Aplisulfamines, New Sulfoxide-Containing Metabolites from an Aplidium Tunicate: Absolute Stereochemistry at Chiral Sulfur and Carbon Atoms Assigned Through an Original Combination of Spectroscopic and Computational Methods




 and
and
Abstract
:1. Introduction
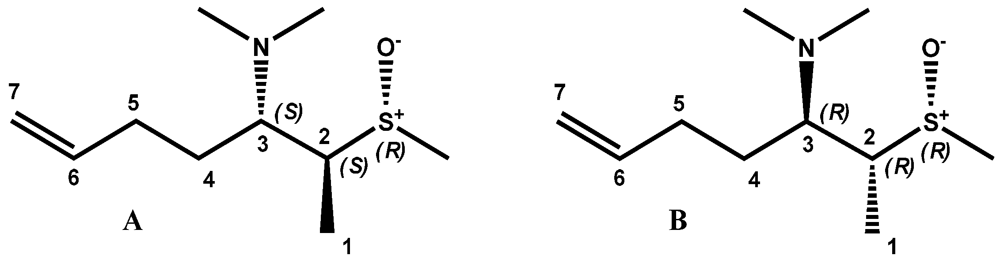
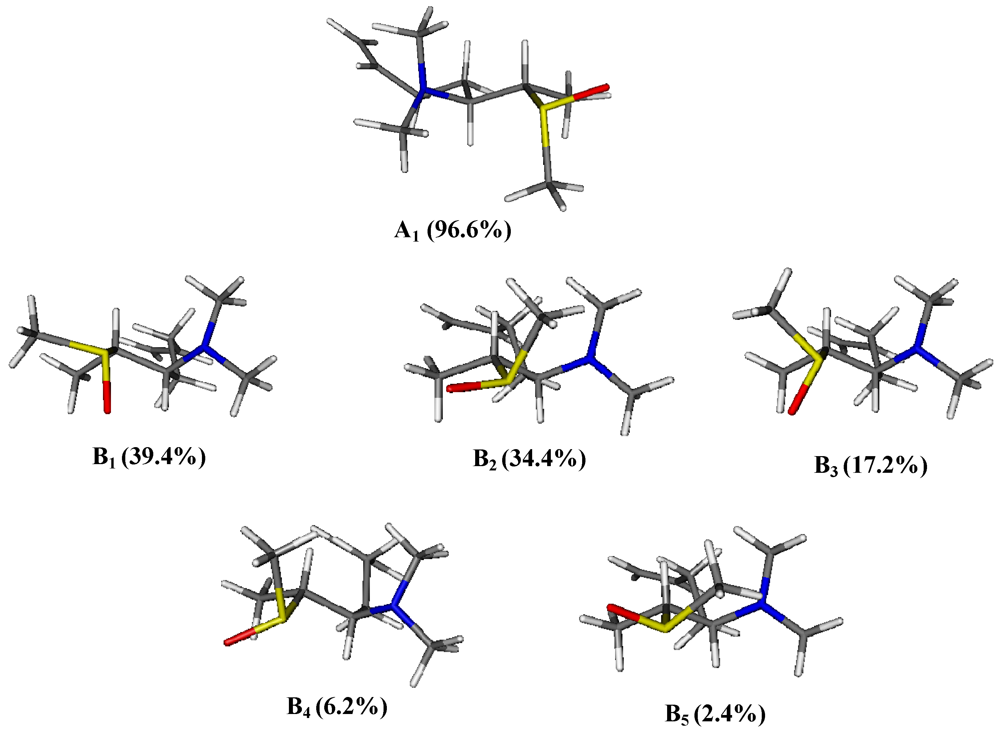
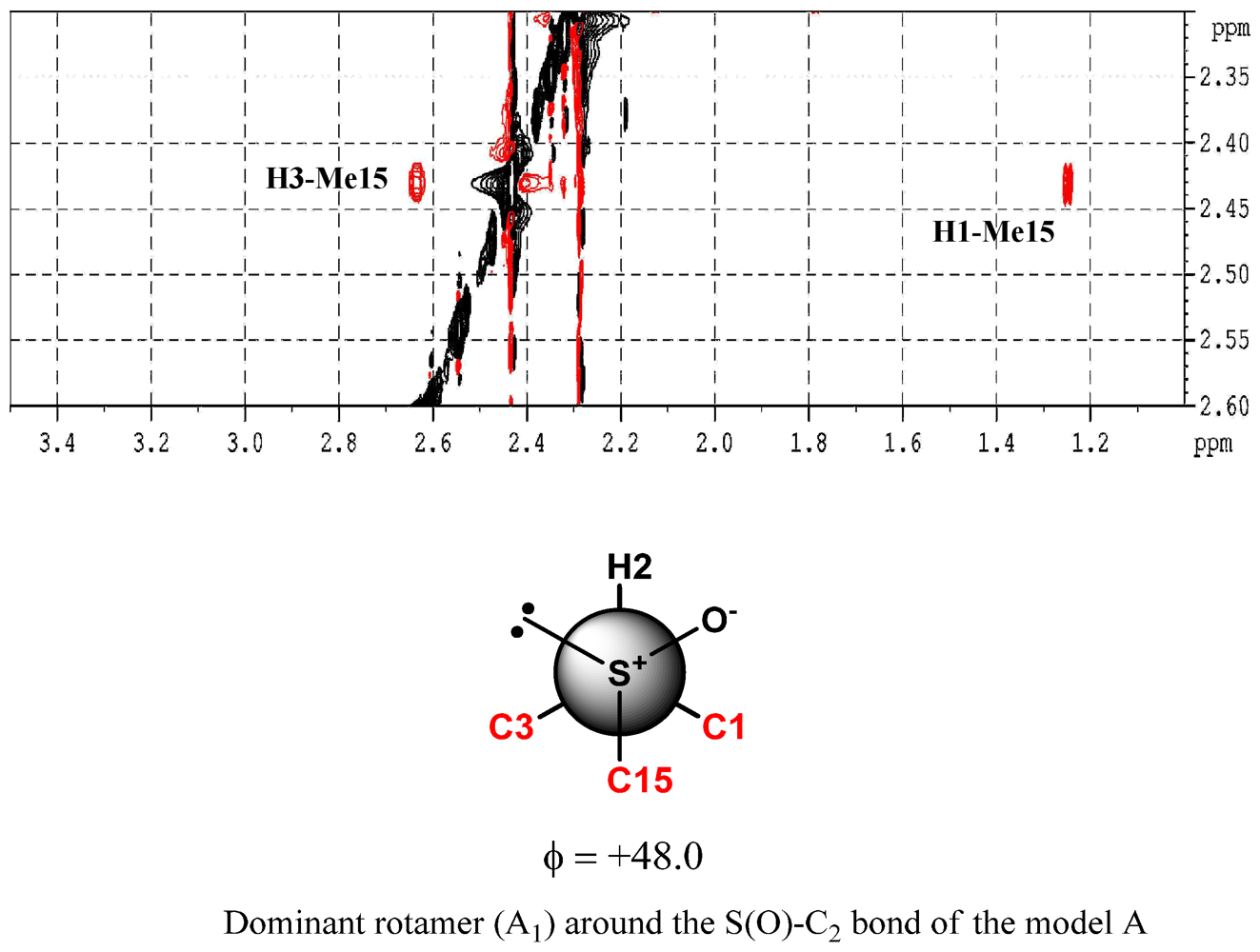
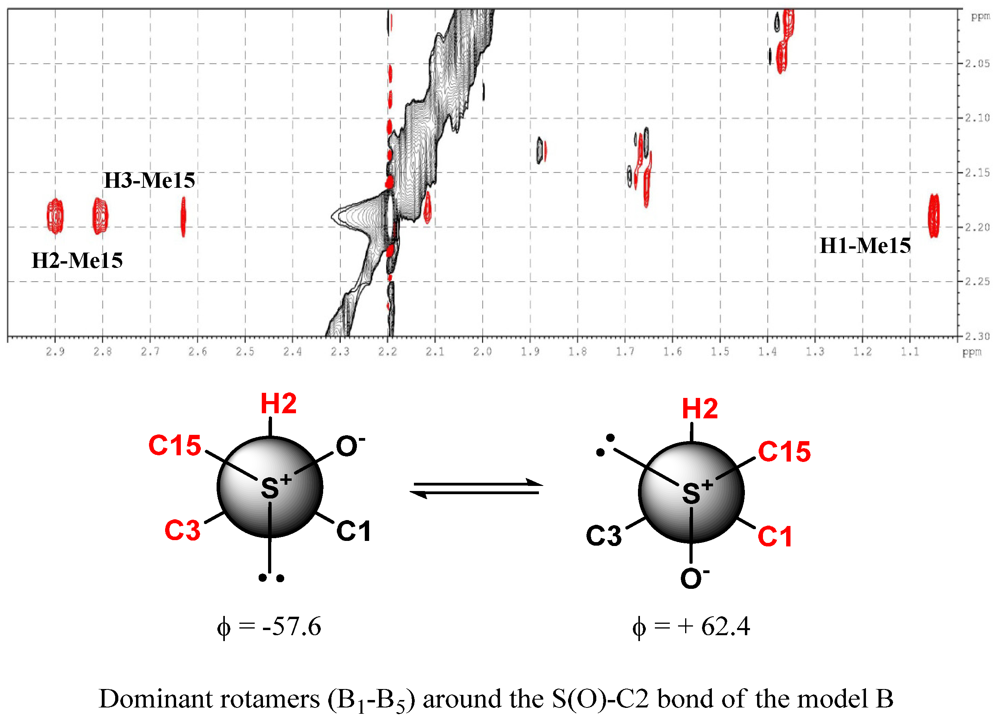
2. Results and Discussion
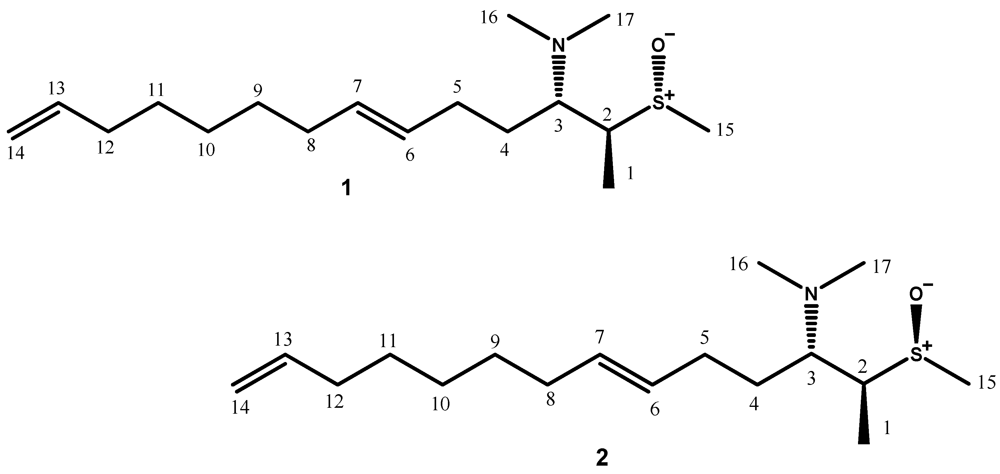

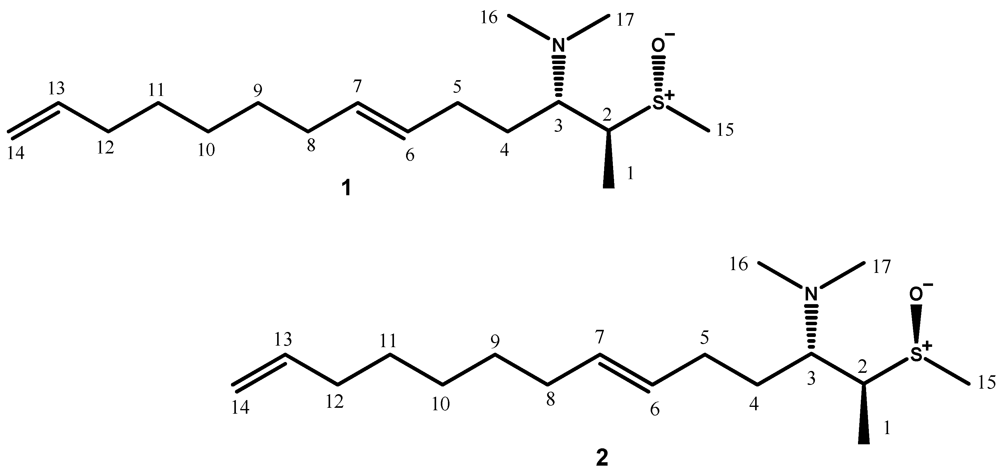{kind=link}
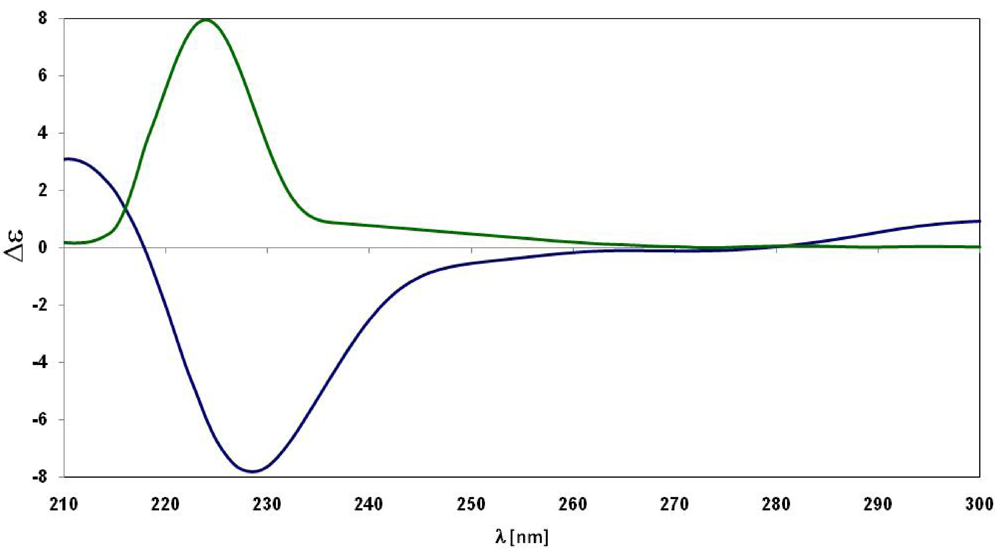{kind=link}
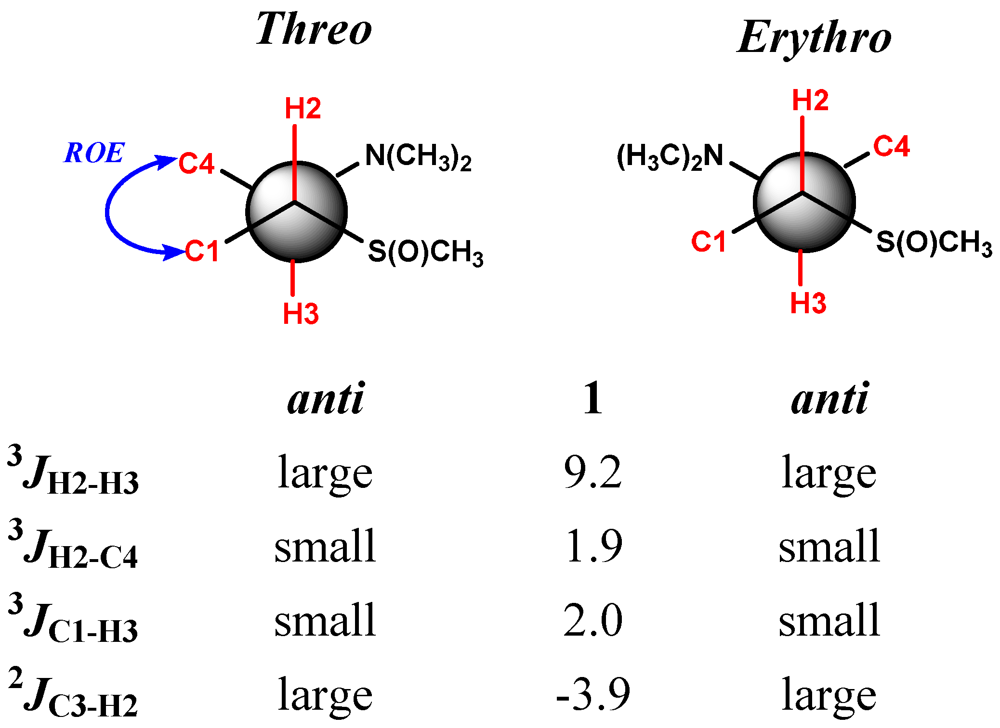{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| POS | 1 | 2 | ||
|---|---|---|---|---|
| δCa | δH (mult, J in Hz)b | δCa | δH (mult, J in Hz)b | |
| 1 | 7.8 | 1.25 (d, 6.8 ) | 9.4 | 1.06 (d, 6.6 ) |
| 2 | 58.2 | 3.31(dq, 9.2,6.8) | 60.0 | 2.91(dq, 10.6,6.6) |
| 3 | 65.2 | 2.64(m) | 65.2 | 2.81 (dt, 10.6,4.4) |
| 4a | 28.8 | 1.43c | 25.2 | 1.70 c |
| 4b | 1.78 (m) | 1.90 (m) | ||
| 5 | 32.5 | 2.12(m) | 31.2 | 2.17 (m) |
| 6 | 130.6 | 5.42(bdt 15.2, 6.2) | 132.7 | 5.41(bdt 15.1, 6.5) |
| 7 | 132.4 | 5.47(bdt 15.1, 6.2) | 132.7 | 5.47(bdt 15.1, 6.5) |
| 8 | 33.5 | 2.00(m) | 34.6 | 2.02 (m) |
| 9 | 30.5 | 1.38 c | 30.0 | 1.40 c |
| 10 | 29.7 | 1.33 c | 30.0 | 1.40 c |
| 11 | 30.0 | 1.39 c | 30.0 | 1.40 c |
| 12 | 34.9 | 2.05(q 6.7) | 31.1 | 2.04 (m) |
| 13 | 140.0 | 5.80(ddt, 17.1, 10.2, 6.7) | 138.8 | 5.80(ddt, 17.1, 10.2, 6.7) |
| 14a | 114.7 | 4.92 (bdd, 10.2, 2.0) | 114.7 | 4.92 c |
| 14b | 4.98 (bdd, 17.1, 2.0) | 4.98(bdd, 17.1, 2.0) | ||
| 15 | 30.4 | 2.43 (s) | 40.0 | 2.20 (s) |
| 16/17 | 41.0 | 2.29 (s) | 38.0 | 2.63 (s) |
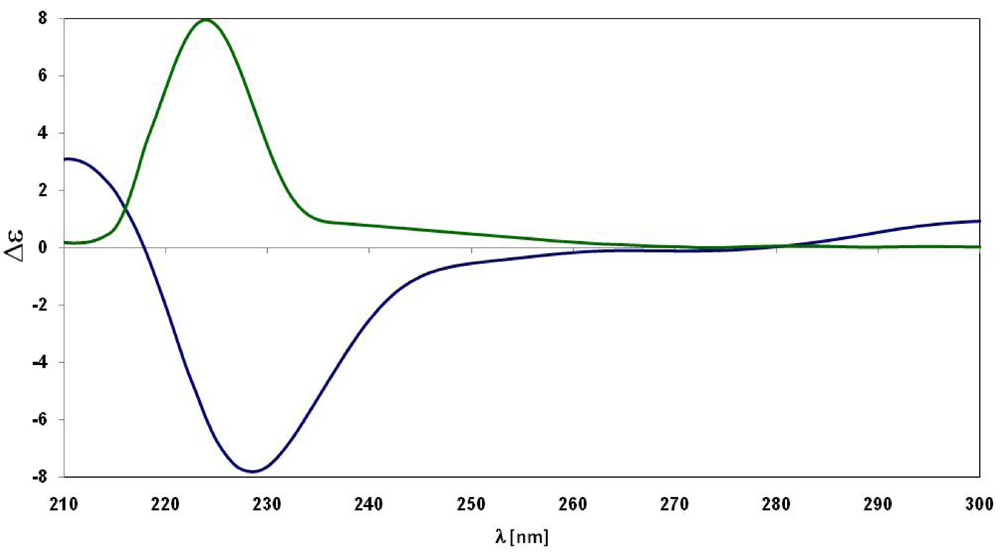
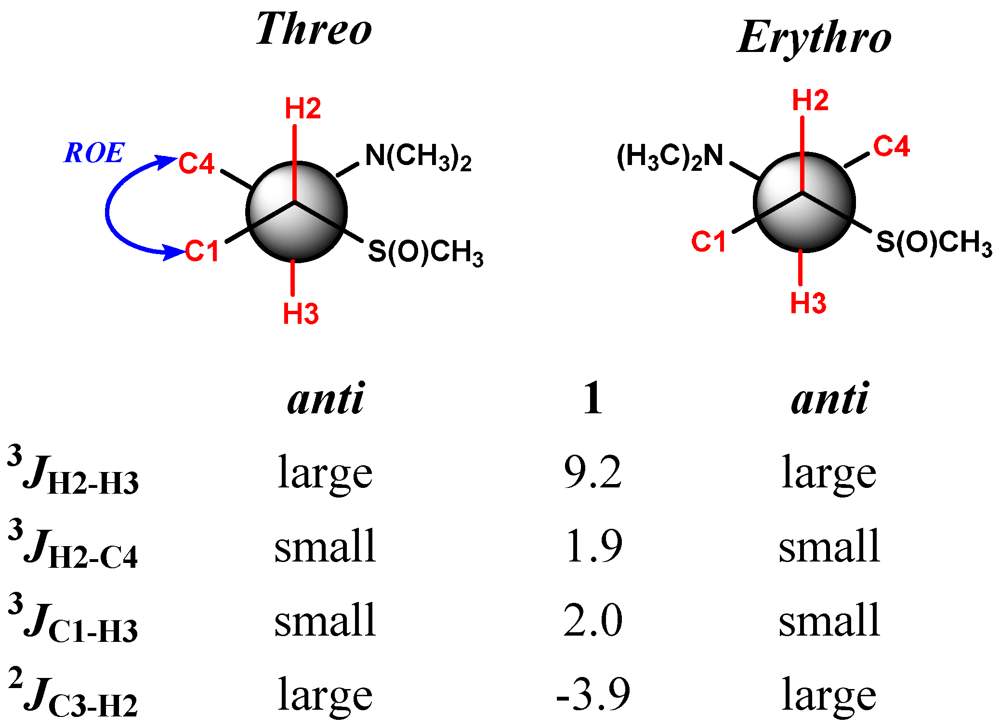
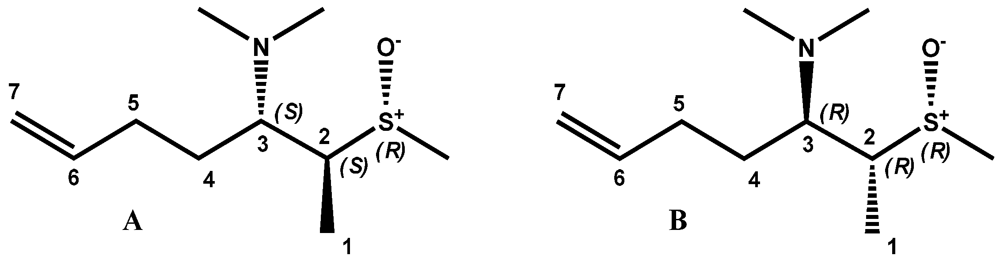
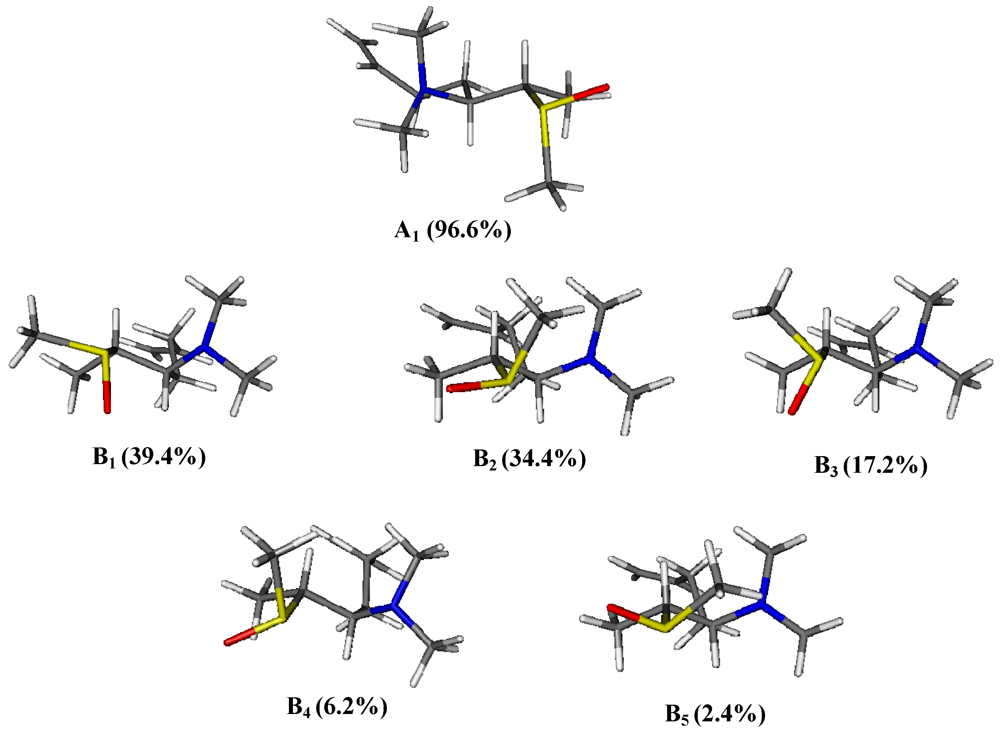
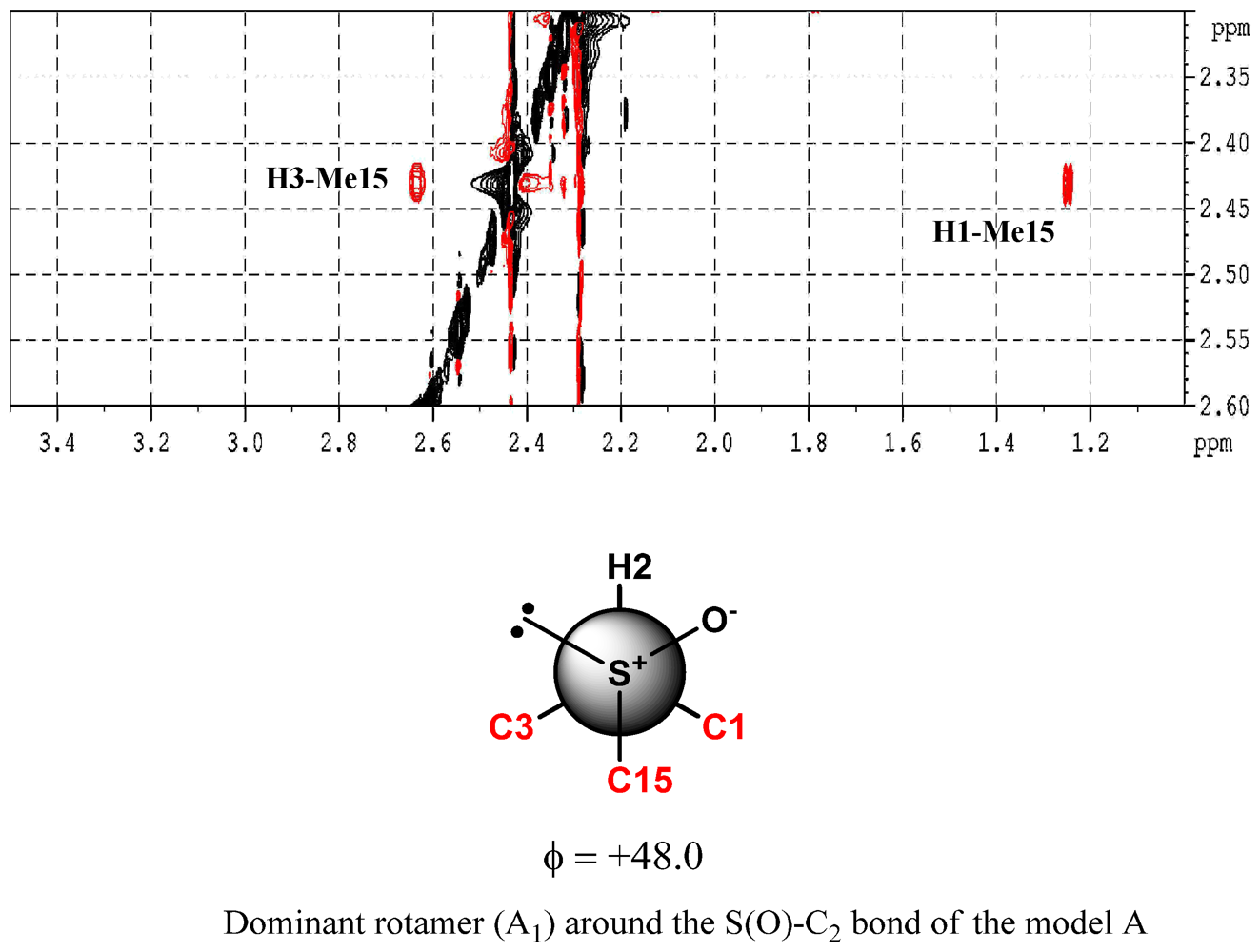
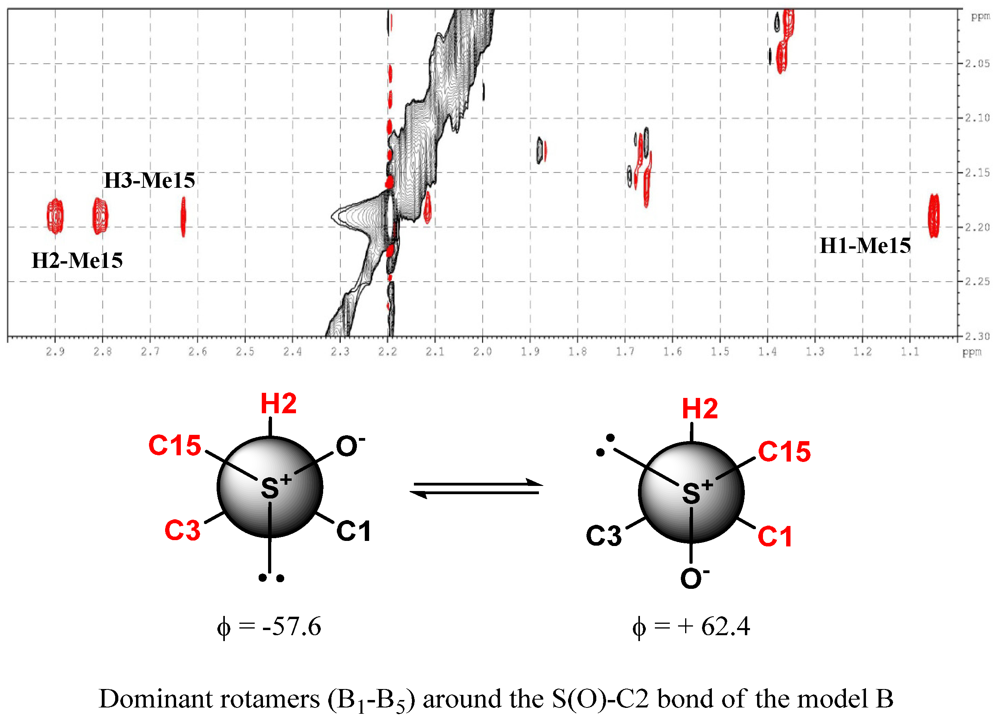
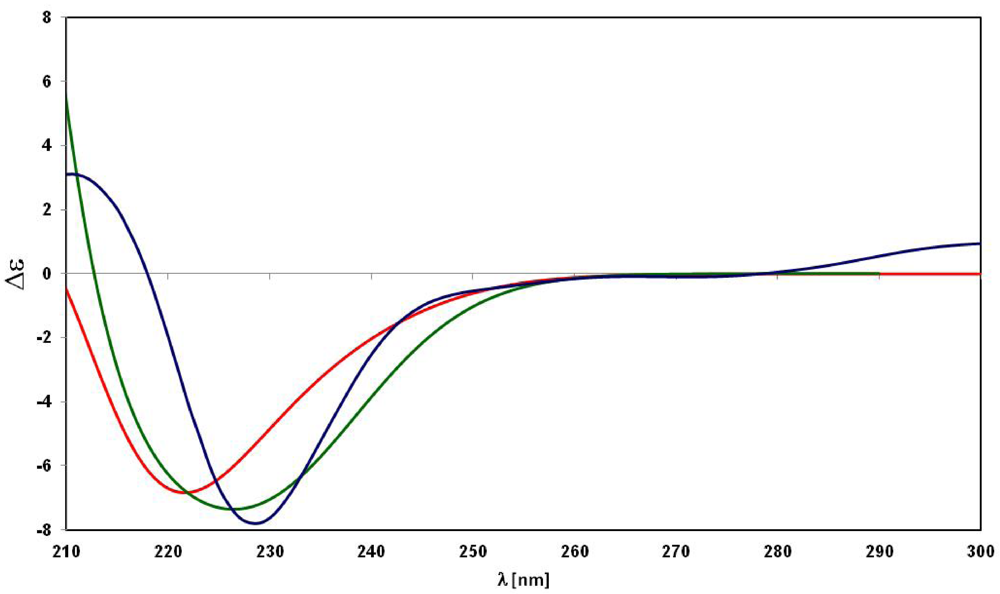
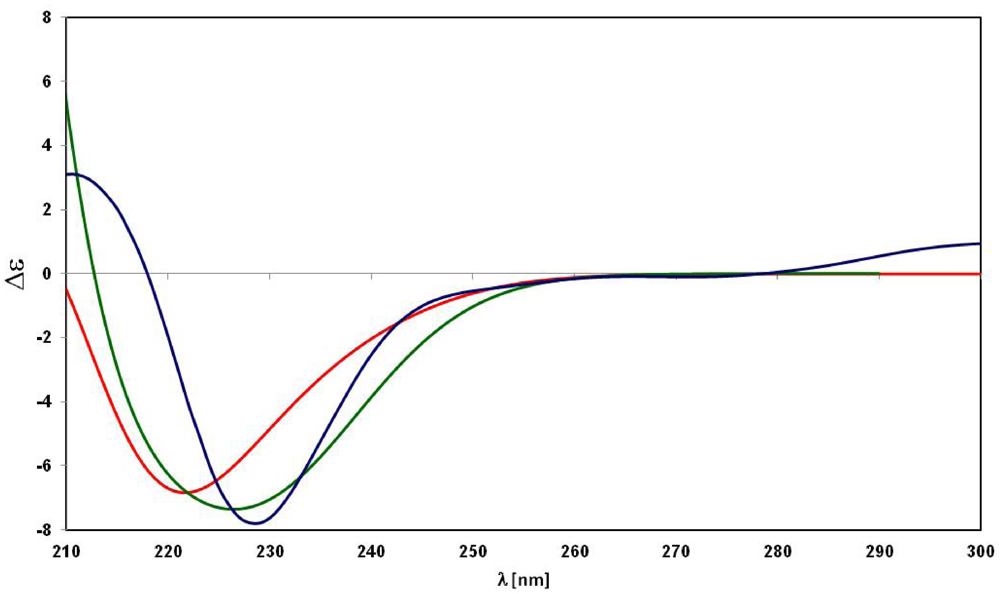
3. Experimental Section
3.1. General Experimental Procedures
3.2. Collection, Extraction and Isolation
3.3. Computational Details
4. Conclusions
Acknowledgments
- Samples Availability: Available from the authors.
References
- Harrigan, G.G.; Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Nagle, D.G.; Paul, V.J. Symplostatin 2: a dolastatin 13 analogue from the marine cyanobacterium Symploca hydnoides. J. Nat. Prod. 1999, 62, 655–658. [Google Scholar]
- Hamada, T.; Matsunaga, S.; Yano, G.; Fusetani, N. Polytheonamides A and B, highly cytotoxic, linear polypeptides with unprecedented structural features, from the marine sponge, Theonella swinhoei. J. Am. Chem. Soc. 2005, 127, 110–118. [Google Scholar]
- Galbraith, M.N.; Horn, D.H.S.; Sasse; Jenneth, M. Podolactones C and D, terpene sulfoxides from Podocarpus neriifolius. J. Chem. Soc. 1971, 21, 1362–1363. [Google Scholar]
- Fenwick, G.R.; Hanley, A.B. The genus Allium--Part 1. Crit. Rev. Food Sci. Nutr. 1985, 22, 199–271. [Google Scholar]
- Kuriyama, M.; Takagi, M.; Murata, K. A new amino acid, chondrine, isolated from a red alga. Nip. Suis. G. 1960, 26, 627. [Google Scholar]
- Lake, R.J.; Brennan, M.M.; Blunt, J.W.; Munro, M.H.G.; Pannell, L.K. Eudistomin K sulfoxide. An antiviral sulfoxide from the New Zealand ascidian Ritterella sigillinoides. Tetrahedron Lett. 1988, 29, 2255–2256. [Google Scholar]
- Murata, O.; Shigemori, H.; Ishibashi, M.; Sugama, K.; Hayashi, K.; Kobayashi, J. Eudistomidins E and F, new β-carboline alkaloids from the Okinawan marine tunicate Eudistoma glaucus. Tetrahedron Lett. 1991, 32, 3539–3542. [Google Scholar]
- Makarieva, T.N.; Stonik, V.A.; Dmitrenok, A.S.; Grebnev, B.B.; Isakov, V.V.; Rebachyk, N.M.; Rashkes, Y.W. Varacin and three new marine antimicrobial polysulfides from the far-eastern ascidian Polycitor sp. J. Nat. Prod. 1995, 58, 254–258. [Google Scholar]
- Dorman, D.E.; Jautelat, M.D.; Roberts, J. Carbon-13 nuclear magnetic resonance spectroscopy. Quantitative correlations of the carbon chemical shifts of acyclic alkenes. J. Org. Chem. 1971, 36, 2757–2766. [Google Scholar] [CrossRef]
- Ivanciuc, O.; Rabine, J.P.; Cabrol-Bass, D.; Panaye, A.; Doucet, P.J. 13C NMR Chemical Shift Prediction of the sp3 Carbon Atoms in the α Position Relative to the Double Bond in Acyclic Alkenes. J. Chem. Inf. Comput. Sci. 1997, 37, 587–598. [Google Scholar]
- Charan, R.D.; Garson, M.J.; Brereton, I.M.; Willis, A.C.; Hooper, J.N.A. Haliclonacyclamines A and B, cytotoxic alkaloids from the tropical marine sponge Haliclona sp. Tetrahedron 1996, 52, 9111–9120. [Google Scholar]
- Mislow, K.; Green, M.M.; Laur, P.; Melillo, J.T.; Simmons, T.; Ternay, A.L., Jr. Absolute configuration and optical rotatory power of sulfoxides and sulfinate esters. J. Am. Chem. Soc. 1965, 87, 1958–1976. [Google Scholar]
- Axelrod, M.; Bickart, P.; Goldstein, M.L.; Green, M.M.; Kjaer, A.; Mislow, K. Absolute configuration and optical rotatory dispersion of methyl alkyl sulfoxides. Tetrahedron Lett. 1968, 29, 3249–3252. [Google Scholar]
- Ottenheijm, H.C.J.; Liskamp, R.M.J.; Helquist, P.; Lather, J.W.; Shekhani, M.S. Absolute configuration of sparsomycin. A chiroptical study of sulfoxides. J. Am. Chem. Soc. 1981, 103, 1720–1723. [Google Scholar]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical Determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [PubMed]
- Bassarello, C.; Bifulco, G.; Evidente, A.; Riccio, R.; Gomez-Paloma, L. Stereochemical studies on ascaulitoxin: a J-based NMR configurational analysis of a nitrogen substituted system. Tetrahedron Lett. 2001, 42, 8611–8613. [Google Scholar]
- Kobayashi, J.; Tsuda, M. Amphidinolides, bioactive macrolides from symbiotic marine dinoflagellates. Nat. Prod. Rep. 2004, 21, 77–93. [Google Scholar]
- Ciminiello, P.; Dell’Aversano, C.; Fattorusso, E.; Forino, M.; Magno, S.; Di Rosa, M.; Ianaro, A.; Poletti, R. Structure and stereochemistry of a new cytotoxic Polychlorinated sulfolipid from adriatic shellfish. J. Am. Chem. Soc. 2002, 124, 13114–13120. [Google Scholar]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar]
- Aiello, A.; Fattorusso, E.; Luciano, P.; Menna, M.; Vitalone, R. Polyaxibetaine, an amino acid derivative from the marine sponge Axinella polypoides. J. Nat. Prod. 2010, 73, 620–622. [Google Scholar]
- Kover, K.E.; Hruby, V.J.; Uhrin, D. Sensitivity- and gradient-enhanced heteronuclear coupled/decoupled HSQC-TOCSY experiments for measuring long-range heteronuclear coupling constants. J. Magn. Res. 1997, 129, 125–129. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Aiello, A.; Fattorusso, E.; Imperatore, C.; Luciano, P.; Menna, M.; Vitalone, R. Aplisulfamines, New Sulfoxide-Containing Metabolites from an Aplidium Tunicate: Absolute Stereochemistry at Chiral Sulfur and Carbon Atoms Assigned Through an Original Combination of Spectroscopic and Computational Methods. Mar. Drugs 2012, 10, 51-63. https://doi.org/10.3390/md10010051
Aiello A, Fattorusso E, Imperatore C, Luciano P, Menna M, Vitalone R. Aplisulfamines, New Sulfoxide-Containing Metabolites from an Aplidium Tunicate: Absolute Stereochemistry at Chiral Sulfur and Carbon Atoms Assigned Through an Original Combination of Spectroscopic and Computational Methods. Marine Drugs. 2012; 10(1):51-63. https://doi.org/10.3390/md10010051
Chicago/Turabian StyleAiello, Anna, Ernesto Fattorusso, Concetta Imperatore, Paolo Luciano, Marialuisa Menna, and Rocco Vitalone. 2012. "Aplisulfamines, New Sulfoxide-Containing Metabolites from an Aplidium Tunicate: Absolute Stereochemistry at Chiral Sulfur and Carbon Atoms Assigned Through an Original Combination of Spectroscopic and Computational Methods" Marine Drugs 10, no. 1: 51-63. https://doi.org/10.3390/md10010051
APA StyleAiello, A., Fattorusso, E., Imperatore, C., Luciano, P., Menna, M., & Vitalone, R. (2012). Aplisulfamines, New Sulfoxide-Containing Metabolites from an Aplidium Tunicate: Absolute Stereochemistry at Chiral Sulfur and Carbon Atoms Assigned Through an Original Combination of Spectroscopic and Computational Methods. Marine Drugs, 10(1), 51-63. https://doi.org/10.3390/md10010051