2. Results and Discussion
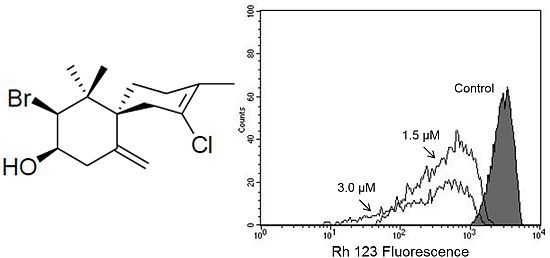
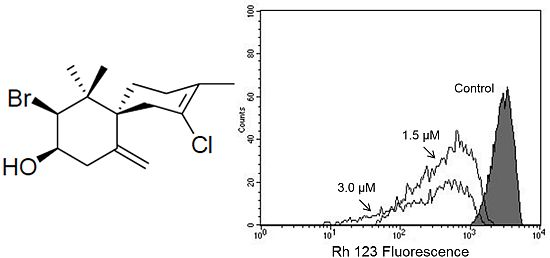
(−)-Elatol (
Figure 1) has previously been reported to have trypanocidal [
8], leishmanicidal [
7], and antibacterial activity [
10,
11,
12] and significantly active roles in ecological interactions, such as antiherbivore activity [
13]. In the present study we focus our efforts on the trypanocidal activity of (−)-elatol in an attempt to delineate the putative mechanism of action of this compound.
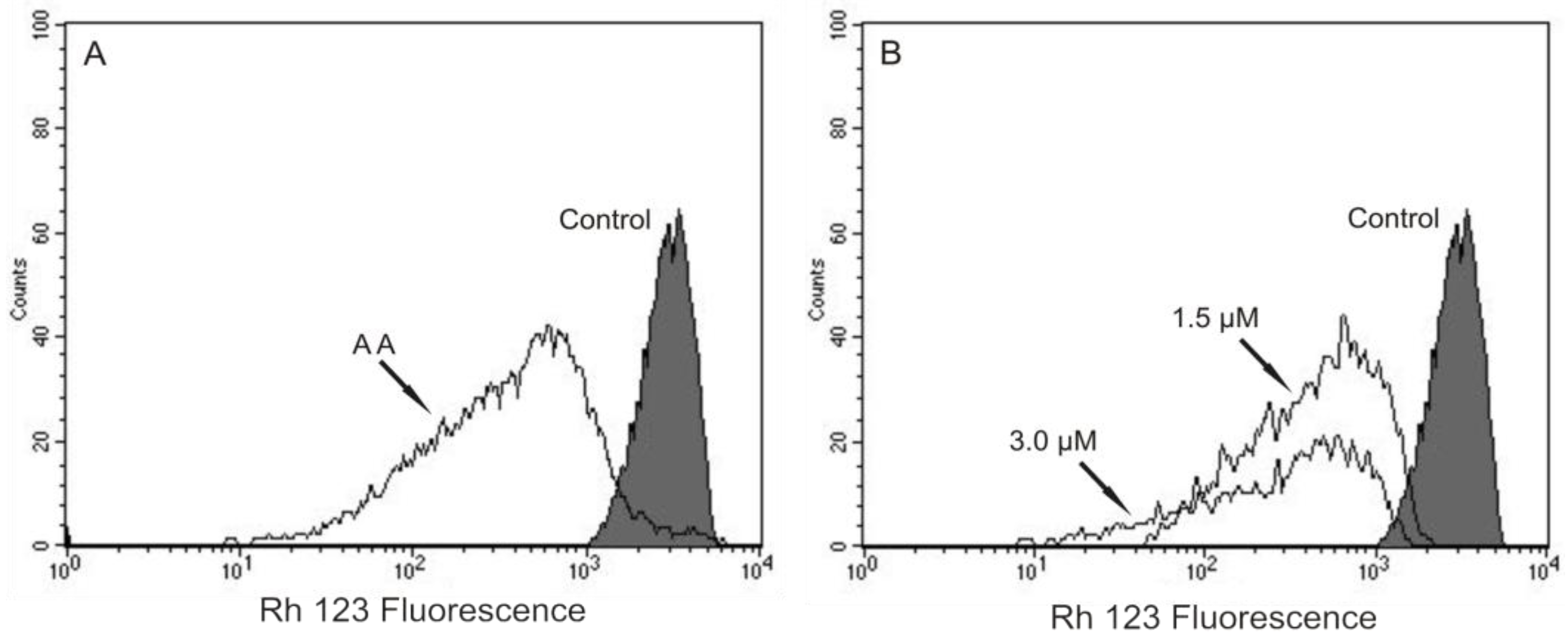
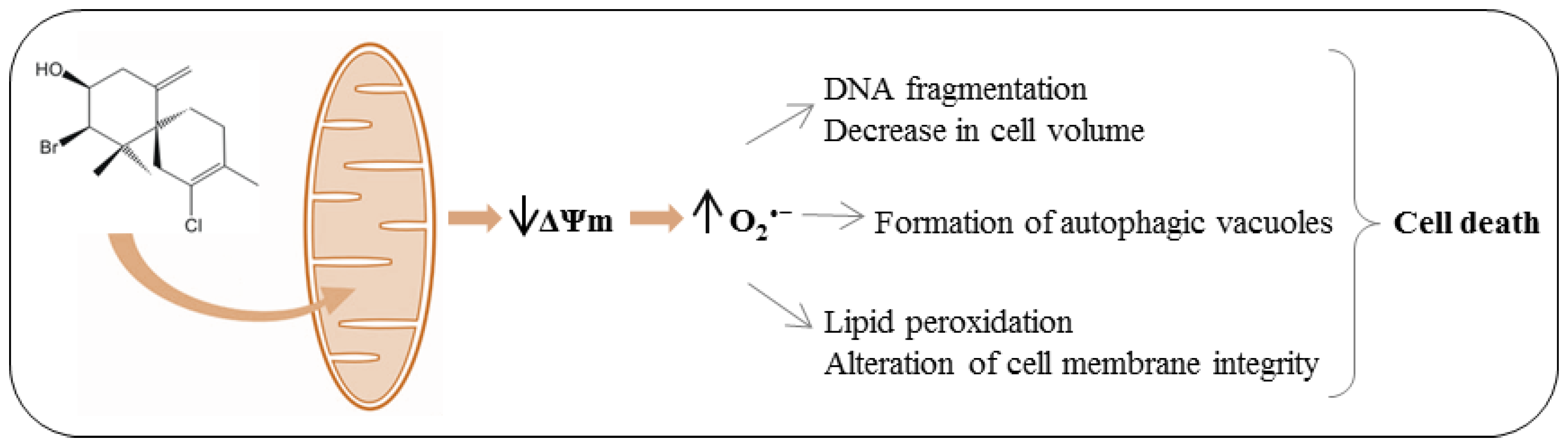
Based on our previous work that indicated, by electron microscopy, the effect of (−)-elatol on
T. cruzi mitochondria and cell membranes [
8], we decided to evaluate the mitochondrial membrane potential (ΔΨm) and the cell membrane integrity in (−)-elatol-treated trypomastigotes by flow cytometry. Histograms show a marked decrease in fluorescence intensity total rhodamine 123 (Rh 123), indicating mitochondrial depolarization in cells treated with 1.5 and 3.0 µM of (−)-elatol for 3 h, with ΔΨm reductions in a range of 80.0% (
Figure 2B) when compared to the control group. A decrease in fluorescence intensity was also observed after 2 h of treatment, however the ΔΨm reductions were almost 3-fold smaller than those observed after 3 h (data not shown). The positive control antimicyn A (AA) induced 81.3% change in mitochondrial membrane potential (
Figure 2A).
Figure 2.
Flow cytometry analysis of trypomastigotes of Trypanosoma cruzi treated with (−)-elatol for 3 h and stained with Rh 123. (A) Trypomastigotes treated with 2.0 µM of antimicyn A (AA) (positive control); (B) Trypomastigotes treated with 1.5 and 3.0 µM of (−)-elatol for 3 h. Control group (untreated parasite) is also shown. Typical histograms of at least three independent experiments.
Figure 2.
Flow cytometry analysis of trypomastigotes of Trypanosoma cruzi treated with (−)-elatol for 3 h and stained with Rh 123. (A) Trypomastigotes treated with 2.0 µM of antimicyn A (AA) (positive control); (B) Trypomastigotes treated with 1.5 and 3.0 µM of (−)-elatol for 3 h. Control group (untreated parasite) is also shown. Typical histograms of at least three independent experiments.
In this context, our data adds further evidences that mitochondria are a target for (−)-elatol action, strengthening the idea introduced in our previous work [
8]. In fact, increasingly well documented papers have described trypanocidal compounds targeting parasite mitochondrial function [
9,
14].
Our results also show that not only the mitochondria, a unique and essential organelle of trypomastigotes [
15], was affected by (−)-elatol, but also the plasma membrane, a selective structure that controls the movement of substances in and out of cells essential for the maintenance of the parasite homeostasis. This effect was evidenced by propidium iodide (PI)-stained cells.
Figure 3 shows an increase in the intensity of PI fluorescence in trypomastigotes treated with (−)-elatol at 1.5 and 3.0 µM for 2 h around 90.0% which is noticeably higher than the PI fluorescence of the control group, indicating alteration of cell membrane integrity. The positive control (B) with digitonin also shows an increase in fluorescence (of 55.0%).
Figure 3.
Flow cytometry analysis of trypomastigotes of Trypanosoma cruzi treated with (−)-elatol for 2 h and stained with propidium iodide (PI). (A) Control group (untreated cells);(B) Trypomastigotes treated with digitonin 40.0 µM (positive control); (C) Trypomastigotes treated with 1.5 µM (−)-elatol; (D) Trypomastigotes treated with 3.0 µM (−)-elatol. The numbers shows the percentage of PI-stained positive cells in upper right and left quadrant. Typical histograms of at least three independent experiments.
Figure 3.
Flow cytometry analysis of trypomastigotes of Trypanosoma cruzi treated with (−)-elatol for 2 h and stained with propidium iodide (PI). (A) Control group (untreated cells);(B) Trypomastigotes treated with digitonin 40.0 µM (positive control); (C) Trypomastigotes treated with 1.5 µM (−)-elatol; (D) Trypomastigotes treated with 3.0 µM (−)-elatol. The numbers shows the percentage of PI-stained positive cells in upper right and left quadrant. Typical histograms of at least three independent experiments.
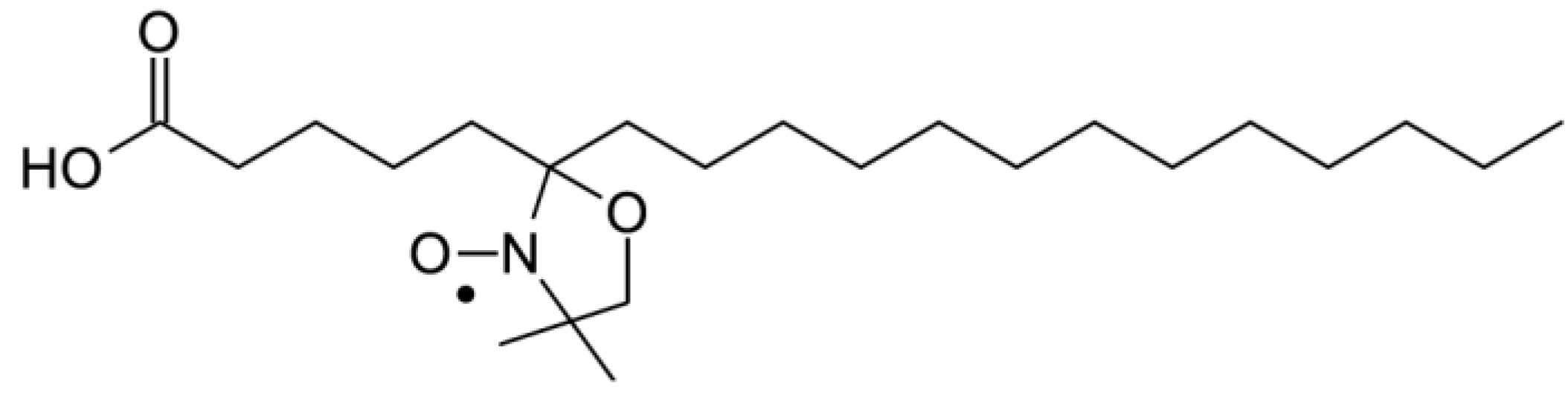
To confirm the effect of (−)-elatol on the cell membrane the experimental and best-fit electron paramagnetic resonance (EPR) spectra of spin label 5-doxyl stearic acid (5-DSA) (
Figure 4) structured in the plasmatic membrane of trypomastigotes was made and are shown in
Figure 5. These EPR spectra are typical for cellular membranes containing an appreciable amount of integral proteins. The treatment with (−)-elatol increased two EPR parameters, the outer hyperfine splitting, 2A
//, and the rotational correlation time, τ
C, indicating significant reduction in membrane lipid dynamics. 2A
// is a practice parameter measured directly in the EPR spectra (
Figure 5). This has been widely used to monitor membrane fluidity even though, in principle, it is a static parameter associated with the orientation distribution of the spin labels in the membrane.
Figure 4.
Chemical structure of spin label 5-doxyl stearic acid (5-DSA) used in this work.
Figure 4.
Chemical structure of spin label 5-doxyl stearic acid (5-DSA) used in this work.
The presence of the sesquiterpene (−)-elatol significantly increased the rigidity of the membrane of T. cruzi as evidenced by EPR spectra. The spin probe used in EPR is sparsely distributed in the membrane and, therefore, the spin probe spectroscopy only detects changes in membrane fluidity when a widespread change occurs.
Figure 5.
Experimental (black line) and best-fit (red line) electron paramagnetic resonance (EPR) spectra of spin label 5-DSA of trypomastigotes of Trypanosoma cruzi treated with (−)-elatol. The EPR spectra (a) and (d) were obtained from trypomastigotes without treatment (control samples); spectra (b) and (e) are from samples treated with 5.4 × 109 (−)-elatol molecules/cell and the spectrum (c) is from a sample treated with 1.6 × 1010 (−)-elatol molecules/cell. EPR spectra were simulated with the fitting program NLLS and the values of the parameter rotational correlation time, τC, obtained from the fit for each spectrum are indicated in nanosecond scale. The EPR parameter 2A// is the separation in magnetic-field units between the first and last resonance lines (indicated by vertical lines) of the spectrum. The estimated experimental error for 2A// and τC parameters are 0.5 G and 1.0 ns, respectively. Typical spectra of two independent experiments.
Figure 5.
Experimental (black line) and best-fit (red line) electron paramagnetic resonance (EPR) spectra of spin label 5-DSA of trypomastigotes of Trypanosoma cruzi treated with (−)-elatol. The EPR spectra (a) and (d) were obtained from trypomastigotes without treatment (control samples); spectra (b) and (e) are from samples treated with 5.4 × 109 (−)-elatol molecules/cell and the spectrum (c) is from a sample treated with 1.6 × 1010 (−)-elatol molecules/cell. EPR spectra were simulated with the fitting program NLLS and the values of the parameter rotational correlation time, τC, obtained from the fit for each spectrum are indicated in nanosecond scale. The EPR parameter 2A// is the separation in magnetic-field units between the first and last resonance lines (indicated by vertical lines) of the spectrum. The estimated experimental error for 2A// and τC parameters are 0.5 G and 1.0 ns, respectively. Typical spectra of two independent experiments.
All these cell membrane alterations induced by (−)-elatol could be a result of oxidative damage induced by reactive oxygen species (ROS) production. The increase of ROS can lead to destructive effects through the reaction with biological macromolecules such as lipids, proteins and DNA [
16]. It is well known that mitochondria play an important role in the production of ROS through oxidative phosphorylation involving the electron transport chain. In certain situations, for example changes in mitochondrial membrane potential, an increase in the production of ROS through the electron transport chain is observed [
9]. This increase contributes to mitochondrial damage followed by an increase of permeability of their membranes resulting in the release of apoptosis activating factors such as ROS toward the cytosol [
17,
18].
Thus, based on our results of cell membrane and mitochondrial membrane potential alterations we decided to evaluate the superoxide anion production (O2•−), by a very sensitive fluorimetric assay, in mitochondria of (−)-elatol-treated trypomastigotes.
As shown in
Figure 6, (−)-elatol induced an increase in the O
2•− production in all concentrations assayed starting from 1 h of incubation. However, 15.0 and 30.0 µM after 2 and 3 h were the most effective concentrations of (−)-elatol displaying a significant increase (about 20.0%) of mitochondrial O
2•− production when compared to the control group. The positive control with AA also induced an increase of mitochondrial O
2•− production (data not shown).
Figure 6.
Mitochondrial O2•− production in trypomastigote forms of Trypanosoma cruzi treated with (−)-elatol for up to 3 h. Mitochondrial O2•− production was evaluated using the fluorescent probe MitoSOX. At the indicated times, parasites were used to fluorimetrically measure oxidized MitoSOX (oxMitoSOX). Results are expressed in arbitrary units as means ± SD of at least three independent experiments. Asterisks indicate significant differences relative to the control group (untreated cells) as identified by variance analysis (two-way) with Tukey post-test (p ≤ 0.05).
Figure 6.
Mitochondrial O2•− production in trypomastigote forms of Trypanosoma cruzi treated with (−)-elatol for up to 3 h. Mitochondrial O2•− production was evaluated using the fluorescent probe MitoSOX. At the indicated times, parasites were used to fluorimetrically measure oxidized MitoSOX (oxMitoSOX). Results are expressed in arbitrary units as means ± SD of at least three independent experiments. Asterisks indicate significant differences relative to the control group (untreated cells) as identified by variance analysis (two-way) with Tukey post-test (p ≤ 0.05).
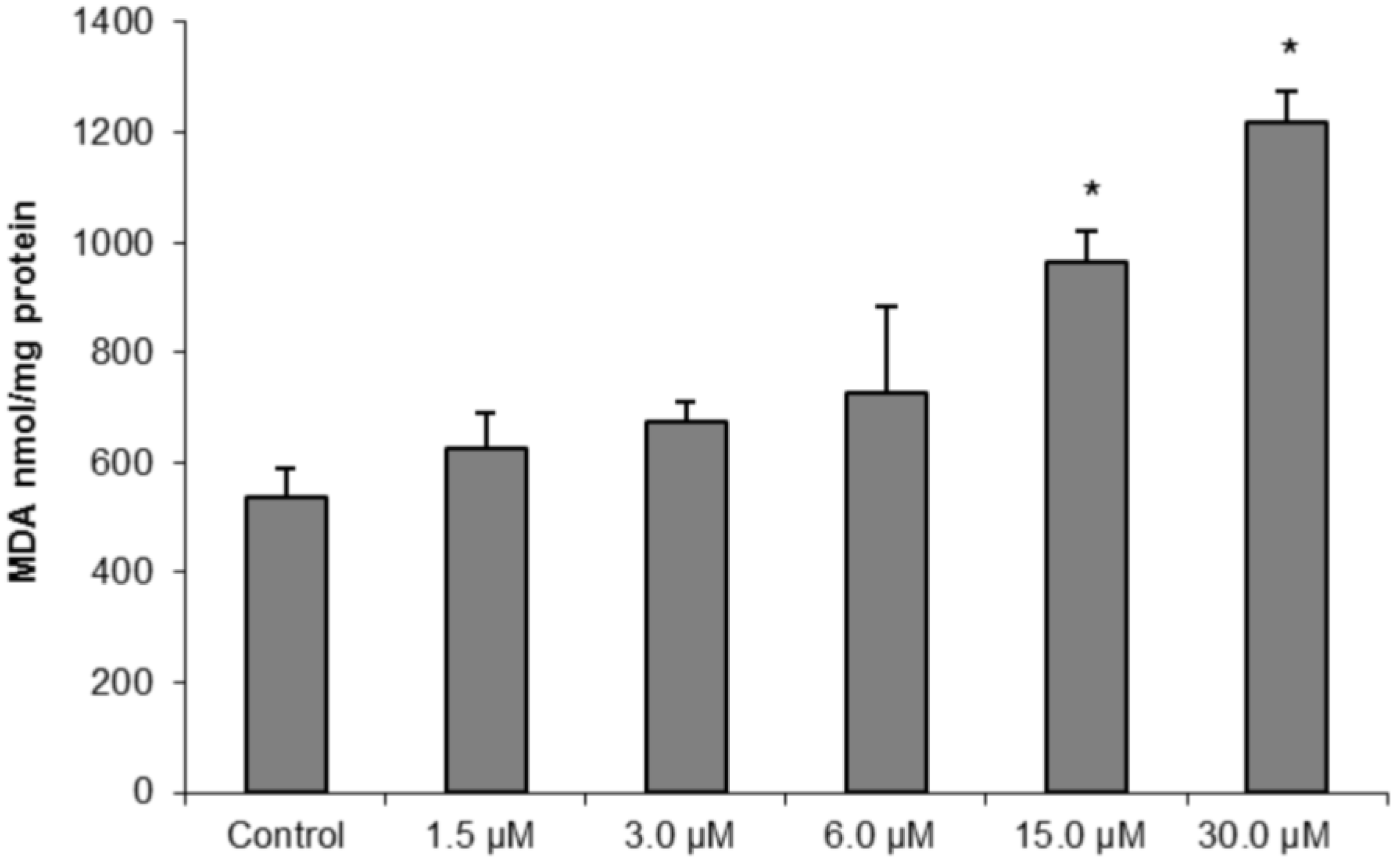
To further confirm our hypothesis of oxidative damage induced by (−)-elatol-treated trypomastigotes, we measured the production of thiobarbituric acid-reactive substances (TBARS) (which is frequently used to quantify lipoperoxidation of the cell membrane and is expressed by the production of malondialdehyde (MDA)). The measurement of TBARS in trypomastigotes treated with 15.0 and 30.0 µM of (−)-elatol revealed a significant increase in lipid peroxidation after 3 h when compared to the control group (
Figure 7). The lipid peroxidation data also gave more evidence of an increase in membrane rigidity of (−)-elatol-treated parasites. Lipid peroxidation alters essential structural components of cell membranes affecting cell membrane permeability and fluidity [
19].
The increase of O
2•− production induced by (−)-elatol might lead to a DNA break as well. As shown in
Figure 8, bright fluorescence was observed in trypomastigotes treated with 1.5 and 3.0 µM of (−)-elatol for 24 h and staining with TUNEL (
Figure 8D,F). Additionally, the counterstaining with PI (
Figure 8J,L) denotes that (−)-elatol induced the condensation and margination of chromatin. The control without treatment was TUNEL and PI negative (
Figure 8B,H). In addition, bright fluorescence was also observed with actinomycin D, a known apoptosis inducer (data not shown).
Figure 7.
Determination of lipid peroxidation in trypomastigote forms of Trypanosoma cruzi treated with different concentrations of (−)-elatol for 3 h. The malondialdehyde (MDA) concentration was measured by thiobarbituric acid-reactive substances (TBARS) production. The results are expressed as means ± SD of at least three independent experiments. Asterisks indicate significant differences relative to the control group (untreated cells) as identified by variance analysis (one-way) with Tukey post-test (p ≤ 0.05).
Figure 7.
Determination of lipid peroxidation in trypomastigote forms of Trypanosoma cruzi treated with different concentrations of (−)-elatol for 3 h. The malondialdehyde (MDA) concentration was measured by thiobarbituric acid-reactive substances (TBARS) production. The results are expressed as means ± SD of at least three independent experiments. Asterisks indicate significant differences relative to the control group (untreated cells) as identified by variance analysis (one-way) with Tukey post-test (p ≤ 0.05).
Figure 8.
DNA fragmentation in trypomastigote forms of Trypanosoma cruzi treated with (−)-elatol for 24 h. TUNEL (panels A–F) and PI (panels G–L) were analyzed by fluorescence microscope. Gray column is differential interference contrast (DIC) and black column is fluorescence; (A,B,G,H) Representative images of untreated cells; (C,D,I,J) Representative images of trypomastigotes treated with 1.5 µM (−)-elatol; (E,F,K,L) Representative images of trypomastigotes treated with 3.0 µM (−)-elatol. Arrows indicate DNA fragmentation (green) and condensation and margination of chromatin (red). Bars: 10 µm.
Figure 8.
DNA fragmentation in trypomastigote forms of Trypanosoma cruzi treated with (−)-elatol for 24 h. TUNEL (panels A–F) and PI (panels G–L) were analyzed by fluorescence microscope. Gray column is differential interference contrast (DIC) and black column is fluorescence; (A,B,G,H) Representative images of untreated cells; (C,D,I,J) Representative images of trypomastigotes treated with 1.5 µM (−)-elatol; (E,F,K,L) Representative images of trypomastigotes treated with 3.0 µM (−)-elatol. Arrows indicate DNA fragmentation (green) and condensation and margination of chromatin (red). Bars: 10 µm.
In this context, and based on well-established literature, we can state that (−)-elatol induces an oxidative stress condition leading to cumulative oxidative damage in the parasite macromolecules. Besides lipid peroxidation, we showed that (−)-elatol-treated trypomastigotes can also trigger destructive effects on DNA evidenced by TUNEL and PI staining.
The DNA fragmentation is one of the final steps in the apoptotic process and could be evidence of apoptosis in trypomastigotes treated with (−)-elatol. Therefore, we performed additional experiments to evaluate the cell shrinkage, a hallmark of apoptotic death as well. As shown in
Figure 9, there was a decrease in cell volume in the presence of concentrations of 1.5 and 3.0 µM of (−)-elatol after 24 h, where reductions of 20.0% and 23.8% were observed, respectively. The positive control actinomycin D induced a decrease of 79.7% in the cell volume.
Figure 9.
Flow cytometry analysis of trypomastigote forms of Trypanosoma cruzi treated with (−)-elatol for 24 h. Forward light scatter (FSC-H) was considered a function of cell size. Representative FACS histogram showing FSC-H of trypomastigotes treated with 1.5 µM and 3.0 µM (−)-elatol and the control group (untreated cells, gray full histogram). Typical histogram of at least three independent experiments.
Figure 9.
Flow cytometry analysis of trypomastigote forms of Trypanosoma cruzi treated with (−)-elatol for 24 h. Forward light scatter (FSC-H) was considered a function of cell size. Representative FACS histogram showing FSC-H of trypomastigotes treated with 1.5 µM and 3.0 µM (−)-elatol and the control group (untreated cells, gray full histogram). Typical histogram of at least three independent experiments.
Based on our previous work that showed by TEM the extensive cytoplasmic vacuolization on
T. cruzi treated with (−)-elatol [
8] we decided to evaluate if autophagy could also be a death pathway induced by (−)-elatol. For this, we evaluated autophagy by staining trypomastigotes treated with (−)-elatol with monodansylcadaverine (MDC), a fluorescent probe that accumulates in autophagic vacuoles [
20]. As shown in
Figure 10 the presence of fluorescence in rounded structures in cells treated with (−)-elatol revealed the formation of autophagic vacuoles (
Figure 10D,H), unlike the untreated cells (
Figure 10B). This effect could be partially prevented in trypomastigotes pre-treated with wortmannin (
Figure 10F).
Figure 10.
Determination of autophagy in trypomastigote forms of Trypanosoma cruzi treated with (−)-elatol for 24 h and stained with MDC. Gray column is DIC and black column is fluorescence. (A,B) Representative image of untreated cells; (C,D) Representativeimage of trypomastigotes treated with 1.5 µM (−)-elatol; (E,F) Representative image of trypomastigotes treated with 1.5 µM (−)-elatol + wortmannin; (G,H) Representative image of trypomastigotes treated with 3.0 µM (−)-elatol. Arrows indicate the stained autophagic structures. Bars: 10 µm.
Figure 10.
Determination of autophagy in trypomastigote forms of Trypanosoma cruzi treated with (−)-elatol for 24 h and stained with MDC. Gray column is DIC and black column is fluorescence. (A,B) Representative image of untreated cells; (C,D) Representativeimage of trypomastigotes treated with 1.5 µM (−)-elatol; (E,F) Representative image of trypomastigotes treated with 1.5 µM (−)-elatol + wortmannin; (G,H) Representative image of trypomastigotes treated with 3.0 µM (−)-elatol. Arrows indicate the stained autophagic structures. Bars: 10 µm.
Up to here our results indicate that (−)-elatol induced alterations that might be responsible for different types of cell death. For example, the alterations in mitochondria and the breakdown of the plasma membrane observed here and the distortion in the cell body described before [
8] are all hallmarks of necrosis. The DNA fragmentation, one of the final steps in the apoptotic process, could be evidence of apoptosis in (−)-elatol-treated trypomastigotes. Additionally, the decrease in cell volume observed here in treated parasites is one more indicator of apoptosis [
21]. Another type of cell death described for
T. cruzi is autophagy, which is characterized by an increase in cytoplasmic vacuolization [
22]. Our previous TEM data [
8] point to the formation of cytoplasmic vacuoles in
T. cruzi treated with (−)-elatol which suggest autophagic death. In this work we confirm autophagic death. This result is based on a fluorescent assay where the autophagic vacuoles were partially reduced by wortmannin, a PI3-K inhibitor, which is an enzyme of the signaling pathway involved in autophagy regulation [
21,
23].
3. Experimental Section
3.1. Chemicals and Materials
Actinomycin D, antimicyn A, bovine serum albumin, digitonin, dimethylsulfoxide, monodansylcadaverine, rhodamine 123, thiobarbituric acid, wortmannin and spin label 5-doxyl stearic acid, having the nitroxide radical moiety (doxyl) in the 5th carbon atom of the acyl chain, were purchased from Sigma-Aldrich (St. Louis, MO, USA); Dulbecco’s modified Eagle’s medium, fetal bovine serum was from Invitrogen (Grand Island, NY, USA); MitoSOX kit, propidium iodide, and TUNEL kit was from Invitrogen (Eugene, OR, USA) and protein assay kit was from Bio-Rad (Hercules, CA, USA). All other reagents were of analytical grade.
3.2. Isolation of (−)-Elatol from Laurencia dendroidea
(−)-Elatol was isolated from specimens of L. dendroidea collected by hand during low tide, in the midlittoral zone on the rocky coast of Cabo Frio Island (22°59′ S, 42°59′ W), Rio de Janeiro State, Brazil. The seaweed was stored in plastic bags and chilled on ice during transport to the laboratory. The specimens of L. dendroidea used in this study were identified by Dr. Mutue Toyota Fujii, and voucher specimens were deposited in the herbaria SP, Instituto de Botânica, São Paulo State, Brazil (SP number: 399789). L. dendroidea was dried in the dark at room temperature in order to avoid photolysis and thermal degradation.
The air-dried algal material (300.0 g) giving 50 mg of (−)-elatol was successive and exhaustively extracted in
n-hexane at room temperature for 15 days. The solvent was eliminated in a rotary evaporator, at low temperature (<50 °C), yielding 3.64 g of a dark green extract containing the sesquiterpene (−)-elatol, which was detected as a blue spot on TLC plates after spraying with a solution of ceric sulfate and sulfuric acid (2.1 g of Ce
2(SO
4)
3·4H
2O; 21 mL of H
2SO
4 and 300 mL of H
2O), followed by heating at 100 °C for 3 min. An aliquot of HE (0.35 g) was submitted to preparative thin layer chromatography (PTLC) (Merck, silica gel 60 F
254, 20 × 20 cm, mobile phase:
n-hexane/ethyl acetate 8:2), to afford a yellowish oil (50 mg) which was identified as the sesquitepene (−)-elatol. The purity was confirmed by TLC (
Rf = 0.45), using
n-hexane/AcOEt 8:2 as mobile phase, and by
1H-NMR spectroscopy (300 MHz), and comparison with the literature [
24,
25].
(−)-Elatol stock solutions (1 mg/mL) were prepared in dimethyl sulfoxide, stored at 4 °C. All groups (including controls) were tested at final concentrations of less than 1% dimethylsulfoxide (DMSO), a concentration found not to affect trypomastigotes (data not shown). The tested concentrations were based on effective concentration (EC
50) about 1.5 µM [
8].
3.3. Parasites and Cells Cultures
T. cruzi trypomastigote forms (Y strain) (95% of purity) were obtained from the supernatant of an infected LLCMK2 cells monolayer (epithelial cell of monkey kidney; Macaca mulatta) in DMEM medium supplemented with 2 mM L-glutamine, 10% heat-inactivated fetal bovine serum (FBS), 50 mg/L gentamicin, and buffered with sodium bicarbonate in a 5% CO2 air mixture at 37 °C. Sub-confluent cultures of LLCMK2 cells were infected with 5 × 106 trypomastigotes. Extracellular parasites were removed after 24 h, the cells washed, and these cultures were maintained in DMEM medium containing 10% FBS, until trypomastigotes emerged from the infected cells.
3.4. Mitochondrial Membrane Potential and Cell Membrane Integrity Assays
Trypomastigotes (1 × 107 cells/mL) treated or untreated with 1.5 and 3.0 µM of (−)-elatol, for 2 and 3 h at 37 °C, were washed and incubated with 5.0 µg/mL of Rh 123 for 15 min to evaluate the ΔΨm and 0.2 µg/mL of PI for 10 min to verify possible alteration in cell membrane integrity. The compound AA at a concentration of 2.0 µM was used as a positive control for measurement of mitochondrial membrane potential and digitonin at 40.0 µM for cell membrane integrity. Data acquisition and analysis were performed using a FACSCalibur flow cytometer (Becton-Dickinson, Rutherford, NJ, USA) equipped with the CellQuest software (Joseph Trotter, Scripps Research Institute, La Jolla, CA, USA). A total of 10,000 events were acquired in the region previously established as that corresponding to the parasites. Alterations in the fluorescence of Rh 123 were quantified as the percent of reduction of the fluorescence compared with the control (untreated parasites).
3.5. Spin Labeling
A small aliquot (3 µL) of stock solution of spin label 5-DSA in ethanol (2 mg/mL) was transferred to an eppendorf tube. After that, the solvent was evaporated and about 1 × 108 trypomastigotes/mL, suspended in 30 µL of phosphate-buffered saline (PBS), was added on the film of spin label and gentle agitation applied. After spin labeling, 1 or 3 µL of a stock solution of (−)-elatol in ethanol (300 mg/mL) was applied to the cell suspension and gently mixed. The cells were then introduced into 1-mm i.d. capillary for EPR measurements, which were sealed by flame.
3.6. EPR Spectroscopy
EPR spectroscopy was performed with a Bruker ESP 300 spectrometer (Rheinstetten, Germany) equipped with an ER 4102 ST resonator. The instrument settings were: microwave power of 10 mW; modulation frequency of 100 KHz; modulation amplitude of 1.0 G; magnetic field scan of 100 G; sweep time of 168 s; and detector time constant of 41 ms. EPR spectra simulations were performed using the NLLS program (nonlinear least-squares fitting program) developed by Freed and coworkers [
26]. In the spectral calculations, the NLLS program includes the magnetic
g- and
A-tensors and the rotational diffusion tensor, R, which are expressed in a system of Cartesian axes fixed in the spin-labeled molecule. To reduce the number of parameters in the fittings and to simplify the simulation, the average rotational diffusion rate,
Rbar, was calculated by the fitting program using the relation
Rbar = (
Rper2Rpar)
1/3, where
Rper is the perpendicular and
Rpar is the parallel component of the rotational diffusion [
26].
Rbar was converted to the parameter rotational correlation time, τ
c, following the relationship τ
c = 1/6
Rbar. In this work, the spectra were simulated with a model of a single spectral component. Similar to previous studies [
27,
28], the magnetic parameters were determined based on a global analysis of the overall spectra obtained in this work, and all of the EPR spectra were simulated using the same predetermined parameters. Input parameters of tensors
g and
A were:
gxx = 2.0082;
gyy = 2.0060;
gzz = 2.0022;
Axx = 7.5;
Ayy = 7.0 G and
Azz = 31.5 G.
3.7. Fluorimetric Detection of Mitochondrial-Derived O2•−
Mitochondrial production of superoxide anion was evaluated during the exposure of trypomastigotes to 1.5, 3.0, 6.0, 15.0 and 30.0 µM of (−)-elatol using the fluorescent O
2•− sensitive, mitochondrial-targeted probe MitoSOX [3,8-phenanthridinediamine, 5-(6-triphenylphosphoniumhexyl)-5,6-dihydro-6-phenyl]. Trypomastigotes (2 × 10
7 cells/mL) were loaded with 5.0 µM MitoSOX for 10 min at room temperature (22 °C) and then washed with the KH (Krebs-Henseleit) buffer (pH 7.3) containing 15 mM NaHCO
3, 5 mM KCl, 120 mM NaCl, 0.7 mM Na
2HPO
4 and 1.5 mM NaH
2PO
4 before the assays. Loaded cells were exposed to the stimuli, and after different times the fluorescence was measured in a fluorescence microplate reader (Victor X3, PerkinElmer) at λ
ex = 510 nm and λ
em = 580 nm. The oxMitoSOX becomes highly fluorescent upon binding to nucleic acids. In some of the experiments, cells were exposed to 10.0 µM AA, a stimulus known to induce O
2•− production by mitochondria [
29].
3.8. Lipid Peroxidation Assay
Trypomastigote forms were incubated in the DMEM medium with 1.5, 3.0, 6.0, 15.0 and 30.0 µM of (−)-elatol for 3 h, at 37 °C. The extent of lipid peroxidation was determined as the amount of TBARS in terms of MDA. After incubation, samples (0.5 mg protein) were heated in a solution containing 0.37% thiobarbituric acid, 15% trichloroacetic acid, and 0.25 N HCl at 95 °C for 45 min. After cooling, the absorbance was read at 532 nm and the concentration of TBARS was calculated based on a ε value of 153,000 M
−1cm
−1 [
30].
3.9. DNA Fragmentation
We analyzed DNA double-strand ruptures in situ by TUNEL (Terminal Deoxynucleotide Transferase dUTP Nick End Labeling). For this, trypomastigotes (1 × 107 cells/mL) were treated with 1.5 and 3.0 µM of (−)-elatol for 24 h, after the cells were subjected to the TUNEL assay according to the manufacturer’s instructions. The compound actinomycin D 10.0 µg/mL was used as a positive control. The nuclei were counterstained with propidium iodide. Cells that have undergone DNA double-strand ruptures should fluorescence brightly, unlike the untreated cells. Fluorescence was observed in a fluorescence microscope Olympus BX51 (Olympus®) and pictures were captured with a UC30 camera (Olympus®).
3.10. Cell Volume Determination
Trypomastigotes (1 × 107 cells/mL) treated with 1.5 and 3.0 µM of (−)-elatol for 3 and 24 h, were collected by centrifugation, washed twice in PBS, resuspended in PBS and analyzed by fluorescence-activated cell sorting using a FACSCalibur flow cytometer. The compound actinomycin D 20.0 mM was used as a positive control. A total of 10,000 events were acquired in the region previously established as that corresponding to the parasites. Histograms and analysis were performed in CellQuest software; FSC-H represents the cell volume.
3.11. Evaluation of Autophagic Vacuoles
The autophagic vacuoles were evaluated using MDC labeling [
31]. For this, trypomastigotes (1 × 10
7 cells/mL) were treated with 1.5 and 3.0 µM of (−)-elatol for 24 h at 37 °C. Thus, the cells were incubated with 0.05 mM of MDC in PBS for 15 min at 37 °C. After incubation the cells were washed in PBS two times. MDC stain was analyzed by fluorescence microscope Olympus BX51 (Olympus
®) and images were captured using a UC30 camera (Olympus
®). In some experiments, cells were pre-treated with wortmannin, a potent PI3-kinase inhibitor, before induction of autophagy.
3.12. Statistical Analysis
The data shown in the graphs are expressed as means ± standard deviation of at least three independent experiments. Data were analyzed with one-way and two-way analysis of variance (ANOVA), significant differences among means were identified by Tukey post-test. p ≤ 0.05 was adopted as the minimum criterion of significance. Statistical analyses were performed using the Statistica™ software package.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










