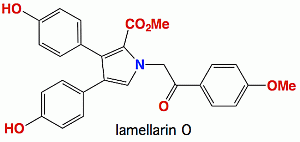
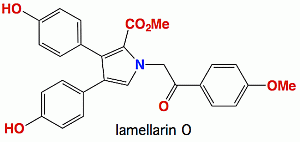
Lamellarin O, a Pyrrole Alkaloid from an Australian Marine Sponge, Ianthella sp., Reverses BCRP Mediated Drug Resistance in Cancer Cells


 and
and 
Abstract
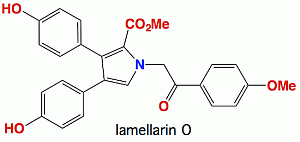
:
1. Introduction
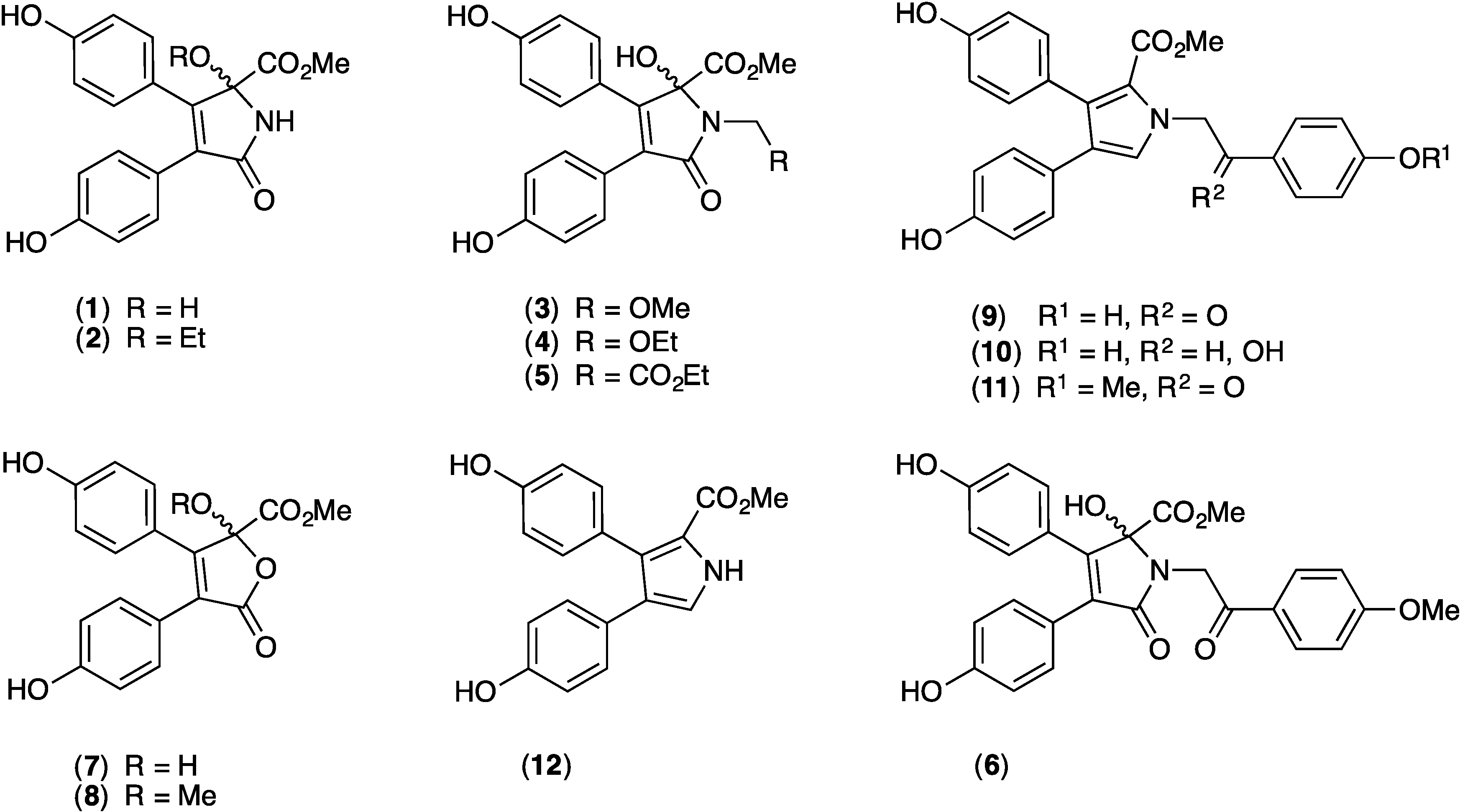
2. Results and Discussion
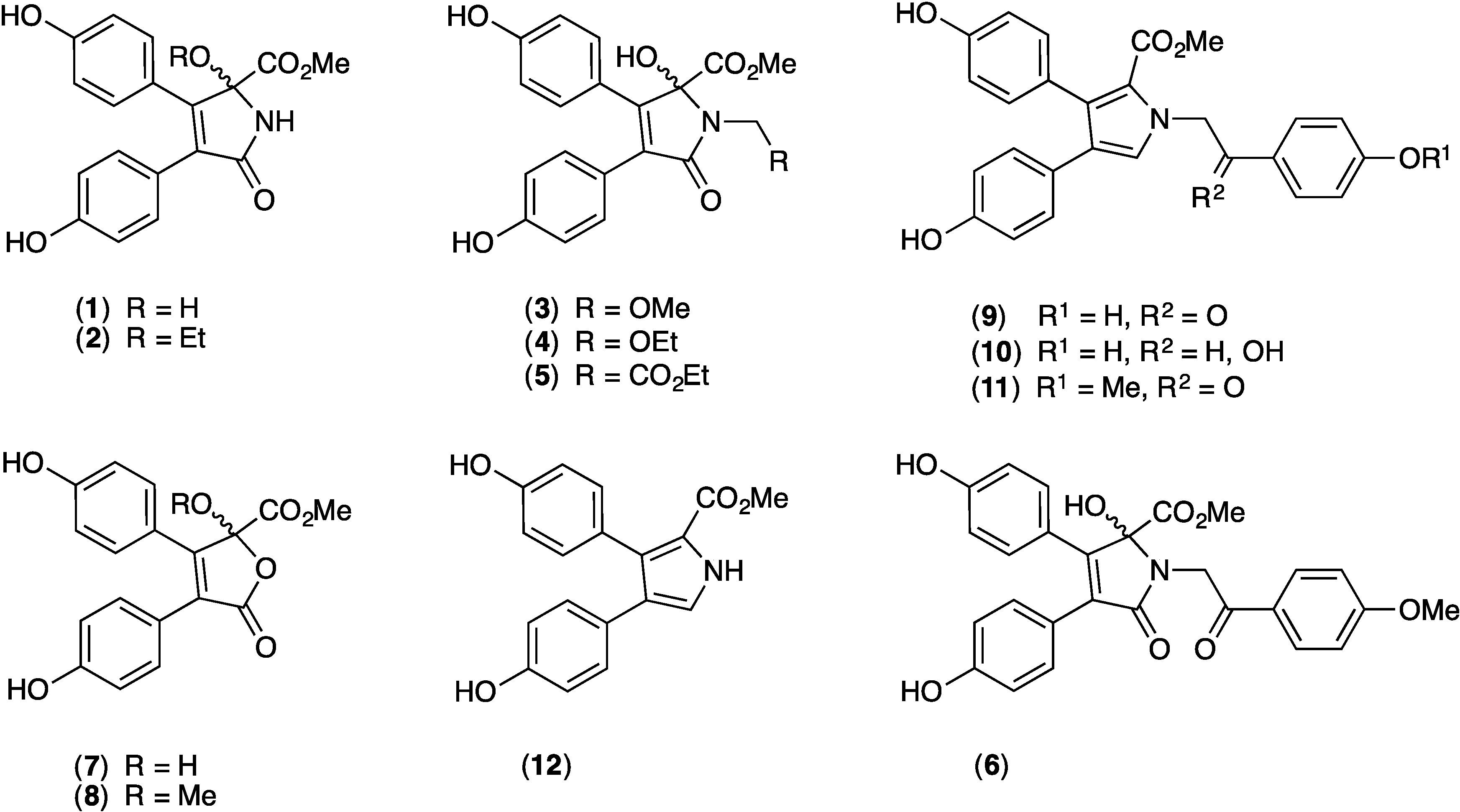
2.1. Cytotoxicity of 1–12 against SW620 and SW620 Ad300
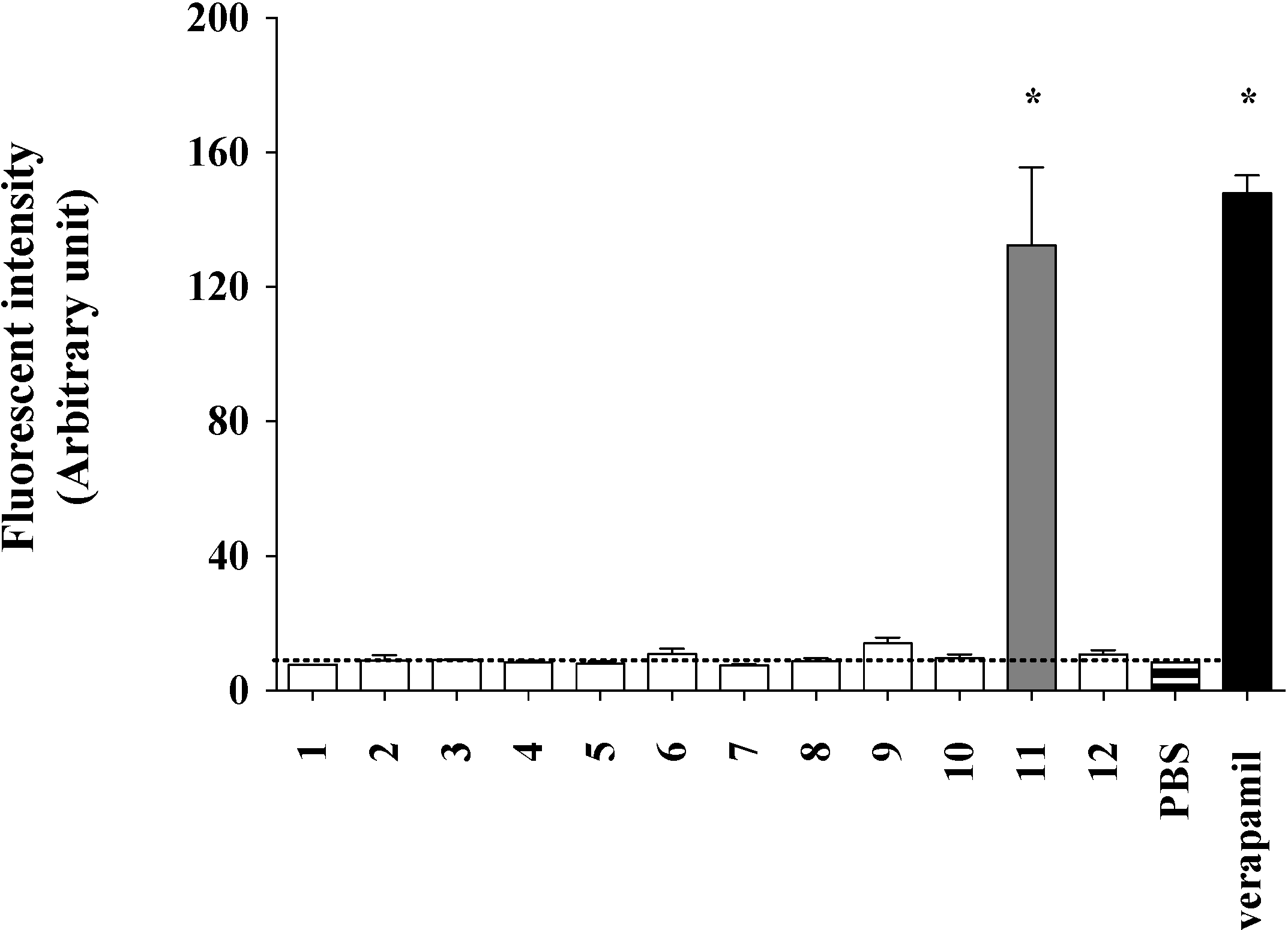
2.2. Lamellarin O (11) as a P-gp Inhibitor in SW620 Ad300 Cancer Cells (Calcein AM Assay)
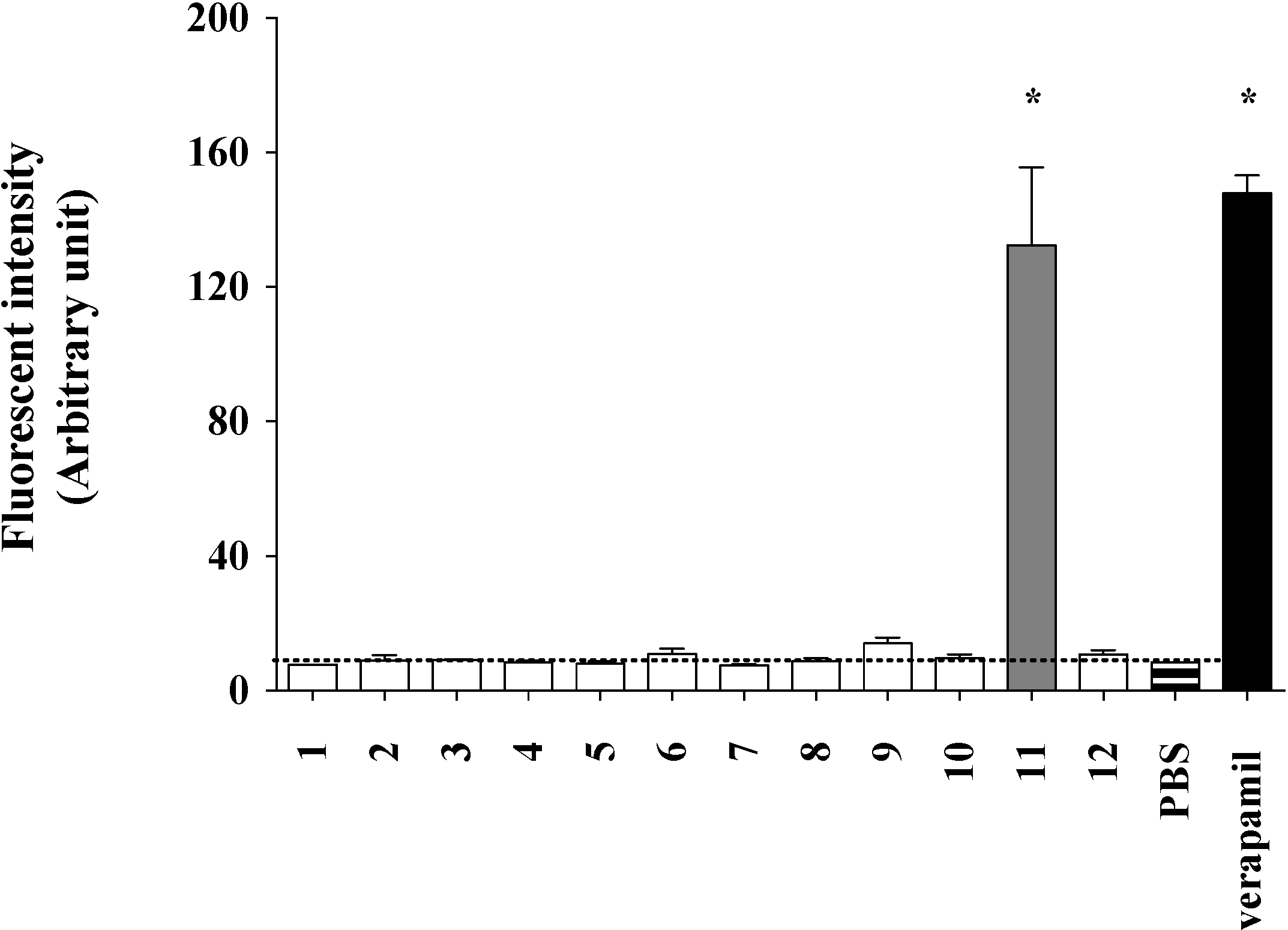

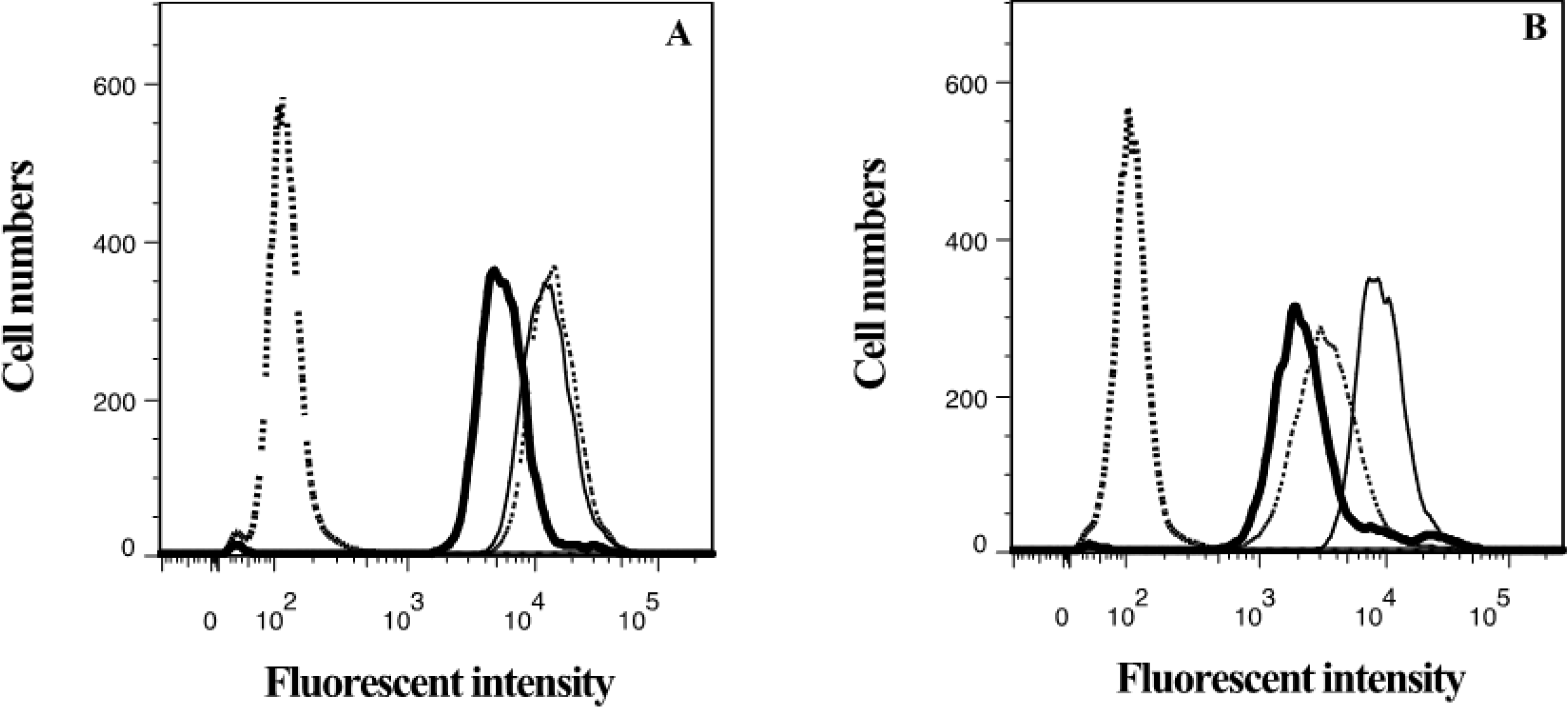
2.3. Lamellarin O (11) as a P-gp Inhibitor in SW620 Ad300 Cells (Calcein AM by Cell Flow Cytometry)
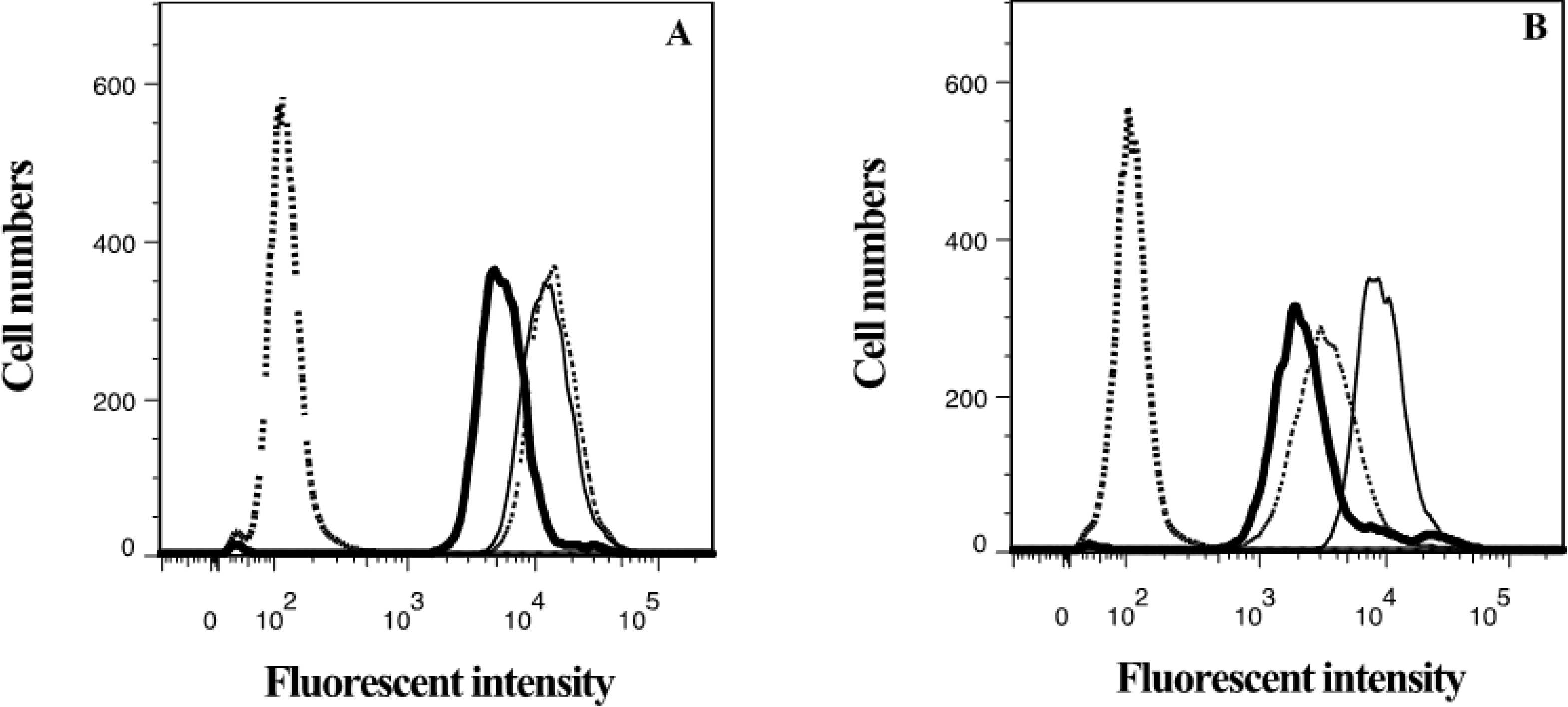
2.4. Lamellarin O (11) as a P-gp Inhibitor in SW620 Ad300 Cells (Hoechst 33342 Accumulation/Efflux)
2.5. Lamellarin O (11) Reverses P-gp Mediated Doxorubicin Resistance in SW620 Ad300 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
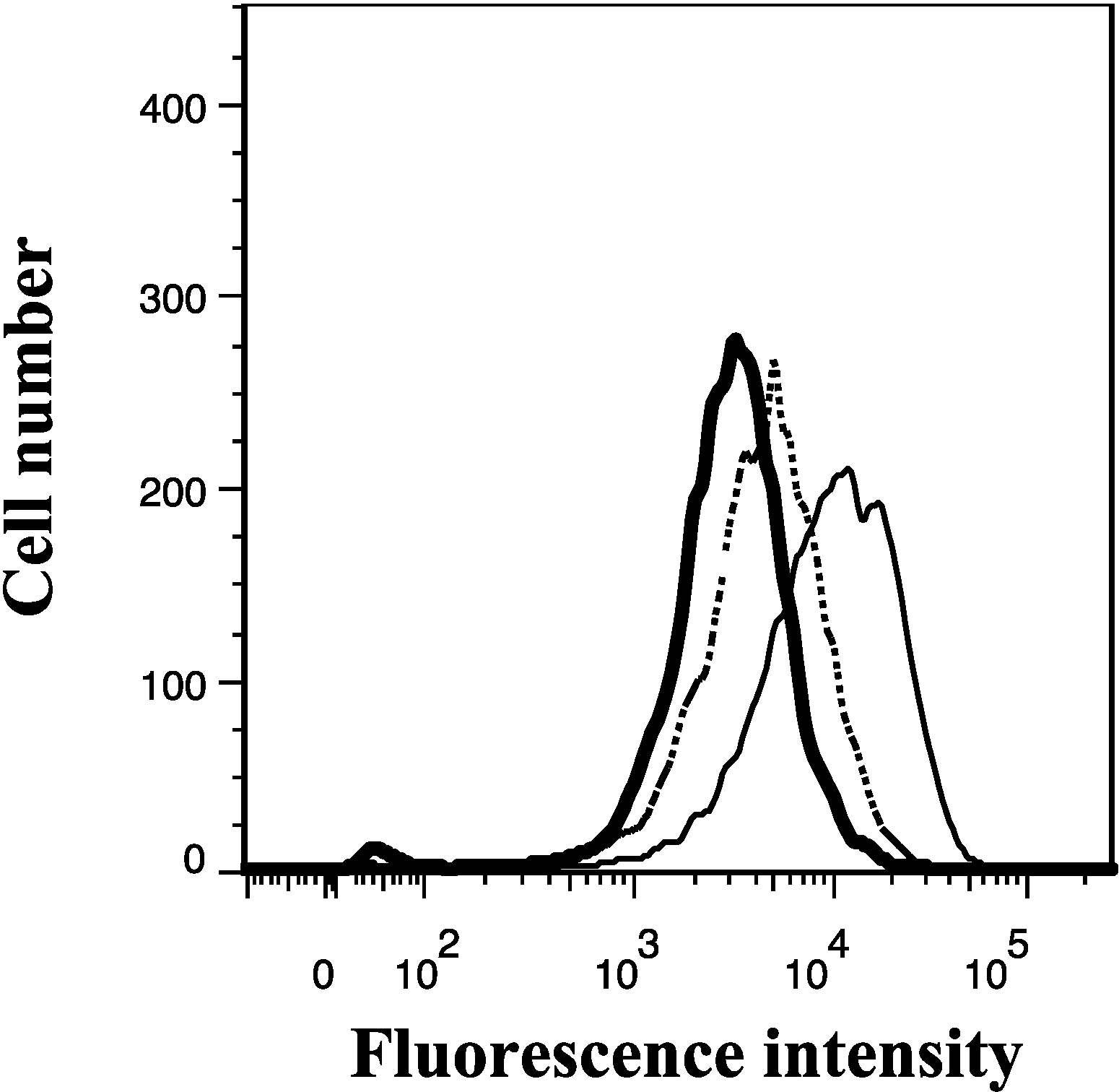{kind=link}
| Treatment | IC50 a (nM) | DMF b |
|---|---|---|
| doxorubicin | 4440 ± 210 | |
| + lamellarin O (11) 5.0 μM | 2910 ± 350 | 1.5 |
| + lamellarin O (11) 10 μM | 1430 ± 160 | 3.1 |
| + lamellarin O (11) 15 μM | 930 ± 120 | 4.8 |
| + verapamil 2.5 μM | 500 ± 230 | 8.8 |
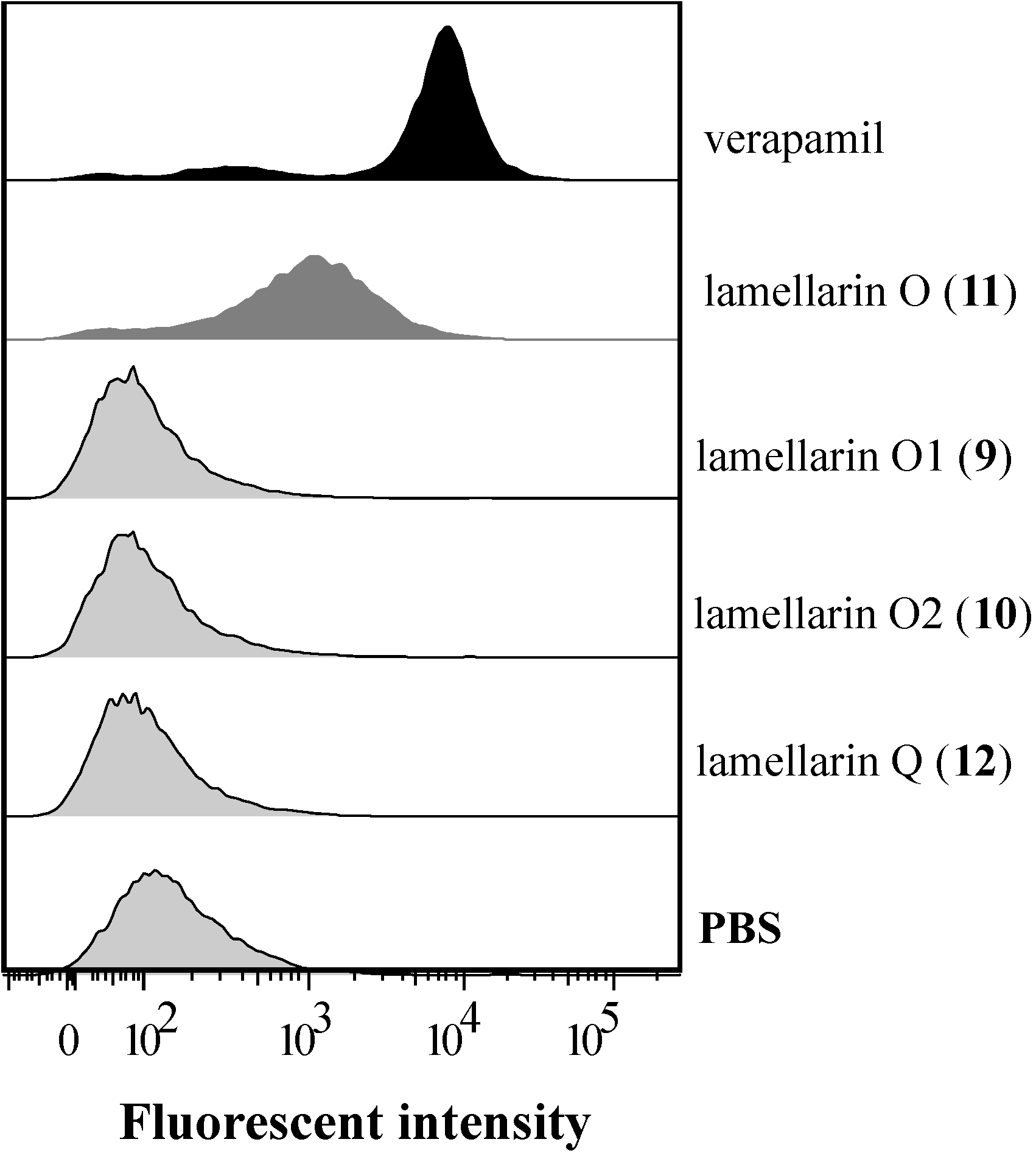
2.6. Lamellarin O (11) Reverses BCRP Mediated Efflux in NCI-H460/MX20 Cells
| Treatment | FAR a | % FTC b |
|---|---|---|
| lamellarin O (11) | 3.05 | 94.5 |
| ianthellidone A (1) | 0.97 | <1.00 |
| ianthellidone B (2) | 1.07 | 3.13 |
| ianthellidone C (3) | 1.12 | 5.60 |
| ianthellidone D (4) | 1.15 | 6.66 |
| ianthellidone E (5) | 1.14 | 6.20 |
| ianthellidone F (6) | 1.21 | 9.79 |
| ianthellidone G (7) | 1.09 | 4.06 |
| ianthellidone H (8) | 1.13 | 6.06 |
| lamellarin O1 (9) | 1.23 | 10.6 |
| lamellarin O2 (10) | 1.11 | 5.13 |
| lamellarin Q (12) | 1.41 | 18.6 |
| PBS | 1.00 | 0.00 |
| FTC 10 μM | 3.17 | 100 |
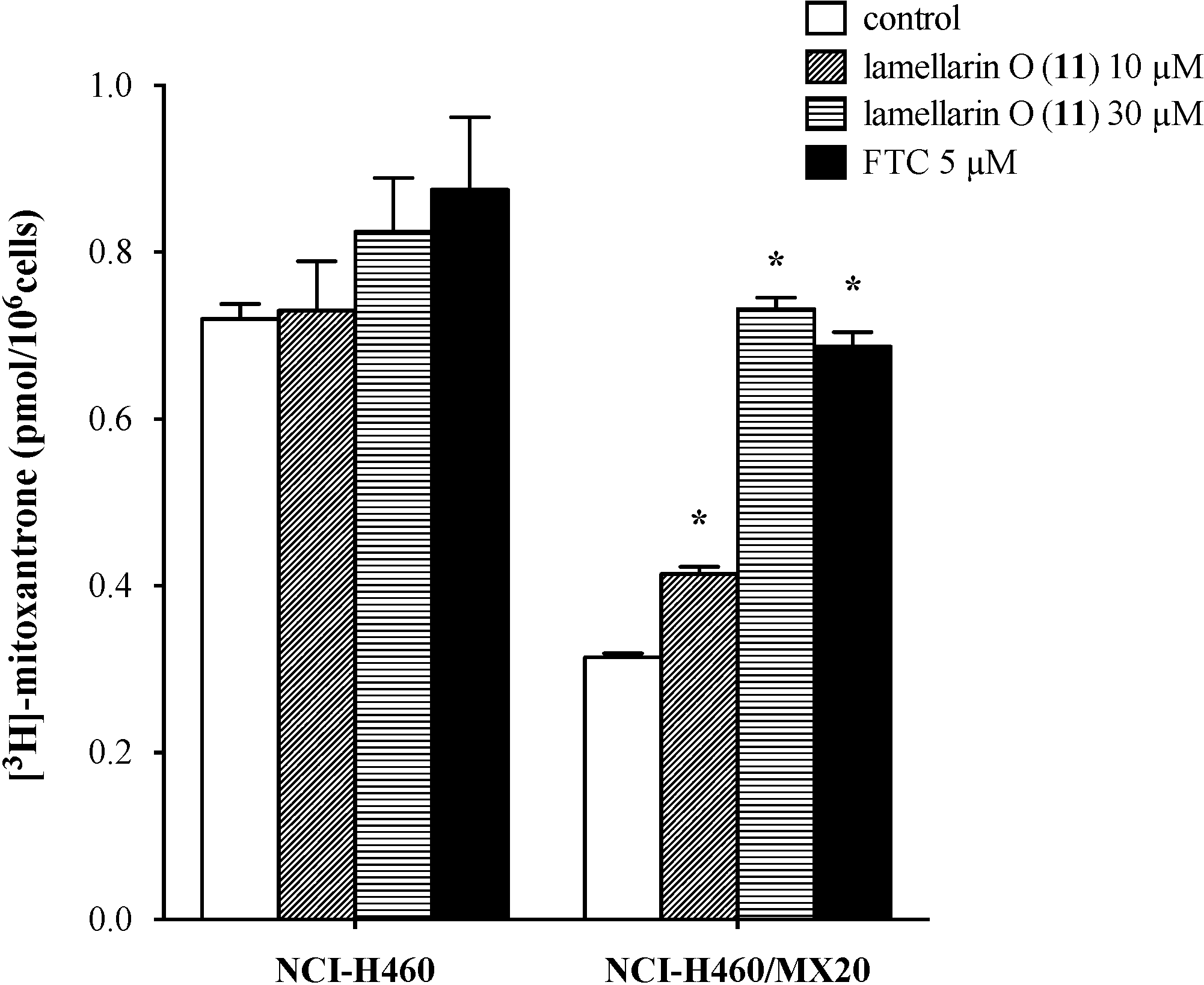
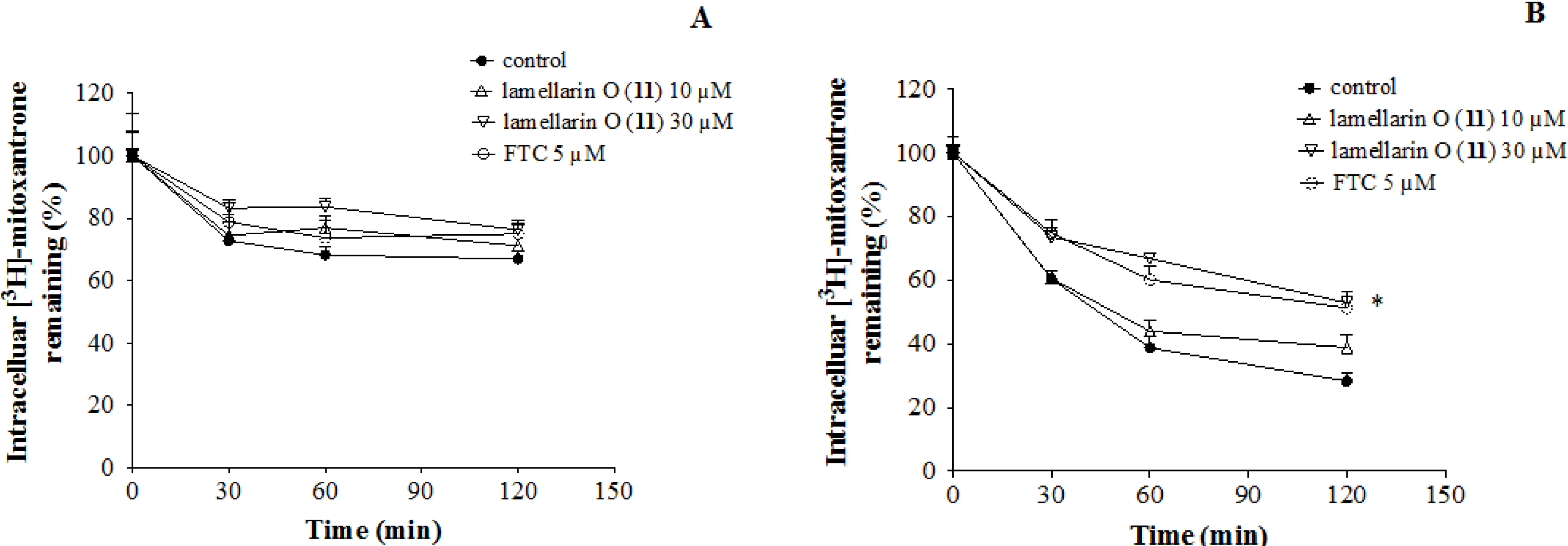
2.7. Lamellarin O (11) Reverses Efflux of [3H]-Mitoxantrone in NCI-H460/MX20 Cells
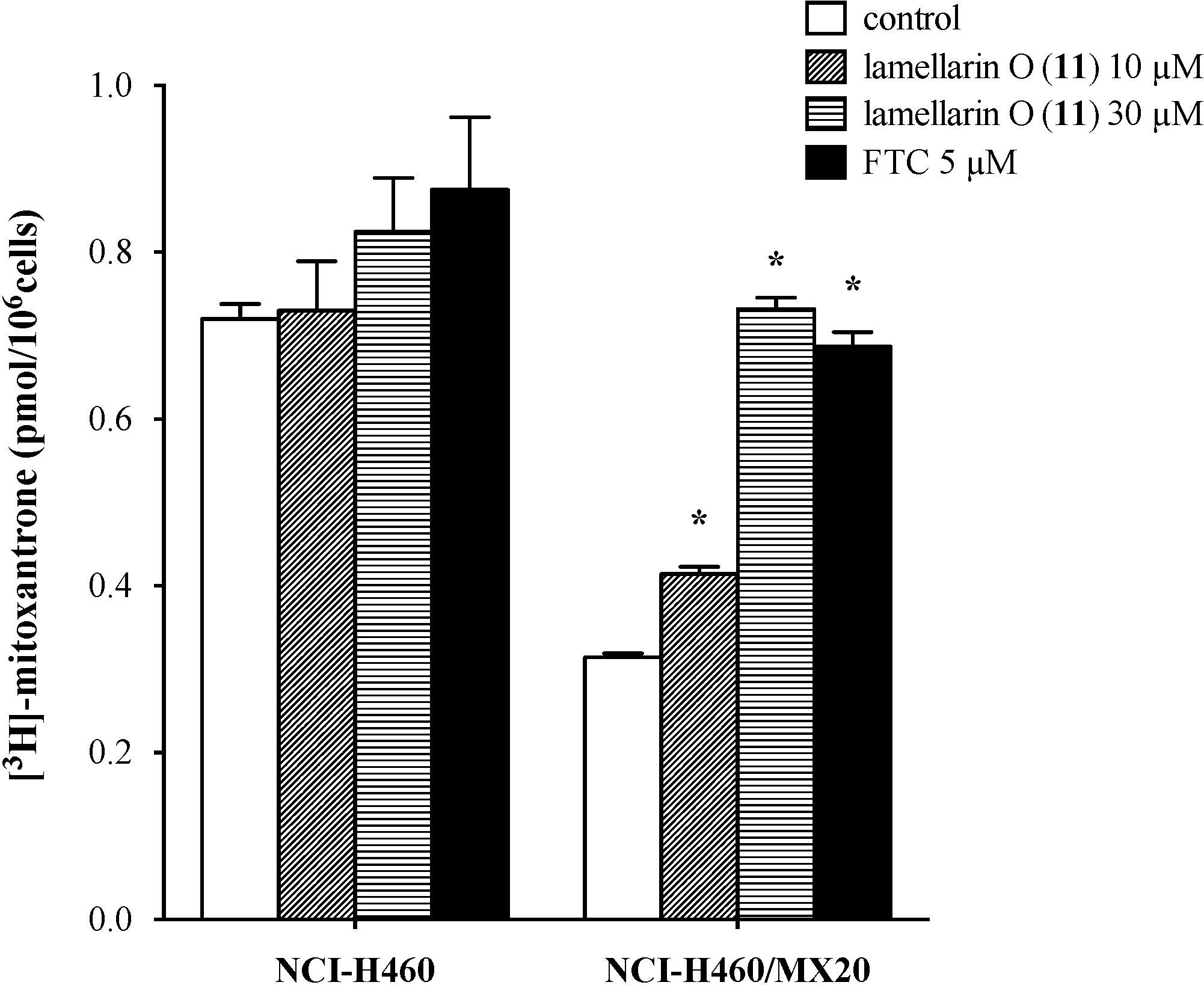
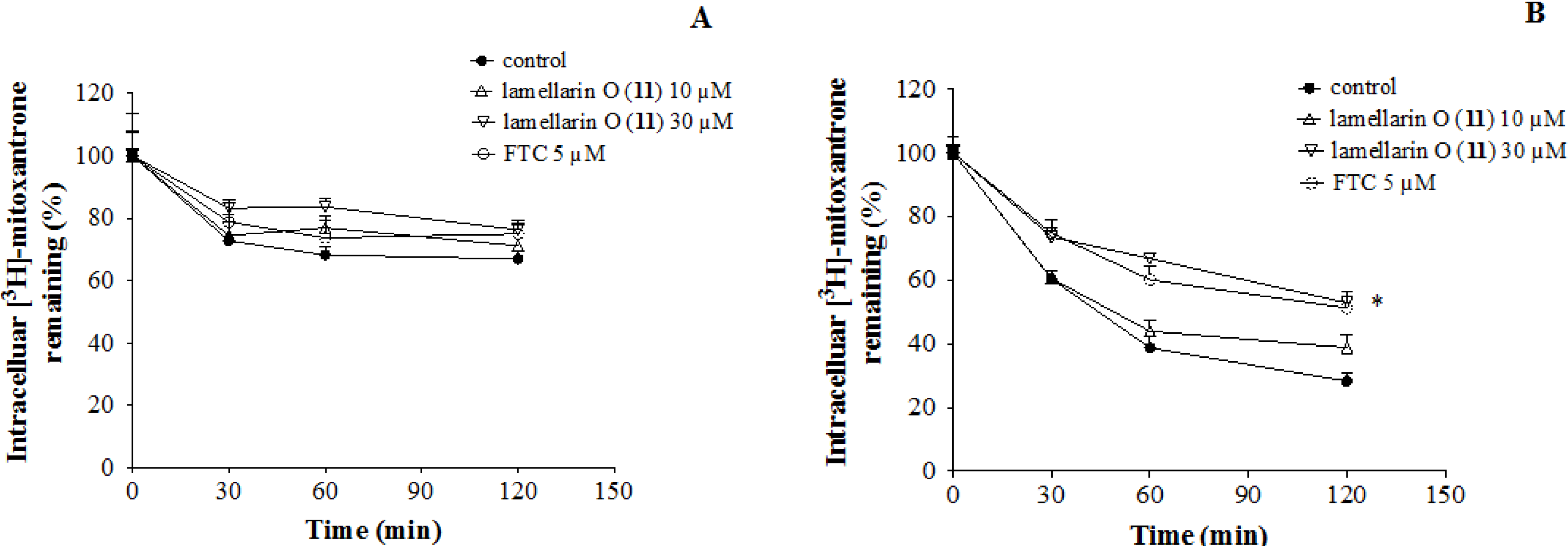
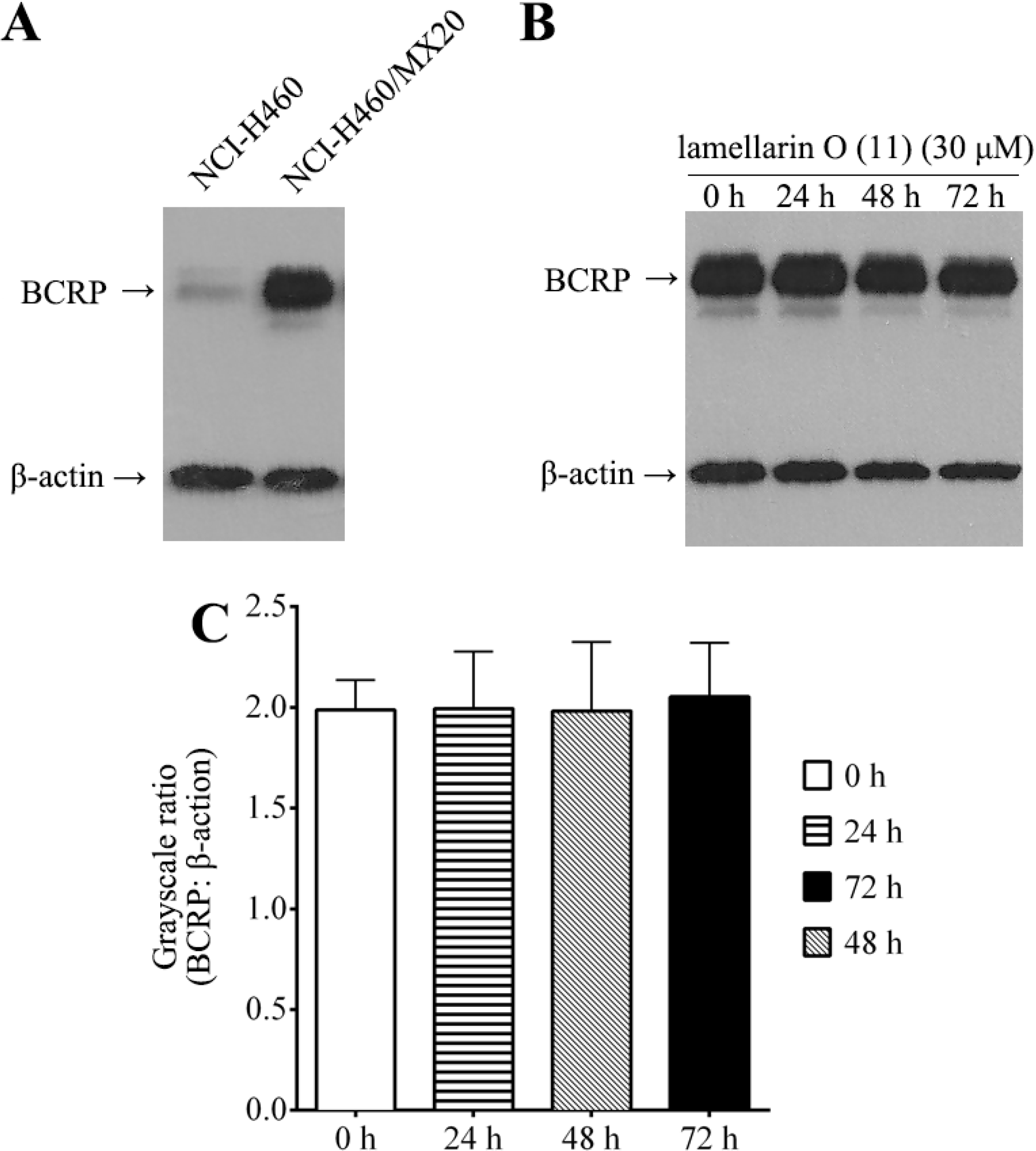
2.8. Effect of Lamellarin O (11) on BCRP Expression in NCI-H460/MX20 Cells
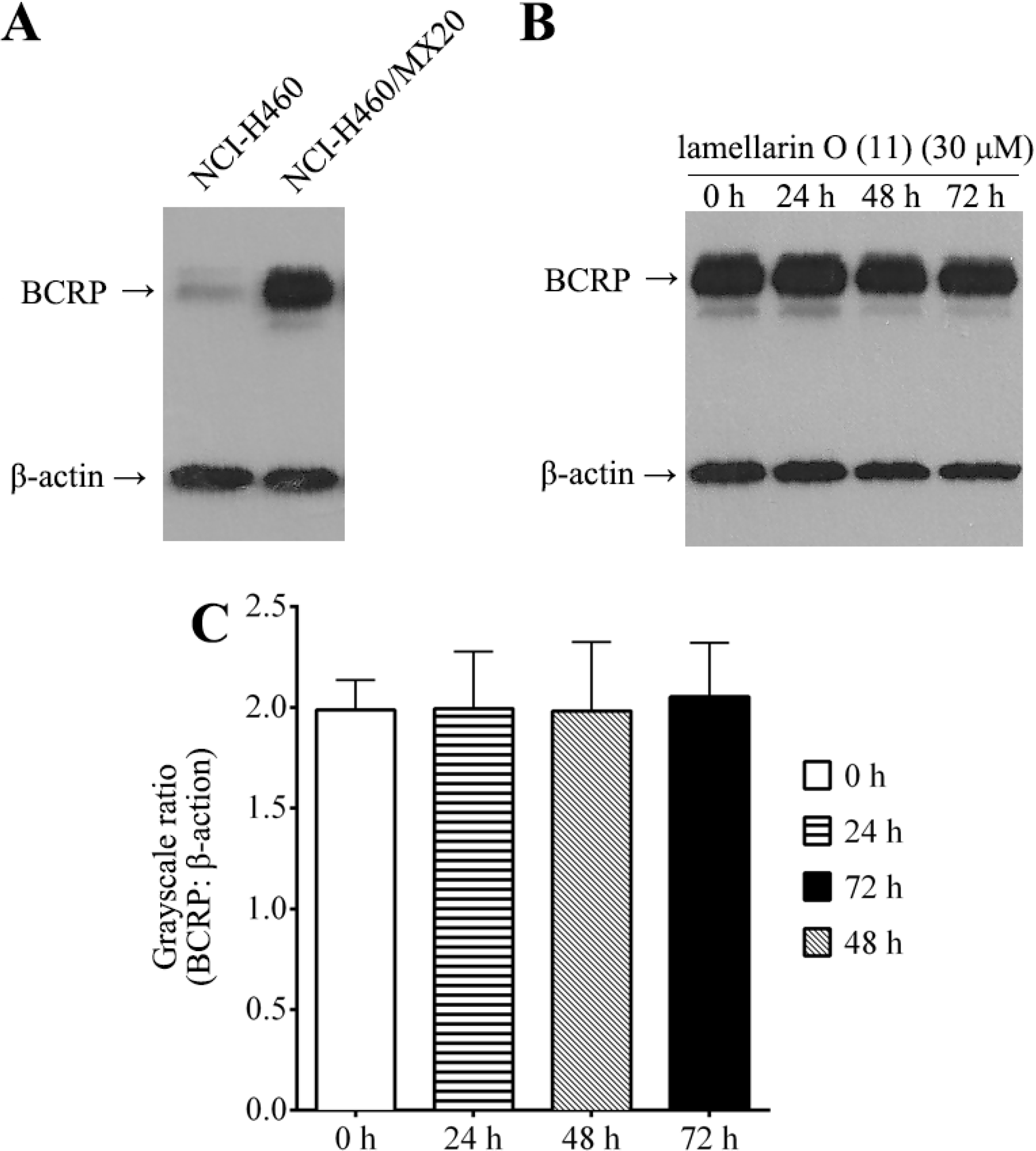
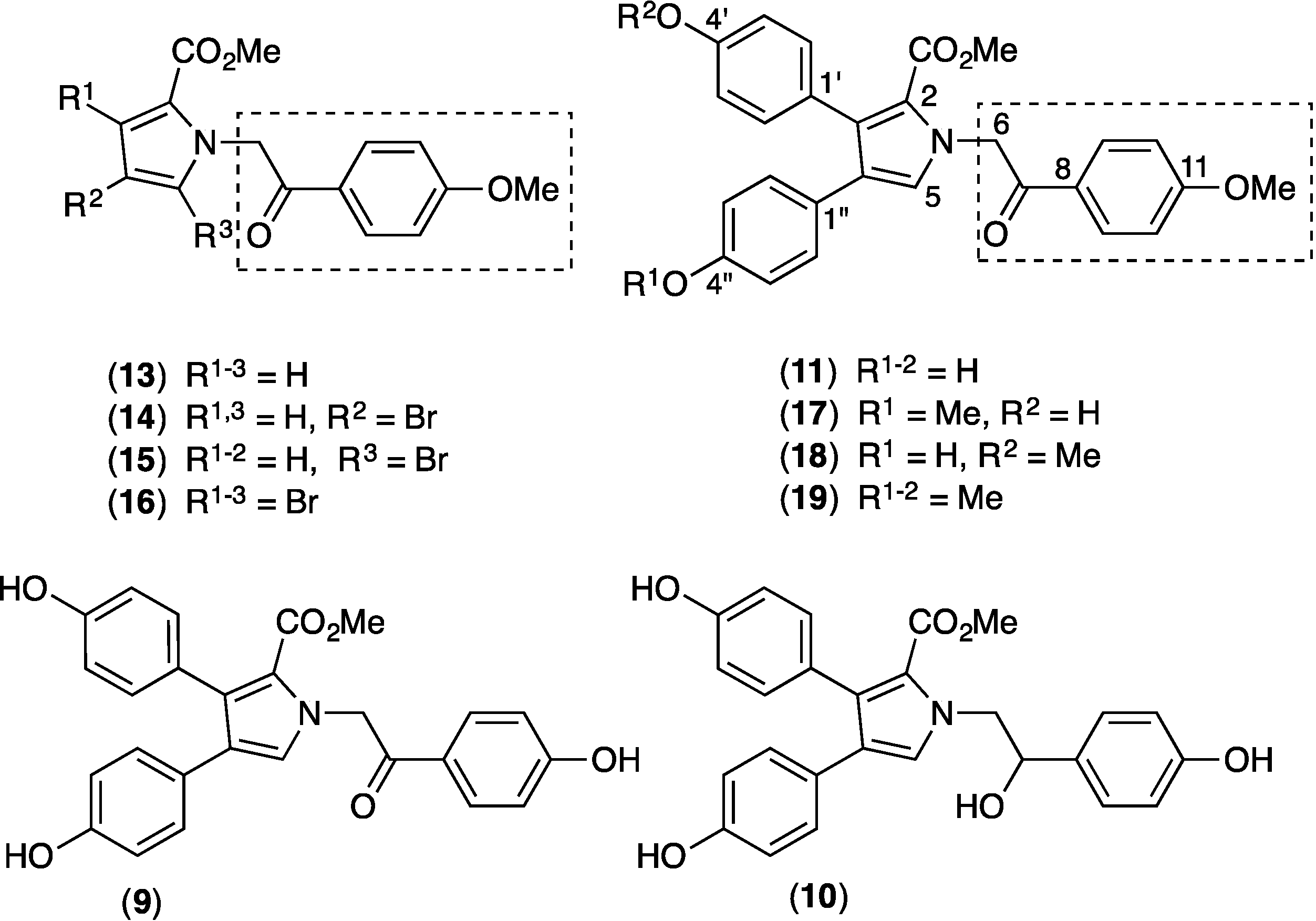
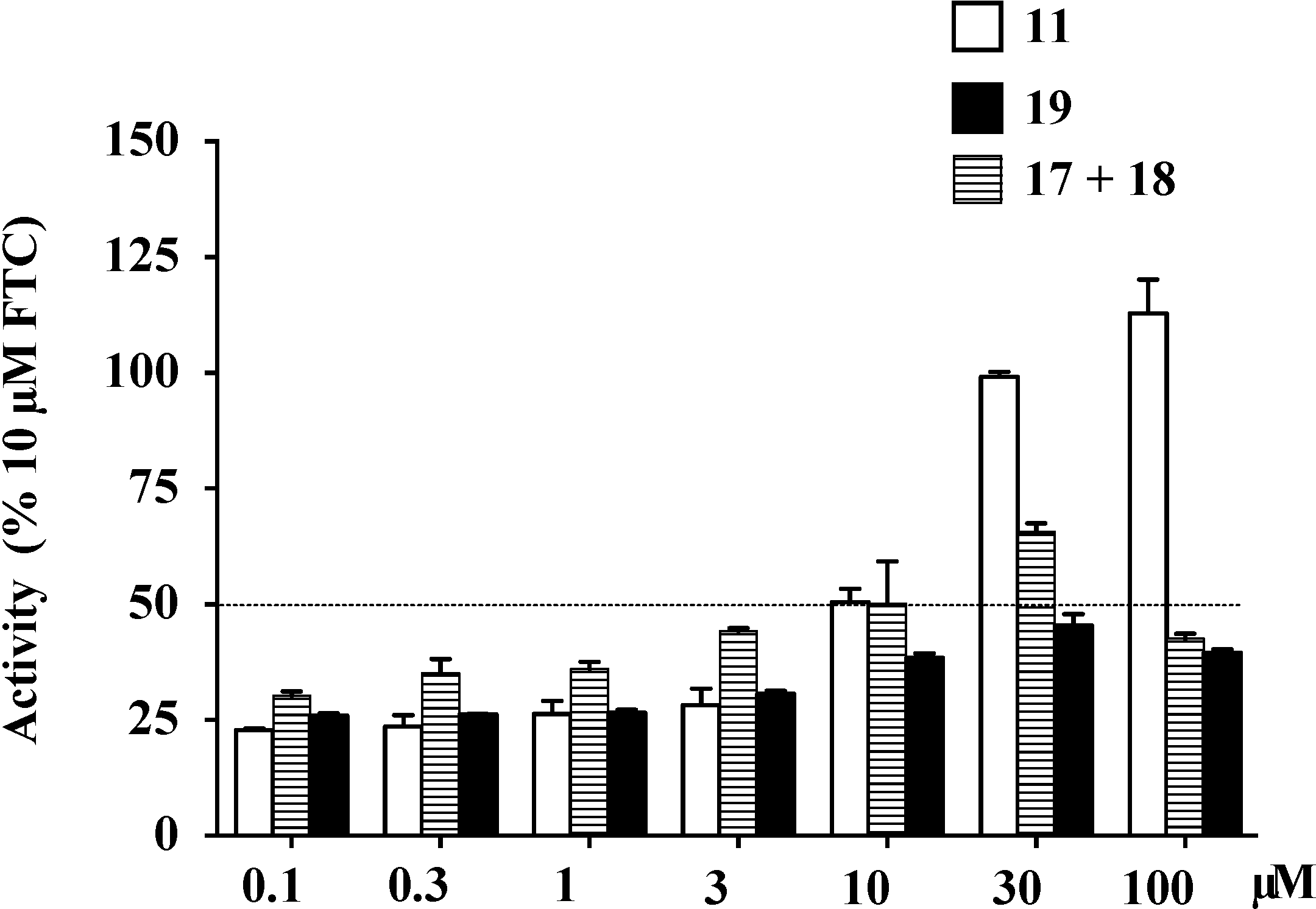
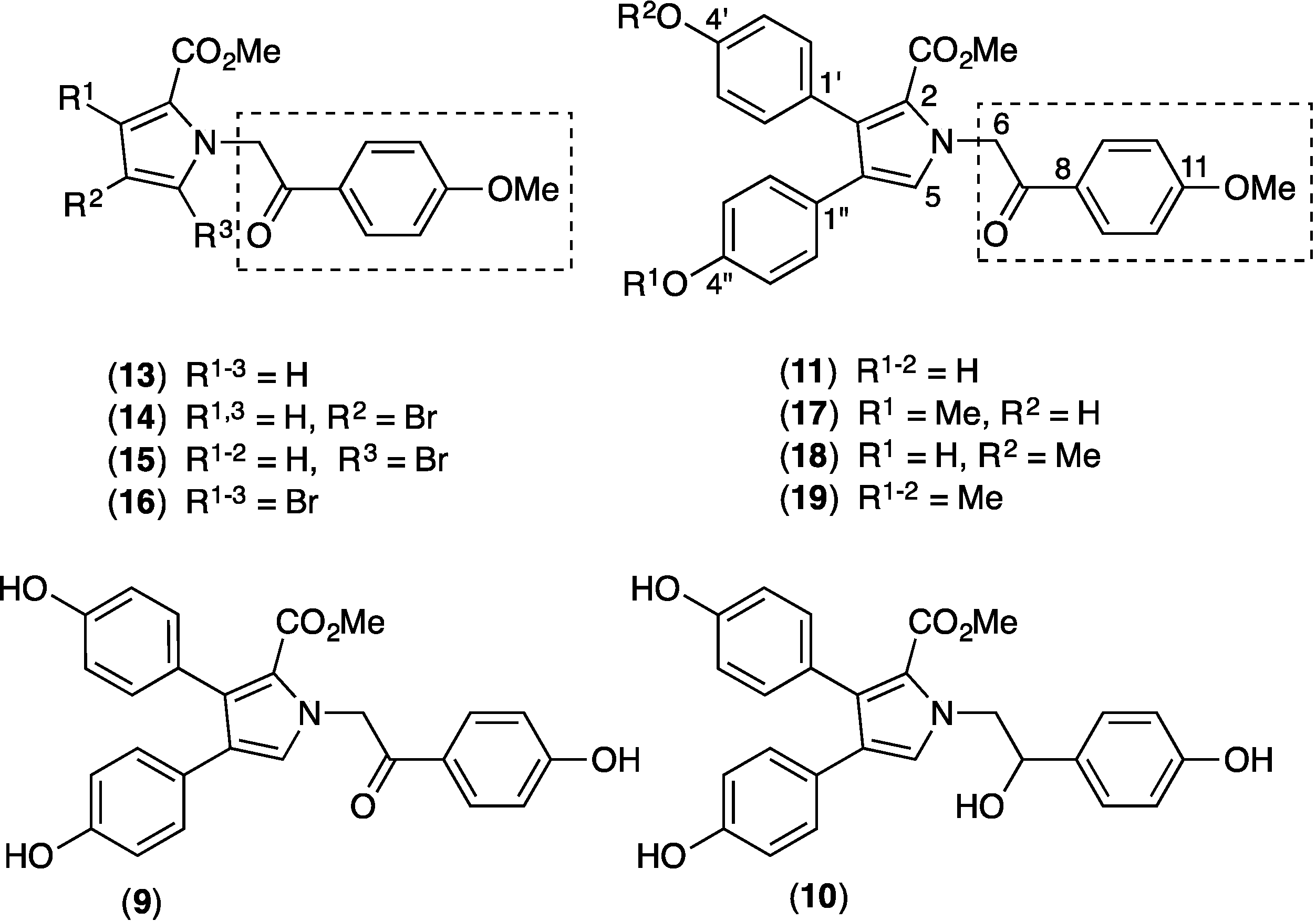
2.9. Structure Activity Relationship (SAR) Studies on Lamellarin O (11)
| IC50 (μM) a | Maximal Activity (% of FTC) | |
|---|---|---|
| 11 | 4.7 ± 0.6 | 112.9 |
| 17+18 | 20.2 ± 2.1 | 65.7 |
| 19 | >100 | 45.4 |
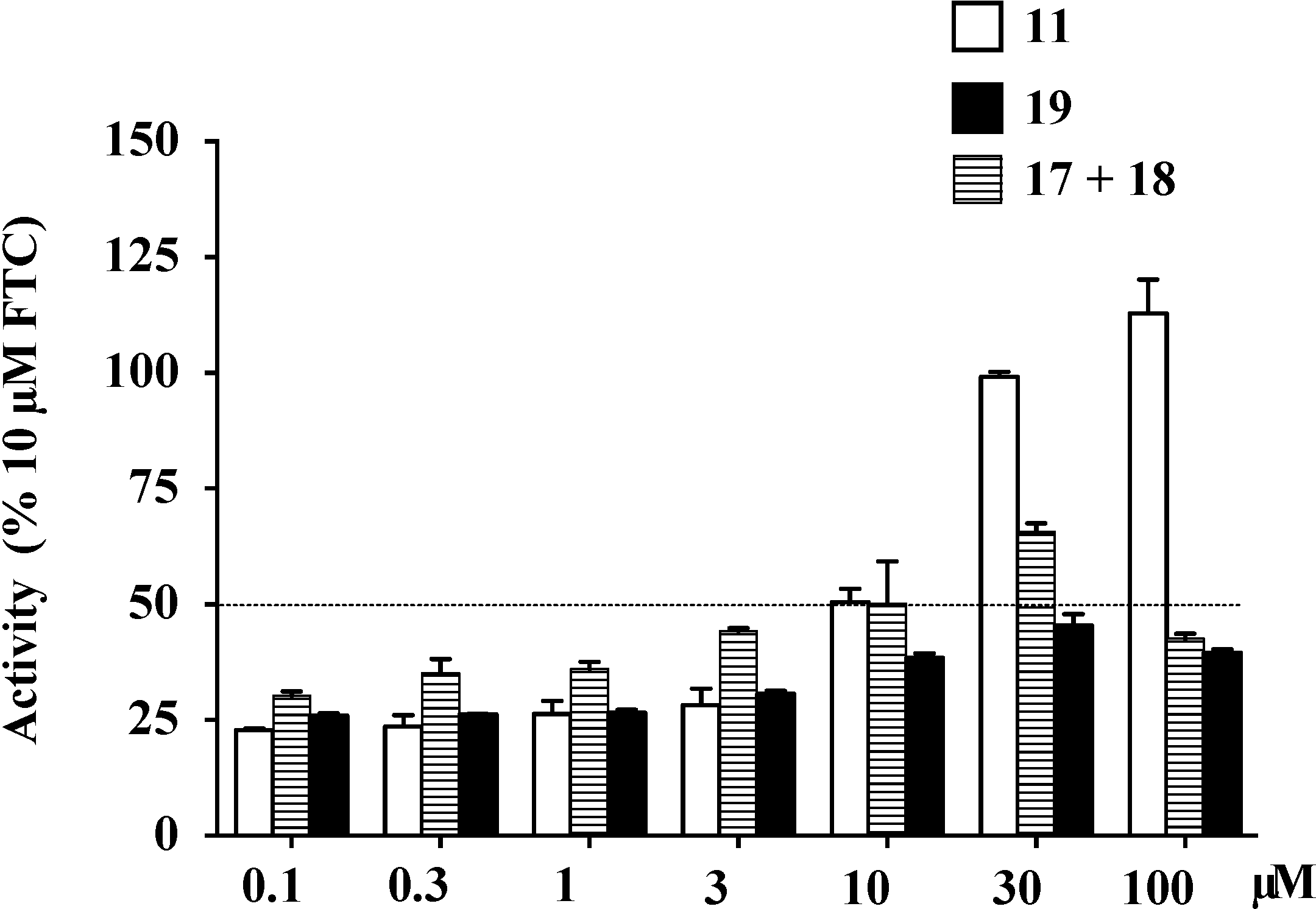
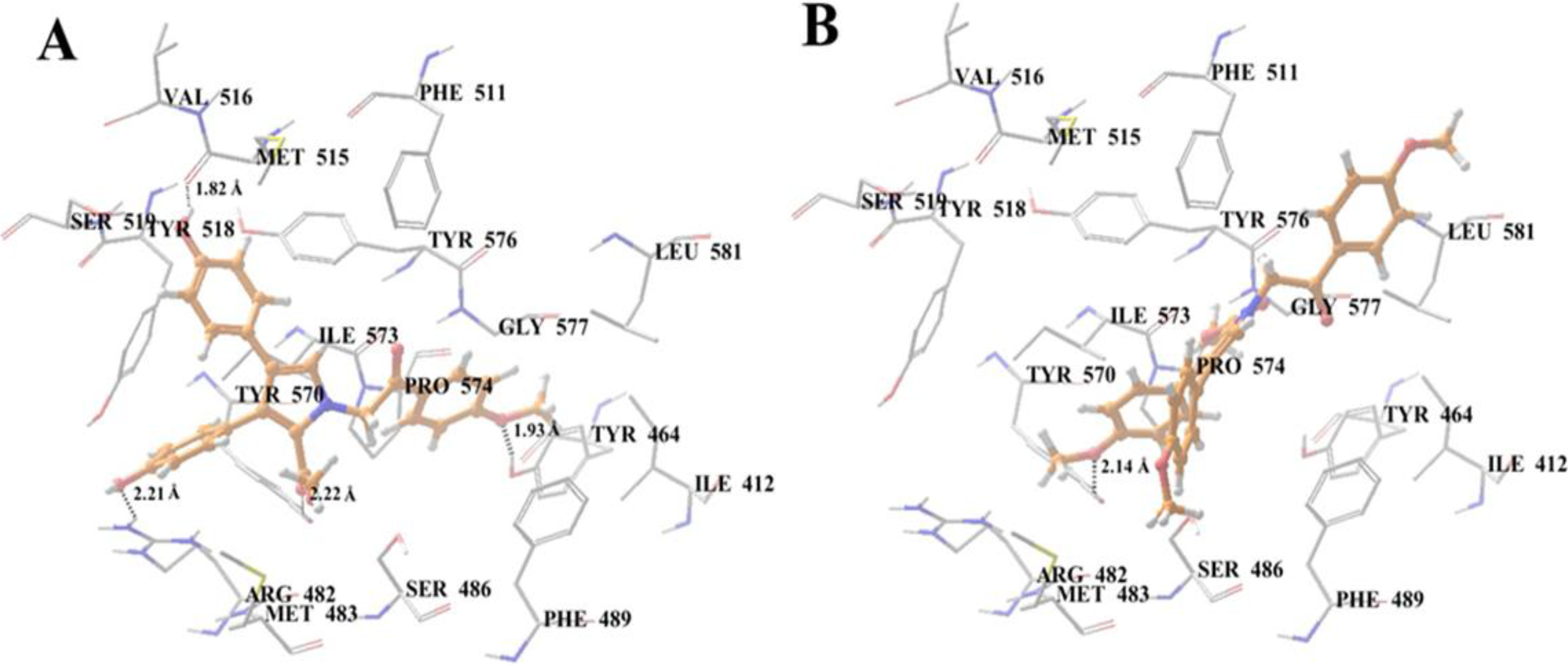
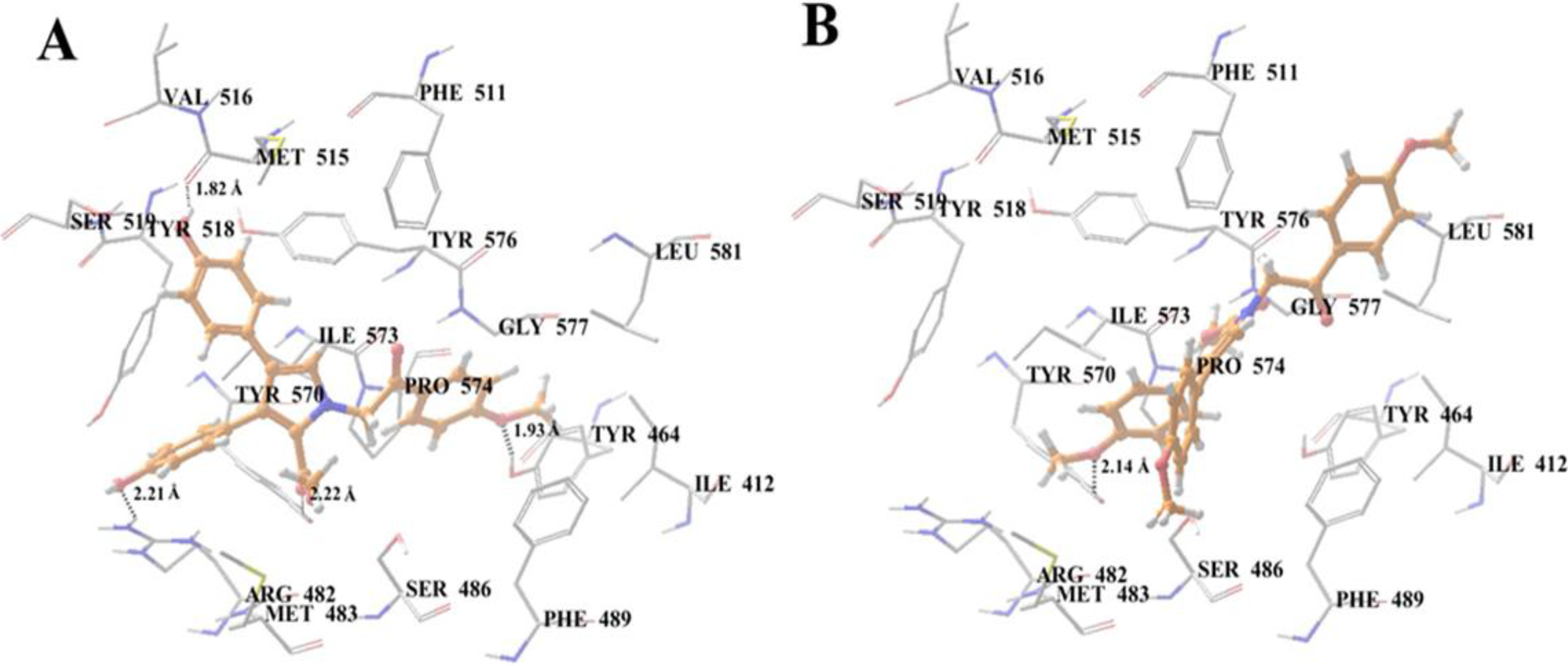
2.10. Docking Analysis of Lamellarin O (11) and 19 with Human ABCG2 Homology Models
2.11. The Effect of Lamellarin O (11) on the MRP1 Mediated Efflux of Calcein in 2008/MRP1 Cells
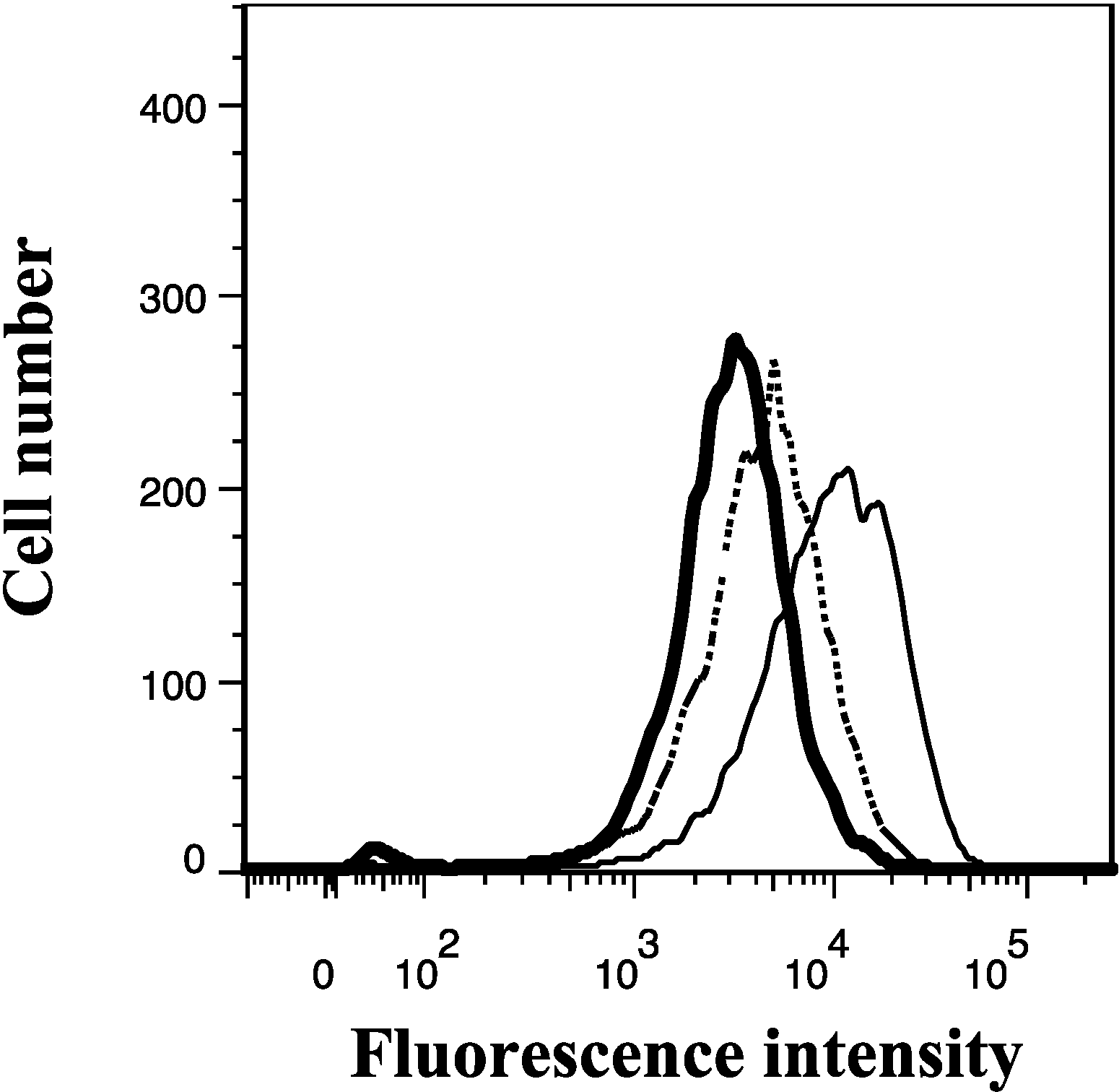
3. Experimental Section
3.1. Chemical Materials
3.2. Cell Lines and Cell Culture
3.3. Calcein AM Accumulation Assay (96-Well Plate Format)
3.4. Flow Cytometry Assays
3.5. MTT Cytotoxicity Assay
3.6. Drug Accumulation and Efflux Assay
3.7. Preparation of Total Cell Lysates and Western Blotting
3.8. Syntheses of Methylated Lamellarin O Derivatives (17, 18 and 19)
3.9. Molecular Modeling
3.10. Statistical Analysis
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dano, K. Active outward transport of daunomycin in resistant ehrlich ascites tumor-cells. Biochim. Biophys. Acta 1973, 323, 466–483. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Hegedus, C.; Ozvegy-Laczka, C.; Apati, A.; Magocsi, M.; Nemet, K.; Orfi, L.; Keri, G.; Katona, M.; Takats, Z.; Varadi, A.; et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: Implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 2009, 158, 1153–1164. [Google Scholar] [CrossRef]
- Cole, S.P.C.; Bhardwaj, G.; Gerlach, J.H.; Mackie, J.E.; Grant, C.E.; Almquist, K.C.; Stewart, A.J.; Kurz, E.U.; Duncan, A.M.V.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung-cancer cell-line. Science 1992, 258, 1650–1654. [Google Scholar]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar]
- Wu, C.P.; Ohnuma, S.; Ambudkar, S.V. Discovering natural product modulators to overcome multidrug resistance in cancer chemotherapy. Curr. Pharm. Biotechnol. 2011, 12, 609–620. [Google Scholar] [CrossRef]
- Prinsep, M.R.; Caplan, F.R.; Moore, R.E.; Patterson, G.M.L.; Smith, C.D. Tolyporphin, a novel multidrug resistance reversing agent from the blue-green-alga tolypothrix-nodosa. J. Am. Chem. Soc. 1992, 114, 385–387. [Google Scholar] [CrossRef]
- Aoki, S.; Yoshioka, Y.; Miyamoto, Y.; Higuchi, K.; Setiawan, A.; Murakami, N.; Chen, Z.S.; Sumizawa, T.; Akiyama, S.; Kobayashi, M. Agosterol a, a novel polyhydroxylated sterol acetate reversing multidrug resistance from a marine sponge of Spongia sp. Tetrahedron Lett. 1998, 39, 6303–6306. [Google Scholar] [CrossRef]
- Chen, Z.S.; Aoki, S.; Komatsu, M.; Ueda, K.; Sumizawa, T.; Furukawa, T.; Okumura, H.; Ren, X.Q.; Belinsky, M.G.; Lee, K.; et al. Reversal of drug resistance mediated by multidrug resistance protein (MRP) 1 by dual effects of agosterol a on mrp1 function. Int. J. Cancer 2001, 93, 107–113. [Google Scholar] [CrossRef]
- Spitaler, M.; Utz, I.; Hilbe, W.; Hofmann, J.; Grunicke, H.H. PKC-independent modulation of multidrug resistance in cells with mutant (v185) but not wild-type (g185) p-glycoprotein by bryosratin 1. Biochem. Pharmacol. 1998, 56, 861–869. [Google Scholar] [CrossRef]
- Aoki, S.; Okano, M.; Matsui, K.; Itoh, T.; Satari, R.; Akiyama, S.; Kobayashi, M. Brianthein A, a novel briarane-type diterpene reversing multidrug resistance in human carcinoma cell line, from the gorgonian briareum excavatum. Tetrahedron 2001, 57, 8951–8957. [Google Scholar] [CrossRef]
- Henrich, C.J.; Robey, R.W.; Takada, K.; Bokesch, H.R.; Bates, S.E.; Shukla, S.; Ambudkar, S.V.; McMahon, J.B.; Gustafson, K.R. Botryllamides: Natural product inhibitors of ABCG2. ACS Chem. Biol. 2009, 4, 637–647. [Google Scholar] [CrossRef]
- Tanaka, C.; Yamamoto, Y.; Otsuka, M.; Tanaka, J.; Ichiba, T.; Marriott, G.; Rachmat, R.; Higa, T. Briarane diterpenes from two species of octocorals, Ellisella sp. and Pteroeides sp. J. Nat. Prod. 2004, 67, 1368–1373. [Google Scholar] [CrossRef]
- Fu, P.; Liu, P.P.; Li, X.; Wang, Y.; Wang, S.X.; Hong, K.; Zhu, W.M. Cyclic bipyridine glycosides from the marine-derived actinomycete actinoalloteichus cyanogriseus WH1-2216-6. Org. Lett. 2011, 13, 5948–5951. [Google Scholar]
- Smith, C.D.; Zilfou, J.T.; Stratmann, K.; Patterson, G.M.L.; Moore, R.E. Welwitindolinone analogs that reverse p-glycoprotein-mediated multiple-drug resistance. Mol. Pharmacol. 1995, 47, 241–247. [Google Scholar]
- Williams, A.B.; Jacobs, R.S. A marine natural product, patellamide-D, reverses multidrug-resistance in a human leukemic-cell line. Cancer Lett. 1993, 71, 97–102. [Google Scholar] [CrossRef]
- Raju, R.; Piggott, A.M.; Huang, X.C.; Capon, R.J. Nocardioazines: A novel bridged diketopiperazine scaffold from a marine-derived bacterium inhibits p-glycoprotein. Org. Lett. 2011, 13, 2770–2773. [Google Scholar] [CrossRef]
- Plisson, F.; Huang, X.C.; Zhang, H.; Khalil, Z.; Capon, R.J. Lamellarins as inhibitors of p-glycoprotein-mediated multidrug resistance in a human colon cancer cell line. Chem. Asian J. 2012, 7, 1616–1623. [Google Scholar] [CrossRef]
- Huang, X.C.; Sun, Y.L.; Salim, A.A.; Chen, Z.S.; Capon, R.J. Parguerenes: Marine red alga bromoditerpenes as inhibitors of P-glycoprotein (ABCB1) in multidrug resistant human cancer cells. Biochem. Pharmacol. 2013, 85, 1257–1268. [Google Scholar] [CrossRef]
- Druley, T.E.; Stein, W.D.; Roninson, I.B. Analysis of MDR1 P-glycoprotein conformational changes in permeabilized cells using differential immunoreactivity. Biochemistry 2001, 40, 4312–4322. [Google Scholar] [CrossRef]
- Henrich, C.J.; Bokesch, H.R.; Dean, M.; Bates, S.E.; Robey, R.W.; Goncharova, E.I.; Wilson, J.A.; McMahon, J.B. A high-throughput cell-based assay for inhibitors of ABCG2 activity. J. Biomol. Screen. 2006, 11, 176–183. [Google Scholar]
- Robey, R.W.; Honjo, Y.; van de Laar, A.; Miyake, K.; Regis, J.T.; Litman, T.; Bates, S.E. A functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2). Biochim. Biophys. Acta 2001, 1512, 171–182. [Google Scholar]
- Zhang, H.; Conte, M.M.; Huang, X.C.; Khalil, Z.; Capon, R.J. A search for bace inhibitors reveals new biosynthetically related pyrrolidones, furanones and pyrroles from a Southern Australian marine sponge, Ianthella sp. Org. Biomol. Chem. 2012, 10, 2656–2663. [Google Scholar] [CrossRef]
- Robey, R.W.; Steadman, K.; Polgar, O.; Morisaki, K.; Blayney, M.; Mistry, P.; Bates, S.E. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004, 64, 1242–1246. [Google Scholar] [CrossRef]
- Hooijberg, J.H.; Broxterman, H.J.; Kool, M.; Assaraf, Y.G.; Peters, G.J.; Noordhuis, P.; Scheper, R.J.; Borst, P.; Pinedo, M.M.; Jansen, G. Antifolate resistance mediated by the multidrug resistance proteins MRP1 and MRP2. Cancer Res. 1999, 59, 2532–2535. [Google Scholar]
- Tiberghien, F.; Loor, F. Ranking of P-glycoprotein substrates and inhibitors by a calcein-AM fluorometry screening assay. Anti-Cancer Drugs 1996, 7, 568–578. [Google Scholar] [CrossRef]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of radiosensitivity. Cancer Res. 1987, 47, 943–946. [Google Scholar]
- Kuang, Y.H.; Patel, J.P.; Sodani, K.; Wu, C.P.; Liao, L.Q.; Patel, A.; Tiwari, A.K.; Dai, C.L.; Chen, X.; Fu, L.W.; et al. OSI-930 analogues as novel reversal agents for ABCG2-mediated multidrug resistance. Biochem. Pharmacol. 2012, 84, 766–774. [Google Scholar] [CrossRef]
- Sodani, K.; Tiwari, A.K.; Singh, S.; Patel, A.; Xiao, Z.J.; Chen, J.J.; Sun, Y.L.; Talele, T.T.; Chen, Z.S. GW583340 and GW2974, human EGFR and HER-2 inhibitors, reverse ABCG2- and ABCB1-mediated drug resistance. Biochem. Pharmacol. 2012, 83, 1613–1622. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Sodani, K.; Dai, C.L.; Abuznait, A.H.; Singh, S.; Xiao, Z.J.; Patel, A.; Talele, T.T.; Fu, L.; Kaddoumi, A.; et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013, 328, 307–317. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, X.-C.; Xiao, X.; Zhang, Y.-K.; Talele, T.T.; Salim, A.A.; Chen, Z.-S.; Capon, R.J. Lamellarin O, a Pyrrole Alkaloid from an Australian Marine Sponge, Ianthella sp., Reverses BCRP Mediated Drug Resistance in Cancer Cells. Mar. Drugs 2014, 12, 3818-3837. https://doi.org/10.3390/md12073818
Huang X-C, Xiao X, Zhang Y-K, Talele TT, Salim AA, Chen Z-S, Capon RJ. Lamellarin O, a Pyrrole Alkaloid from an Australian Marine Sponge, Ianthella sp., Reverses BCRP Mediated Drug Resistance in Cancer Cells. Marine Drugs. 2014; 12(7):3818-3837. https://doi.org/10.3390/md12073818
Chicago/Turabian StyleHuang, Xiao-Cong, Xue Xiao, Yun-Kai Zhang, Tanaji T. Talele, Angela A. Salim, Zhe-Sheng Chen, and Robert J. Capon. 2014. "Lamellarin O, a Pyrrole Alkaloid from an Australian Marine Sponge, Ianthella sp., Reverses BCRP Mediated Drug Resistance in Cancer Cells" Marine Drugs 12, no. 7: 3818-3837. https://doi.org/10.3390/md12073818
APA StyleHuang, X.-C., Xiao, X., Zhang, Y.-K., Talele, T. T., Salim, A. A., Chen, Z.-S., & Capon, R. J. (2014). Lamellarin O, a Pyrrole Alkaloid from an Australian Marine Sponge, Ianthella sp., Reverses BCRP Mediated Drug Resistance in Cancer Cells. Marine Drugs, 12(7), 3818-3837. https://doi.org/10.3390/md12073818