. Lancet 2014, 383, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Sawadogo, W.R.; Schumacher, M.; Teiten, M.-H.; Cerella, C.; Dicato, M.; Diederich, M. A survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2011. Molecules 2013, 18, 3641–3673. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, I.; Kim, S.-K. Marine antitumor drugs: Status, shortfalls and strategies. Mar. Drugs 2010, 8, 2702–2720. [Google Scholar] [CrossRef] [PubMed]
- Sipkema, D.; Franssen, M.C.R.; Osinga, R.; Tramper, J.; Wijffels, R.H. Marine sponges as pharmacy. Mar. Biotechnol. 2005, 7, 142–162. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Martorana, A.; Gia, O.; DallaVia, L.; Cirrincione, G. 3,5-bis(3′-indolyl)pyrazoles, analogues of marine alkaloid nortopsentin: Synthesis and antitumor properties. Bioorg. Med. Chem. Lett. 2007, 17, 6134–6137. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.-H.; Cirrincione, G. Synthesis and antitumor activity of 2,5-bis(3′-indolyl)-furans and 3,5-bis(3′-indolyl)-isoxazoles, nortopsentin analogues. Bioorg. Med. Chem. 2010, 18, 4524–4529. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Montalbano, A.; Parrino, B.; Lopergolo, A.; Pennati, M.; Zaffaroni, N.; Cirrincione, G. Synthesis and antitumor activity of 3-(2-phenyl-1,3-thiazol-4-yl)-1H-indoles and 3-(2-phenyl-1,3-thiazol-4-yl)-1H-7-azaindoles. Chem. Med. Chem. 2011, 6, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Parrino, B.; Barraja, P.; Spanò, V.; Cirrincione, G.; Diana, P.; Maier, A.; Kelter, G.; Fiebig, H.-H. Synthesis and antiproliferative activity of 2,5-bis(3′-indolyl)pyrroles, analogues of the marine alkaloid nortopsentin. Mar. Drugs 2013, 11, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Pennati, M.; Parrino, B.; Lopergolo, A.; Barraja, P.; Montalbano, A.; Spanò, V.; Sbarra, S.; Doldi, V.; de Cesare, M. Novel 1H-pyrrolo[2,3-b]pyridine derivative nortopsentin analogues: Synthesis and antitumor activity in peritoneal mesothelioma experimental models. J. Med. Chem. 2013, 56, 7060–7072. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Pennati, M.; Barraja, P.; Montalbano, A.; Parrino, B.; Spanò, V.; Lopergolo, A.; Sbarra, S.; Doldi, V.; Zaffaroni, N.; et al. Synthesis and antiproliferative activity of substituted 3[2-(1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[3,2-b]pyridines, marine alkaloid nortopsentin analogues. Curr. Med. Chem. 2014, 21, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Stonik, V.; Fedorov, S. Marine low molecular weight natural products as potential cancer preventive compounds. Mar. Drugs 2014, 12, 636–671. [Google Scholar] [CrossRef] [PubMed]
- Youssef, D.T.; Shaala, L.A; Mohamed, G.A; Badr, J.M.; Bamanie, F.H.; Ibrahim, S.R. Theonellamide G, a potent antifungal and cytotoxic bicyclic glycopeptide from the Red Sea marine sponge Theonella swinhoei. Mar. Drugs 2014, 12, 1911–1923. [Google Scholar]
- Kobayashi, J.; Murayama, T.; Ohizumi, Y. Theonelladins A~D,novel antineoplastic pyridine alkaloids from the Okinawan marine sponge Theonella swinhoei. Tetrahedron Lett. 1989, 3, 4833–4836. [Google Scholar] [CrossRef]
- Pereira, J.R.C.S.; Hilário, F.F.; Lima, A.B.; Silveira, M.L.T.; Silva, L.M.; Alves, R.B.; de Freitas, R.P.; Varotti, F.P. Cytotoxicity evaluation of marine alkaloid analogues of viscosaline and theonelladin C. Biomed. Prev. Nutr. 2012, 2, 145–148. [Google Scholar] [CrossRef]
- Hilário, F.F.; de Paula, R.C.; Silveira, M.L.; Viana, G.H.; Alves, R.B.; Pereira, J.R.; Silva, L.M.; de Freitas, R.P.; de Pilla Varotti, F. Synthesis and evaluation of antimalarial activity of oxygenated 3-alkylpyridine marine alkaloid analogues. Chem. Biol. Drug. Des. 2011, 78, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Machado, P.A; Hilário, F.F.; Carvalho, L.O.; Silveira, M.L.; Alves, R.B.; Freitas, R.P.; Coimbra, E.S. Effect of 3-alkylpyridine marine alkaloid analogues in Leishmania species related to American cutaneous leishmaniasis. Chem. Biol. Drug Des. 2012, 80, 745–751. [Google Scholar]
- Iannolo, G.; Conticello, C.; Memeo, L.; de Maria, R. Apoptosis in normal and cancer stem cells. Crit. Rev. Oncol. Hematol. 2008, 66, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Gaidano, G. Messengers of cell death: Apoptotic signaling in health and disease. Haematologica 2003, 88, 212–218. [Google Scholar] [PubMed]
- Beesoo, R.; Neergheen-Bhujun, V.; Bhagooli, R.; Bahorun, T. Apoptosis inducing lead compounds isolated from marine organisms of potential relevance in Cancer Treatment. Mutat. Res. Mol. Mech. Mutagen. 2014. [Google Scholar] [CrossRef]
- Khan, K.H.; Blanco-Codesido, M.; Molife, L.R. Cancer therapeutics: Targeting the apoptotic pathway. Crit. Rev. Oncol. Hematol. 2013, 90, 200–219. [Google Scholar] [CrossRef] [PubMed]
- Kirsch-Volders, M.; Sofuni, T.; Aardema, M.; Albertini, S.; Eastmond, D.; Fenech, M.; Ishidate, M.; Kirchner, S.; Lorge, E.; Morita, T.; et al. Report from the in vitro micronucleus assay working group. Mutat. Res. Toxicol. Environ. Mutagen. 2003, 540, 153–163. [Google Scholar] [CrossRef]
- Decordier, I.; Dillen, L.; Cundari, E.; Kirsch-Volders, M. Elimination of micronucleated cells by apoptosis after treatment with inhibitors of microtubules. Mutagenesis 2002, 17, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Decordier, I.; Cundari, E.; Kirsch-Volders, M. Influence of caspase activity on micronuclei detection: A possible role for caspase-3 in micronucleation. Mutagenesis 2005, 20, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar]
- Nishiguchi, T.; Hayakawa, S.; Saitoh, M. Selective monotetrahydropyranylation of 1,n-diols catalyzed by aqueous acids. 2000, 41, 9843–9846. [Google Scholar]
- Grube, A.; Timm, C.; Kock, M. Synthesis and mass spectrometric analysis of cyclostellettamines H, I, K and L. Eur. J. Org. Chem. 2006, 2006, 1285–1295. [Google Scholar] [CrossRef]
- Dong, S.; Tang, J.-J.; Zhang, C.-C.; Tian, J.-M.; Guo, J.-T.; Zhang, Q.; Li, H.; Gao, J.-M. Semisynthesis and in vitro cytotoxic evaluation of new analogues of 1-O-acetylbritannilactone, a sesquiterpene from Inula britannica. Eur. J. Med. Chem. 2014, 80C, 71–82. [Google Scholar] [CrossRef]
- Jenkins, G.J.S.; Zaïr, Z.; Johnson, G.E.; Doak, S.H. Genotoxic thresholds, DNA repair, and susceptibility in human populations. Toxicology 2010, 278, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Snyder, R. Possible structural and functional determinants contributing to the clastogenicity of pharmaceuticals. Environ. Mol. Mutagen. 2010, 814, 800–814. [Google Scholar] [CrossRef]
- Abbotts, R.; Thompson, N.; Madhusudan, S. DNA repair in cancer: Emerging targets for personalized therapy. Cancer Manag. Res. 2014, 6, 77–92. [Google Scholar] [PubMed]
- Brüsehafer, K.; Rees, B.J.; Manshian, B.B.; Doherty, A.T.; O’Donovan, M.R.; Doak, S.H.; Jenkins, G.J.S. Chromosome breakage induced by the genotoxic agents MMC and araC is concentration and p53 dependent. Toxicol. Sci. Adv. Access 2014, 140, 94–102. [Google Scholar] [CrossRef]
- Honma, M.; Hayashi, M. Comparison of in vitro micronucleus and gene mutation assay results for p53-competent versus p53-deficient human lymphoblastoid cells. Environ. Mol. Mutagen. 2011, 52, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Amundson, S.A; Myers, T.G.; Fornace, A.J. Roles for p53 in growth arrest and apoptosis: Putting on the brakes after genotoxic stress. Oncogene 1998, 17, 3287–3299. [Google Scholar]
- Utani, K.; Kohno, Y.; Okamoto, A.; Shimizu, N. Emergence of micronuclei and their effects on the fate of cells under replication stress. PLoS One 2010, 5, e10089. [Google Scholar] [CrossRef] [PubMed]
- Essack, M.; Bajic, V.B.; Archer, J.A.C. Recently confirmed apoptosis-inducing lead compounds isolated from marine sponge of potential relevance in cancer treatment. Mar. Drugs 2011, 9, 1580–1606. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Uematsu, H.; Tsuchida, N.; Ikeda, M.-A. Essential role of caspase-8 in p53/p73-dependent apoptosis induced by etoposide in head and neck carcinoma cells. Mol. Cancer 2011, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.-J.; Lin, C.-I.; Liou, J.-P.; Kuo, C.-C.; Chang, C.-Y.; Chen, L.-T.; Chang, J.-Y. A novel synthetic microtubule inhibitor, MPT0B214 exhibits antitumor activity in human tumor cells through mitochondria-dependent intrinsic pathway. PLoS One 2013, 8, e58953. [Google Scholar] [CrossRef] [PubMed]
- LaBarbera, D.V; Modzelewska, K.; Glazar, A.I.; Gray, P.D.; Kaur, M.; Liu, T.; Grossman, D.; Harper, M.K.; Kuwada, S.K.; Moghal, N.; et al. The marine alkaloid naamidine a promotes caspase-dependent apoptosis in tumor cells. Anticancer Drugs 2009, 20, 425–436. [Google Scholar]
- Kim, S.-K.; Cho, S.-M.; Kim, H.; Seok, H.; Kim, S.-O.; Kwon, T.K.; Chang, J.-S. The colchicine derivative CT20126 shows a novel microtubule-modulating activity with apoptosis. Exp. Mol. Med. 2013, 45, e19. [Google Scholar] [CrossRef] [PubMed]
- Marzo, I.; Naval, J. Antimitotic drugs in cancer chemotherapy: Promises and pitfalls. Biochem. Pharmacol. 2013, 86, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, M.; Azou-Gros, Y.; Tsai, F.-C.; Alvarado, J.; Bertin, A.; Iv, F.; Kress, A.; Brasselet, S.; Koenderink, G.H.; Lecuit, T. Septins promote F-actin ring formation by crosslinking actin filaments into curved bundles. Nat. Cell Biol. 2014, 16, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Youssef, D.T.A; Mooberry, S.L. Hurghadolide A and swinholide I, potent actin-microfilament disrupters from the Red Sea sponge Theonella swinhoei. J. Nat. Prod. 2006, 69, 154–157. [Google Scholar]
- Topham, C.H.; Taylor, S.S. Mitosis and apoptosis: How is the balance set? Curr. Opin. Cell Biol. 2013, 25, 780–785. [Google Scholar] [CrossRef]
- De Oliveira, M.E.; Cenzi, G.; Nunes, R.R.; Andrighetti, C.R.; de Sousa Valadão, D.M.; Dos Reis, C.; Simões, C.M.O.; Nunes, R.J.; Júnior, M.C.; Taranto, A.G.; et al. Antimalarial activity of 4-metoxychalcones: Docking studies as falcipain/plasmepsin inhibitors,ADMET and lipophilic efficiency analysis to identify a putative oral lead candidate. Molecules 2013, 18, 15276–15287. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2007, 2, 1084–1104. [Google Scholar] [CrossRef] [PubMed]
- Fenech, M. The in vitro micronucleus technique. Mutat. Res. 2000, 455, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Titenko-Holland, N.; Windham, G.; Kolachana, P.; Reinisch, F.; Parvatham, S.; Osorio, A.M.; Smith, M.T. Genotoxicity of malathion in human lymphocytes assessed using the micronucleus assay in vitro and in vivo: A study of malathion-exposed workers. Mutat. Res. 1997, 388, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.C.; Moreira, L.M.; Santos, V.J.S.V.; Ramos, A.S.; Lyon, J.P.; Soares, C.P.; Santos, F.V. Assessment of the genetic risks of a metallic alloy used in medical implants. Genet. Mol. Biol. 2011, 34, 116–121. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gonçalves, A.M.M.N.; De Lima, A.B.; Da Silva Barbosa, M.C.; De Camargos, L.F.; De Oliveira, J.T.; De Souza Barbosa, C.; Villar, J.A.F.P.; Costa, A.C.; Silva, I.V.G.d.; Silva, L.M.; et al. Synthesis and Biological Evaluation of Novel 3-Alkylpyridine Marine Alkaloid Analogs with Promising Anticancer Activity. Mar. Drugs 2014, 12, 4361-4378. https://doi.org/10.3390/md12084361
Gonçalves AMMN, De Lima AB, Da Silva Barbosa MC, De Camargos LF, De Oliveira JT, De Souza Barbosa C, Villar JAFP, Costa AC, Silva IVGd, Silva LM, et al. Synthesis and Biological Evaluation of Novel 3-Alkylpyridine Marine Alkaloid Analogs with Promising Anticancer Activity. Marine Drugs. 2014; 12(8):4361-4378. https://doi.org/10.3390/md12084361
Chicago/Turabian StyleGonçalves, Alessandra Mirtes Marques Neves, Aline Brito De Lima, Maria Cristina Da Silva Barbosa, Luiz Fernando De Camargos, Júlia Teixeira De Oliveira, Camila De Souza Barbosa, José Augusto Ferreira Perez Villar, André Carvalho Costa, Isabella Viana Gomes da Silva, Luciana Maria Silva, and et al. 2014. "Synthesis and Biological Evaluation of Novel 3-Alkylpyridine Marine Alkaloid Analogs with Promising Anticancer Activity" Marine Drugs 12, no. 8: 4361-4378. https://doi.org/10.3390/md12084361
APA StyleGonçalves, A. M. M. N., De Lima, A. B., Da Silva Barbosa, M. C., De Camargos, L. F., De Oliveira, J. T., De Souza Barbosa, C., Villar, J. A. F. P., Costa, A. C., Silva, I. V. G. d., Silva, L. M., De Pilla Varotti, F., Dos Santos, F. V., & Viana, G. H. R. (2014). Synthesis and Biological Evaluation of Novel 3-Alkylpyridine Marine Alkaloid Analogs with Promising Anticancer Activity. Marine Drugs, 12(8), 4361-4378. https://doi.org/10.3390/md12084361