A Marine Anthraquinone SZ-685C Overrides Adriamycin-Resistance in Breast Cancer Cells through Suppressing Akt Signaling

Abstract
:1. Introduction
2. Results
2.1. Growth Inhibition of ADR-Resistant Human Breast Cancer Cell Lines Induced by SZ-685C

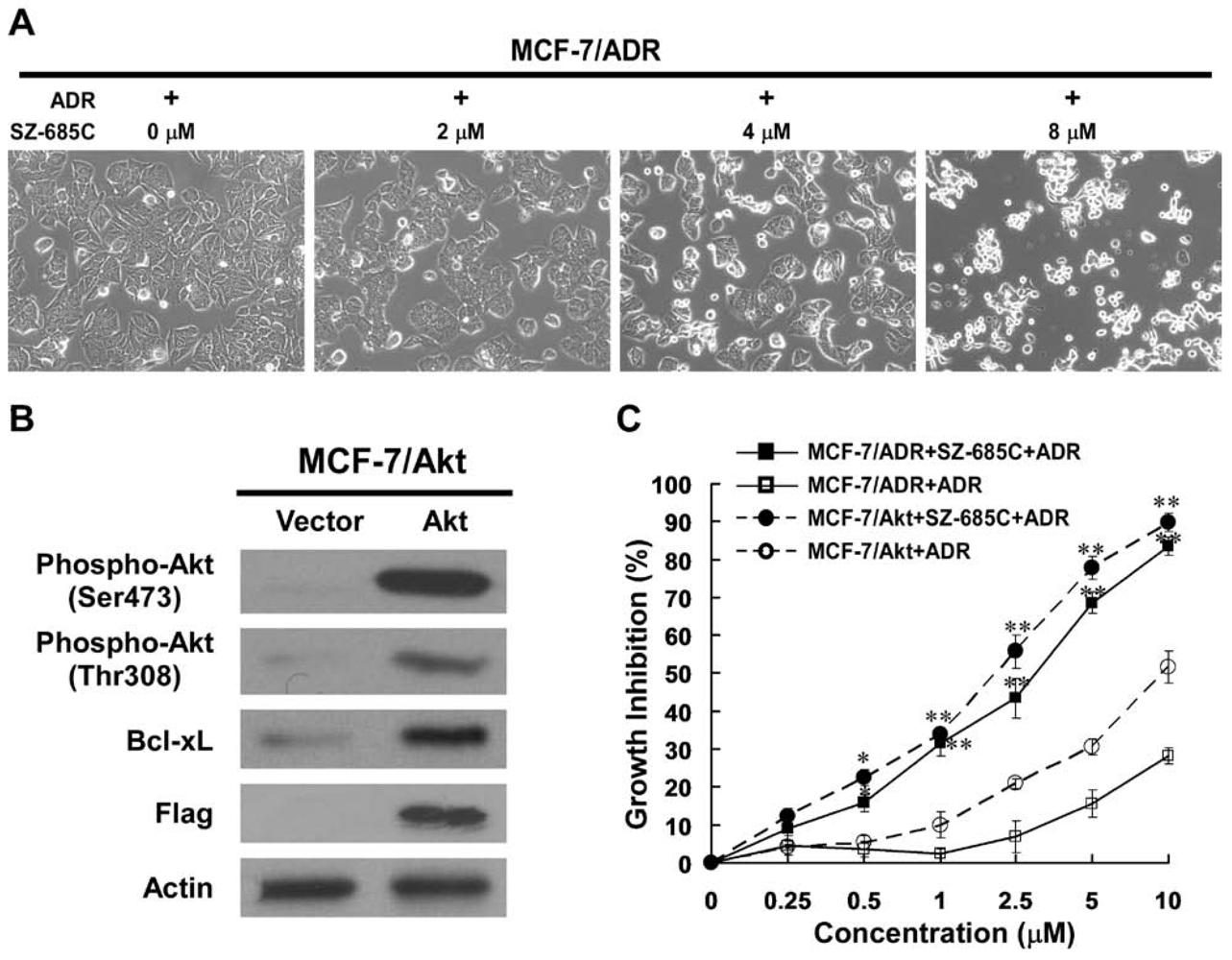{kind=link}
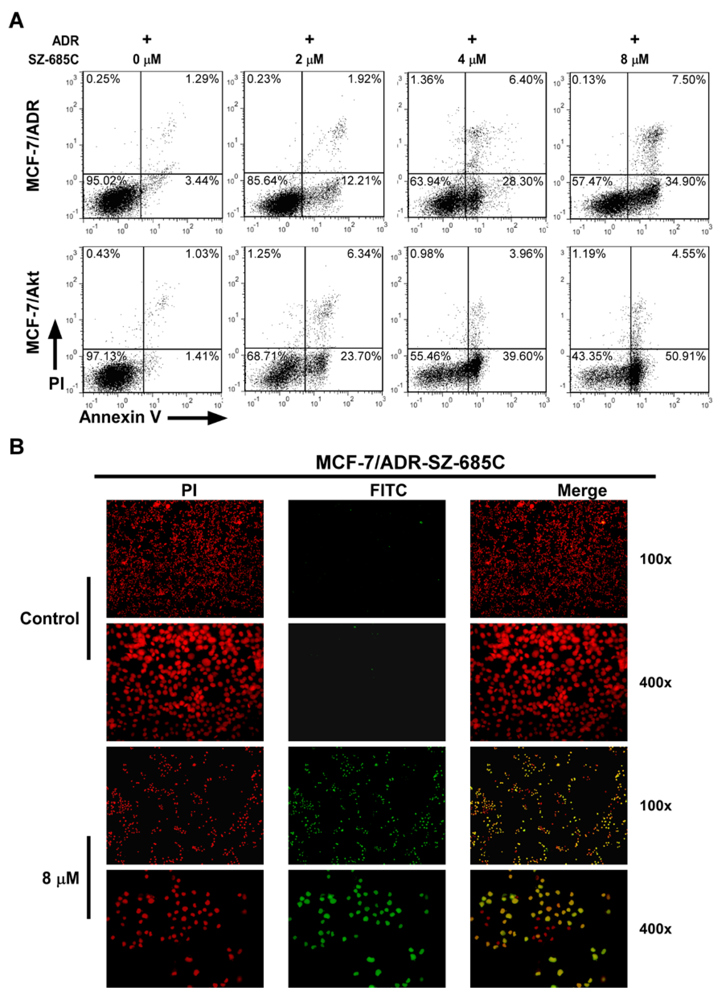{kind=link}
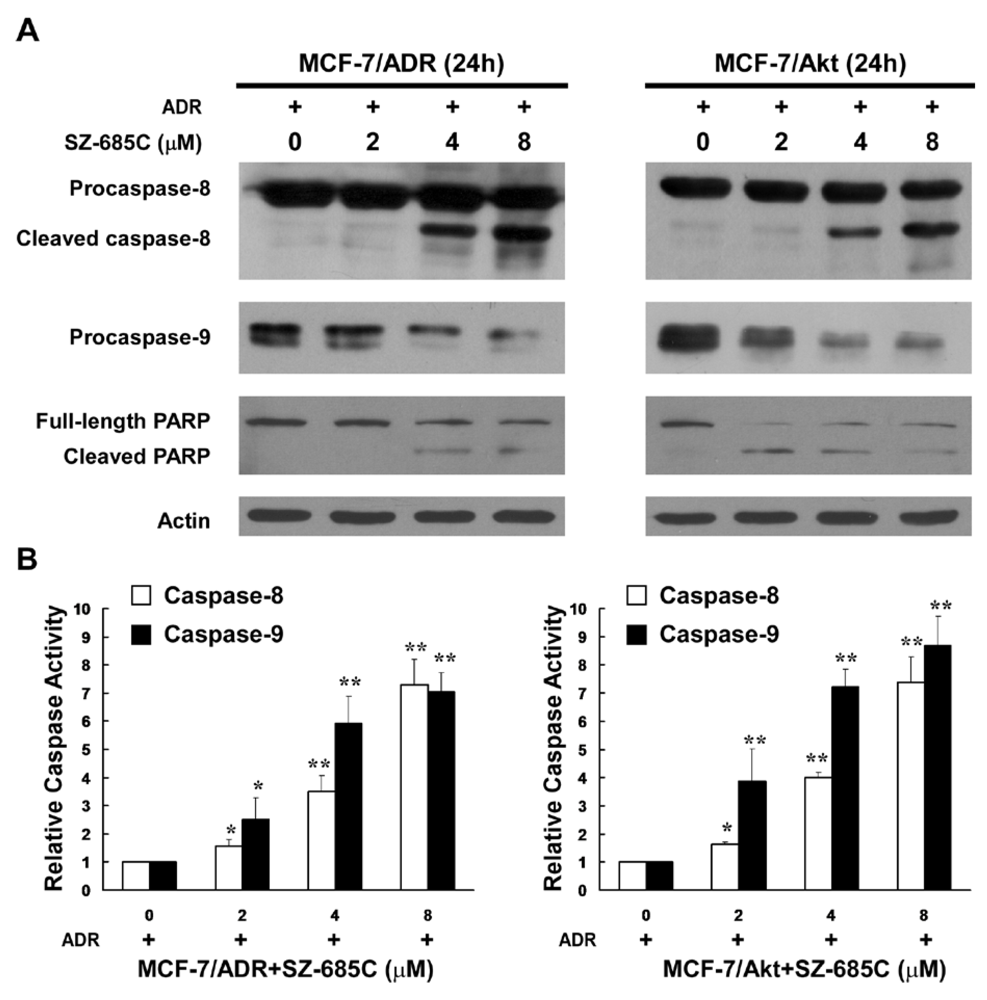{kind=link}
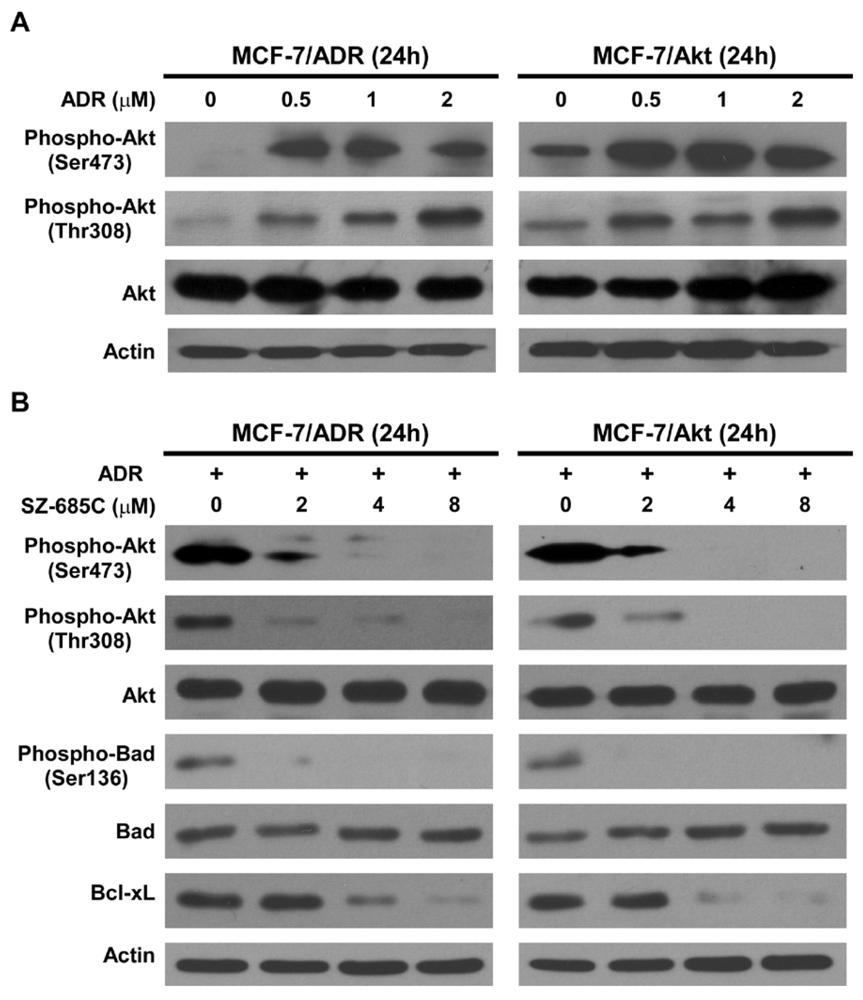{kind=link}
{kind=link}
| Cell Lines | ADR | SZ-685C | ||
|---|---|---|---|---|
| IC50 | RF | IC50 | RF | |
| MCF-7 | 0.96 | - | 7.38 | - |
| MCF-7/ADR | 18.42 | 19.19 | 4.17 | 0.57 |
| MCF-7/Akt | 7.69 | 8.01 | 3.36 | 0.46 |
| K562 | 0.15 | - | 1.09 | - |
| K562/ADR | 8.75 | 58.33 | 1.35 | 1.24 |
| HL-60 | 0.33 | - | 1.94 | - |
| HL-60/ADR | 18.13 | 54.94 | 1.76 | 0.91 |

2.2. SZ-685C Induced Apoptosis in MCF-7/ADR and MCF-7/Akt Cells

2.3. Activation of Both the Extrinsic and Intrinsic Apoptotic Pathways by SZ-685C

2.4. Inhibition of Akt Phosphorylation and Its Downstream Substrates by SZ-685C

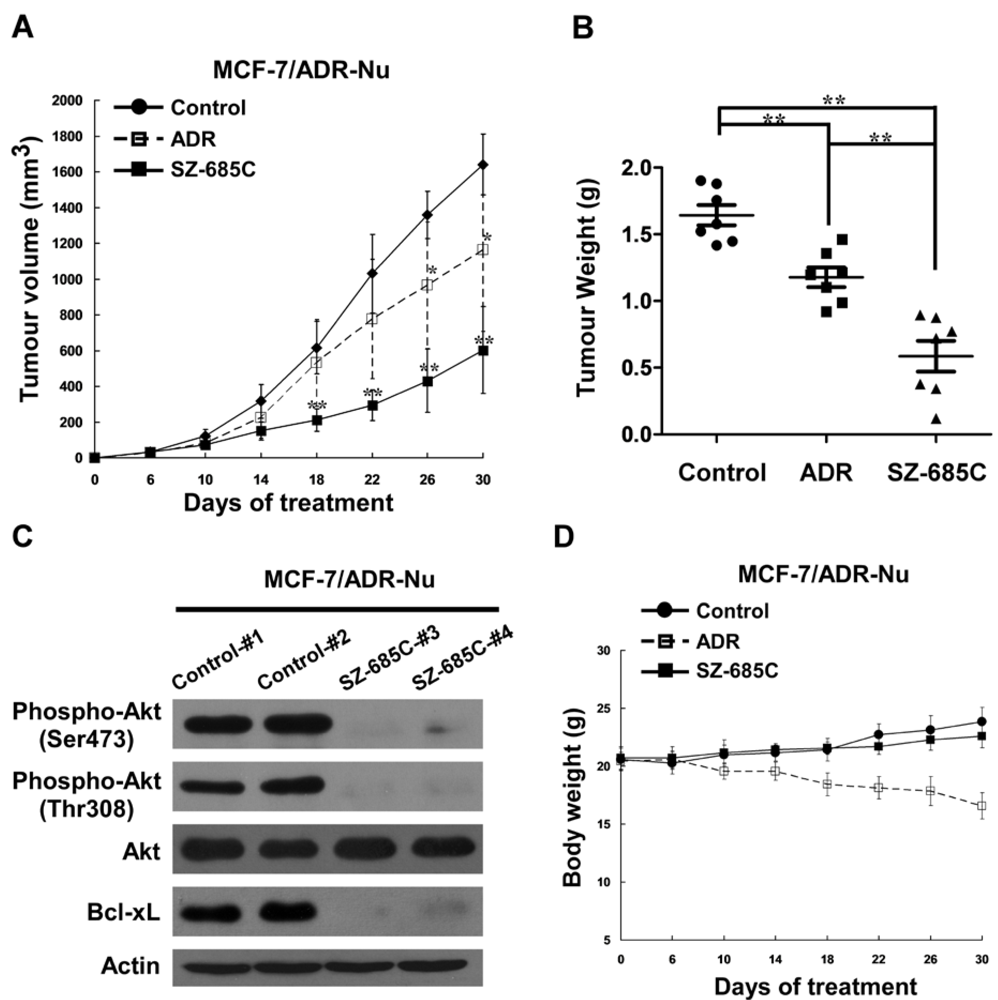
2.5. Anti-Tumor Effect of SZ-685C on ADR-Resistant Breast Cancer Xenografts in Vivo

3. Discussion
4. Experimental Section
4.1. Chemicals
4.2. Cell Culture
4.3. Establishment of Stable Cell Lines Expressing Constitutively Activated Akt
4.4. Cell Viability Analysis
4.5. Annexin V-FITC/Propidium Iodide (PI) Staining Assay
4.6. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling (TUNEL) Assay
4.7. Caspase Activity Assay
4.8. Western Blotting Analysis
4.9. Xenografted Tumor Model and Anti-Tumor Effect of SZ-685C in Vivo
4.10. Statistical Analysis
5. Conclusions
Acknowledgments
References
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar]
- Pritchard, K.I.; Shepherd, L.E.; O’Malley, F.P.; Andrulis, I.L.; Tu, D.; Bramwell, V.H.; Levine, M.N. HER2 and responsiveness of breast cancer to adjuvant chemotherapy. N. Engl. J. Med. 2006, 354, 2103–2111. [Google Scholar]
- Clark, A.S.; West, K.; Streicher, S.; Dennis, P.A. Constitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cells. Mol. Cancer Ther. 2002, 1, 707–717. [Google Scholar]
- Knuefermann, C.; Lu, Y.; Liu, B.; Jin, W.; Liang, K.; Wu, L.; Schmidt, M.; Mills, G.B.; Mendelsohn, J.; Fan, Z. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene 2003, 22, 3205–3212. [Google Scholar]
- Zhang, W.; Ding, W.; Chen, Y.; Feng, M.; Ouyang, Y.; Yu, Y.; He, Z. Up-regulation of breast cancer resistance protein plays a role in HER2-mediated chemoresistance through PI3K/Akt and nuclear factor-kappa B signaling pathways in MCF7 breast cancer cells. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 647–653. [Google Scholar] [CrossRef]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304. [Google Scholar]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar]
- Knight, Z.A.; Shokat, K.M. Chemically targeting the PI3K family. Biochem. Soc. Trans. 2007, 35, 245–249. [Google Scholar]
- Conte, P.F.; Gennari, A. Anthracyclines-paclitaxel combinations in the treatment of breast cancer. Ann. Oncol. 1997, 8, 939–943. [Google Scholar]
- Hortobagyi, G.N. Anthracyclines in the treatment of cancer. An overview. Drugs 1997, 54, 1–7. [Google Scholar] [CrossRef]
- Xie, G.; Zhu, X.; Li, Q.; Gu, M.; He, Z.; Wu, J.; Li, J.; Lin, Y.; Li, M.; She, Z.; Yuan, J. SZ-685C, a marine anthraquinone, is a potent inducer of apoptosis with anticancer activity by suppression of the Akt/FOXO pathway. Br. J. Pharmacol. 2010, 159, 689–697. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar]
- Gianni, L.; Herman, E.H.; Lipshultz, S.E.; Minotti, G.; Sarvazyan, N.; Sawyer, D.B. Anthracycline cardiotoxicity: From bench to bedside. J. Clin. Oncol. 2008, 26, 3777–3784. [Google Scholar]
- Schimmel, K.J.; Richel, D.J.; van den Brink, R.B.; Guchelaar, H.J. Cardiotoxicity of cytotoxic drugs. Cancer Treat. Rev. 2004, 30, 181–191. [Google Scholar]
- Hofland, K.F.; Thougaard, A.V.; Sehested, M.; Jensen, P.B. Dexrazoxane protects against myelosuppression from the DNA cleavage-enhancing drugs etoposide and daunorubicin but not doxorubicin. Clin. Cancer Res. 2005, 11, 3915–3924. [Google Scholar]
- Chien, A.J.; Moasser, M.M. Cellular mechanisms of resistance to anthracyclines and taxanes in cancer: Intrinsic and acquired. Semin Oncol. 2008, 35, S1–S14, quiz S39.. [Google Scholar]
- Batist, G.; Tulpule, A.; Sinha, B.K.; Katki, A.G.; Myers, C.E.; Cowan, K.H. Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells. J. Biol. Chem. 1986, 261, 15544–15549. [Google Scholar]
- Ishida, H.; Okabe, M.; Gomi, K.; Horiuchi, R.; Mikami, K.; Naito, M.; Tsuruo, T. Modulation of adriamycin resistance in human breast carcinoma MCF-7 cells in vitro and in vivo by medroxyprogesterone acetate. Jpn. J. Cancer Res. 1994, 85, 542–549. [Google Scholar] [CrossRef]
- Vickers, P.J.; Dickson, R.B.; Shoemaker, R.; Cowan, K.H. A multidrug-resistant MCF-7 human breast cancer cell line which exhibits cross-resistance to antiestrogens and hormone-independent tumor growth in vivo. Mol. Endocrinol. 1988, 2, 886–892. [Google Scholar] [CrossRef]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar]
- Vanhaesebroeck, B.; Leevers, S.J.; Panayotou, G.; Waterfield, M.D. Phosphoinositide 3-kinases: A conserved family of signal transducers. Trends Biochem. Sci. 1997, 22, 267–272. [Google Scholar]
- WinMdi, version 2.8, windows multiple document interface for flow cytometry, Scripps Research Institute: La Jolla, CA, USA, 2000.
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhu, X.; He, Z.; Wu, J.; Yuan, J.; Wen, W.; Hu, Y.; Jiang, Y.; Lin, C.; Zhang, Q.; Lin, M.; et al. A Marine Anthraquinone SZ-685C Overrides Adriamycin-Resistance in Breast Cancer Cells through Suppressing Akt Signaling. Mar. Drugs 2012, 10, 694-711. https://doi.org/10.3390/md10040694
Zhu X, He Z, Wu J, Yuan J, Wen W, Hu Y, Jiang Y, Lin C, Zhang Q, Lin M, et al. A Marine Anthraquinone SZ-685C Overrides Adriamycin-Resistance in Breast Cancer Cells through Suppressing Akt Signaling. Marine Drugs. 2012; 10(4):694-711. https://doi.org/10.3390/md10040694
Chicago/Turabian StyleZhu, Xun, Zhenjian He, Jueheng Wu, Jie Yuan, Weitao Wen, Yiwen Hu, Yi Jiang, Cuiji Lin, Qianhui Zhang, Min Lin, and et al. 2012. "A Marine Anthraquinone SZ-685C Overrides Adriamycin-Resistance in Breast Cancer Cells through Suppressing Akt Signaling" Marine Drugs 10, no. 4: 694-711. https://doi.org/10.3390/md10040694
APA StyleZhu, X., He, Z., Wu, J., Yuan, J., Wen, W., Hu, Y., Jiang, Y., Lin, C., Zhang, Q., Lin, M., Zhang, H., Yang, W., Chen, H., Zhong, L., She, Z., Chen, S., Lin, Y., & Li, M. (2012). A Marine Anthraquinone SZ-685C Overrides Adriamycin-Resistance in Breast Cancer Cells through Suppressing Akt Signaling. Marine Drugs, 10(4), 694-711. https://doi.org/10.3390/md10040694