Sarcophine-Diol, a Skin Cancer Chemopreventive Agent, Inhibits Proliferation and Stimulates Apoptosis in Mouse Melanoma B16F10 Cell Line

Abstract
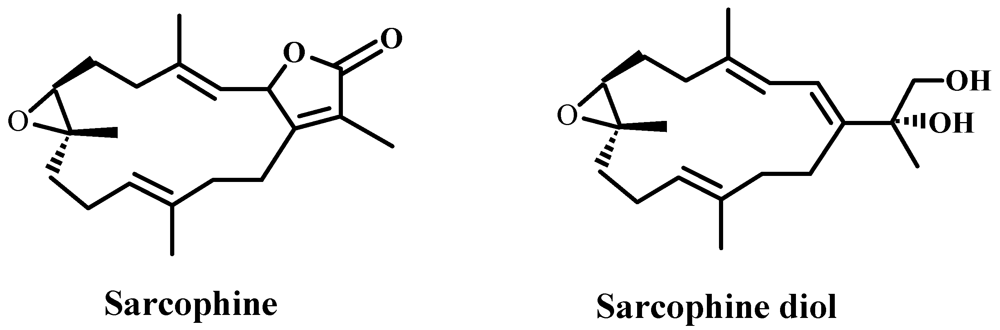
:1. Introduction
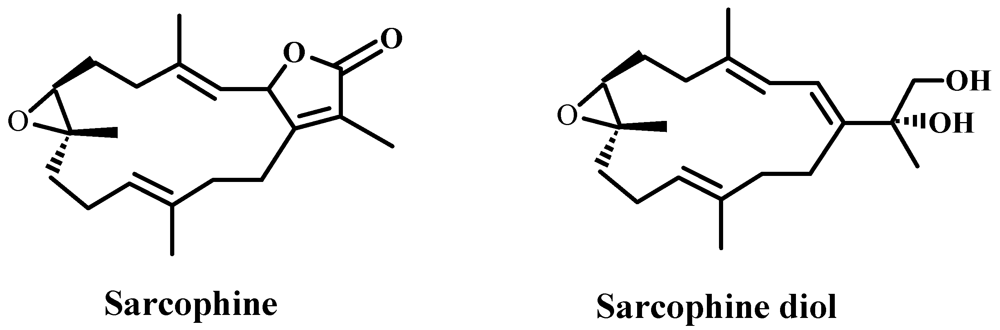
2. Results
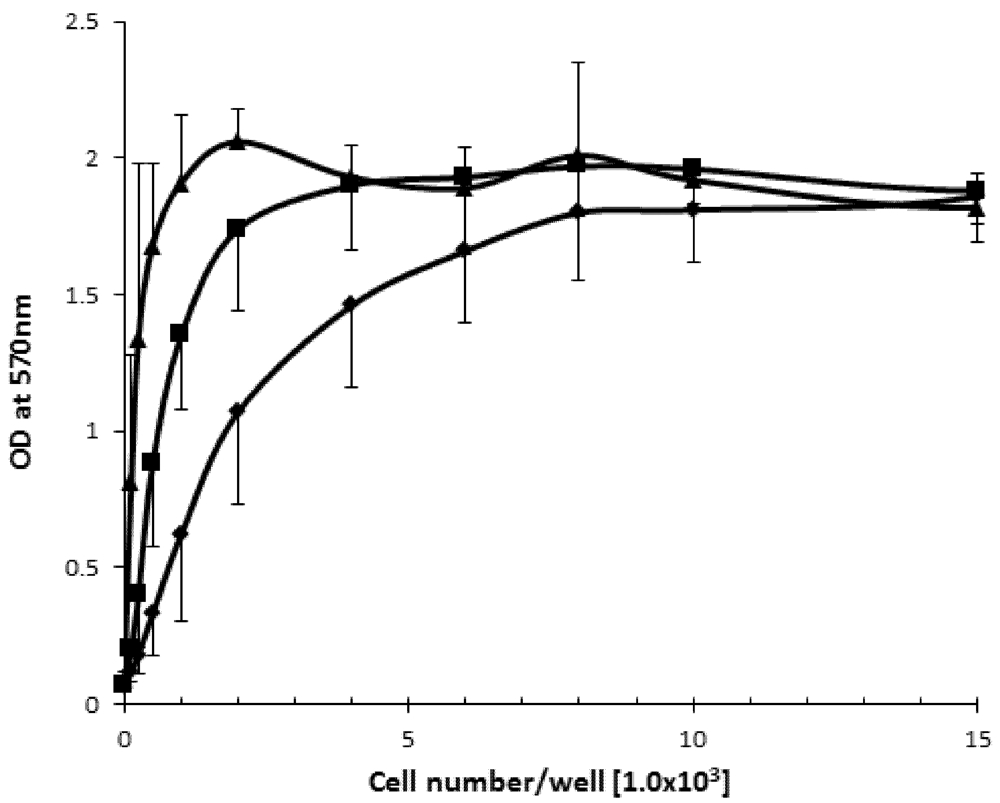
2.1. The Effect of Cell Density on Cell Viability
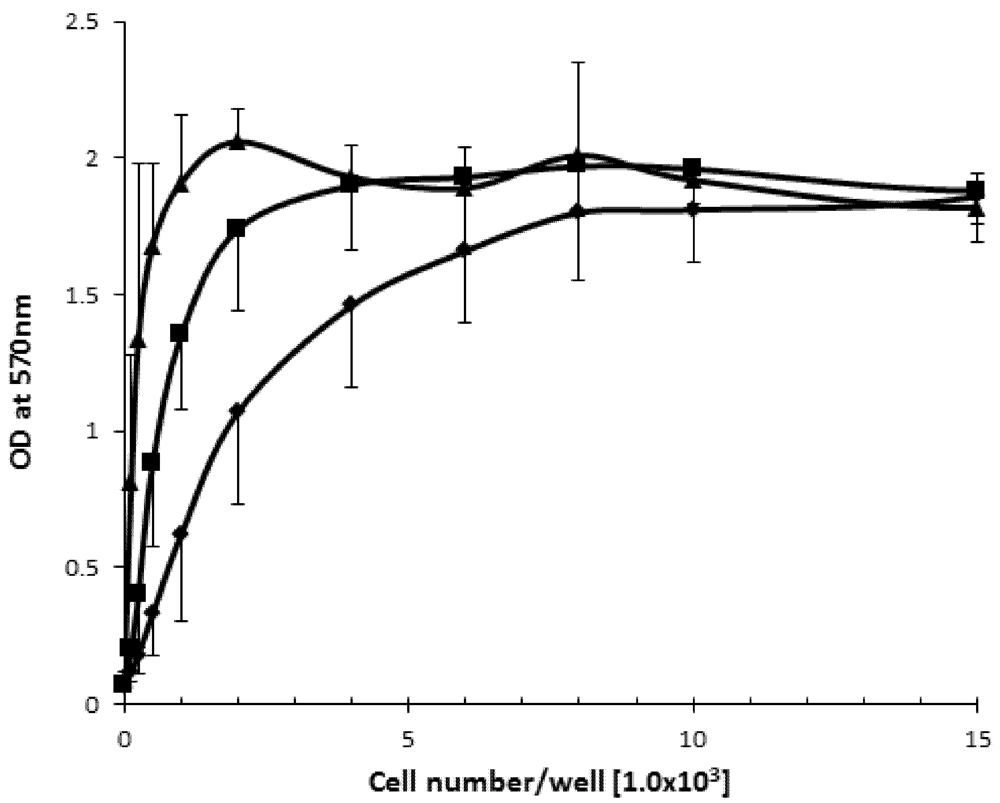
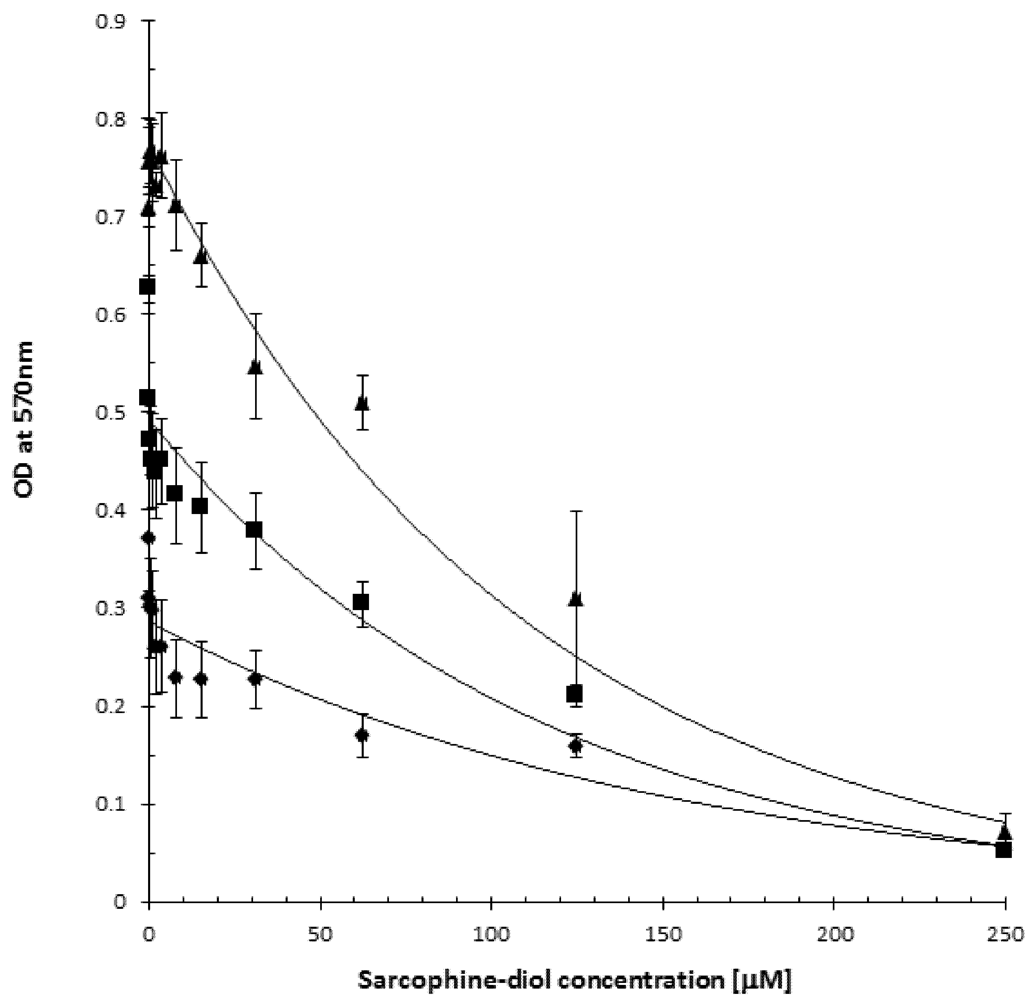
2.2. SD Reduces Viability of the B16F10 Melanoma Cells
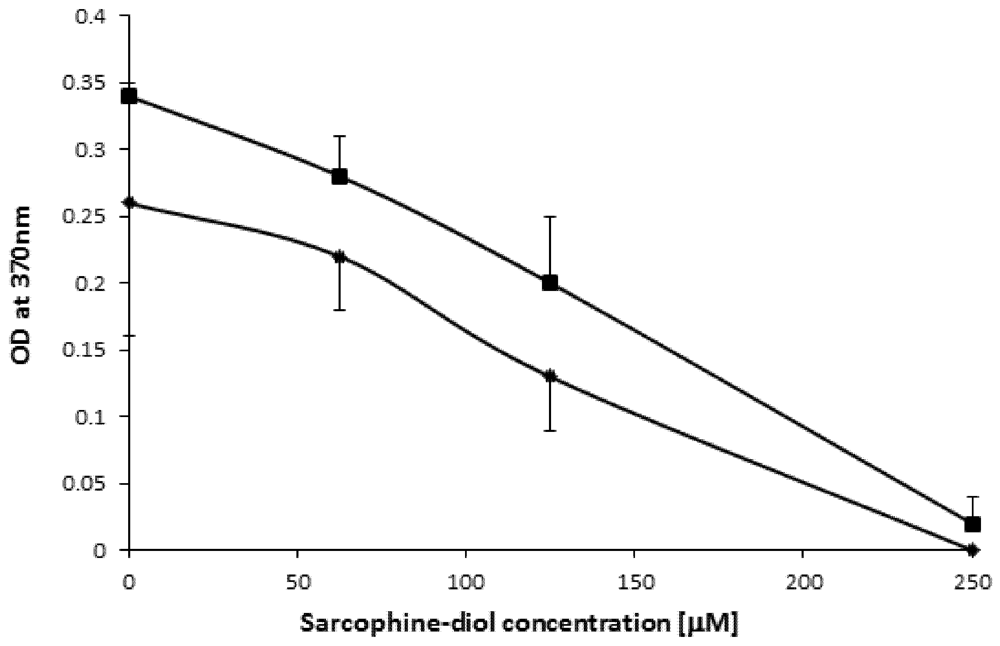
2.3. SD Inhibits de Novo DNA Synthesis
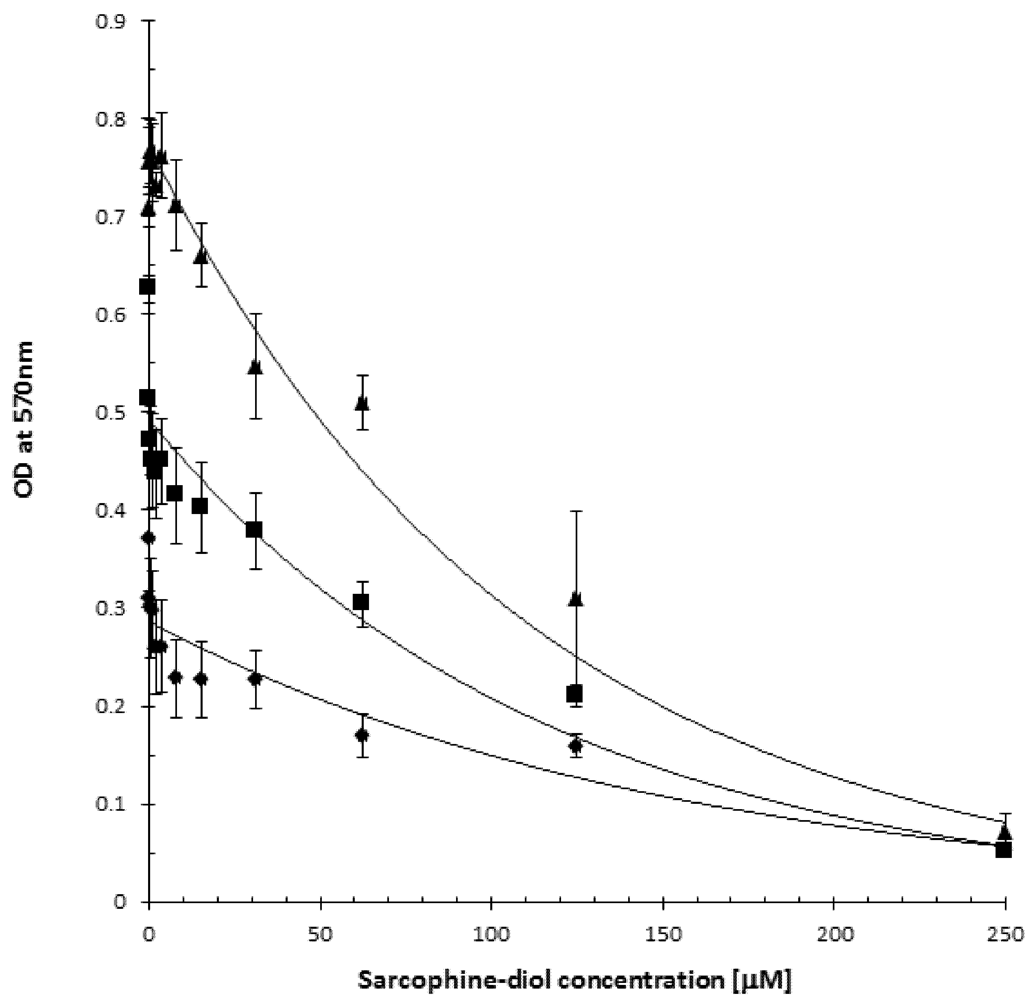
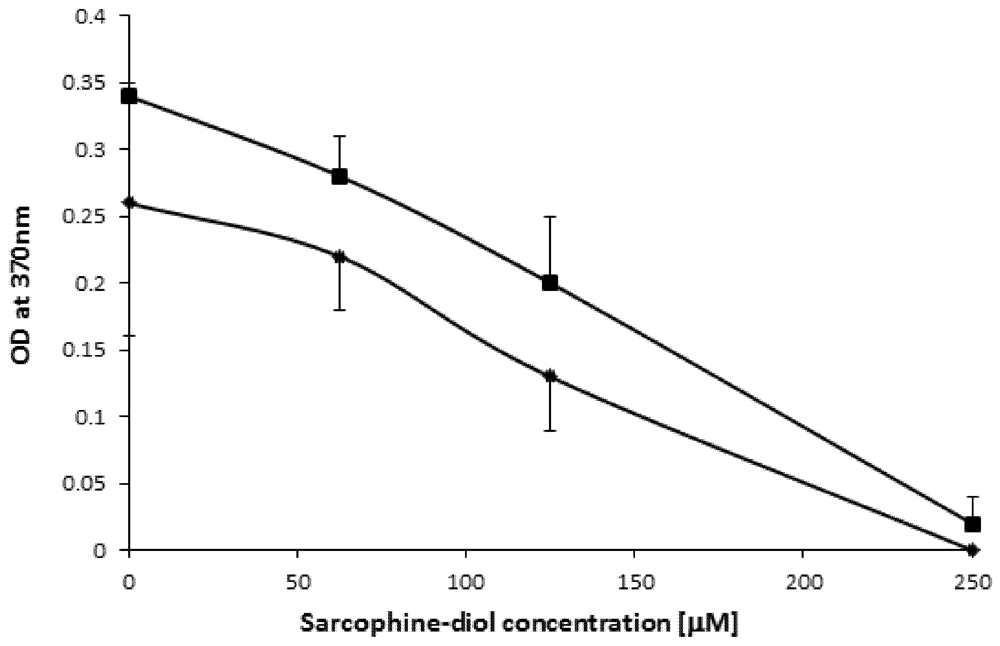
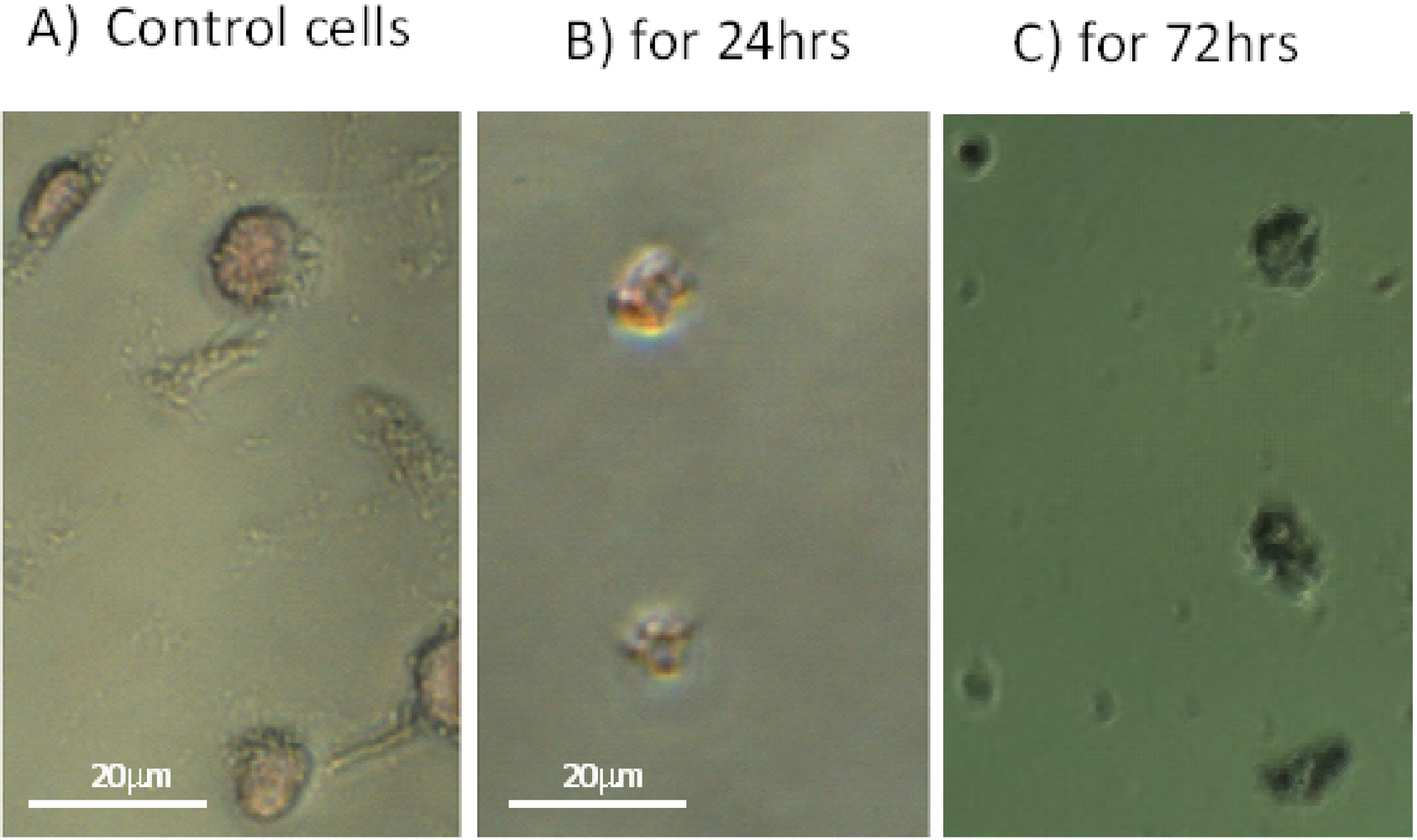
2.4. SD Enhances DNA Fragmentation
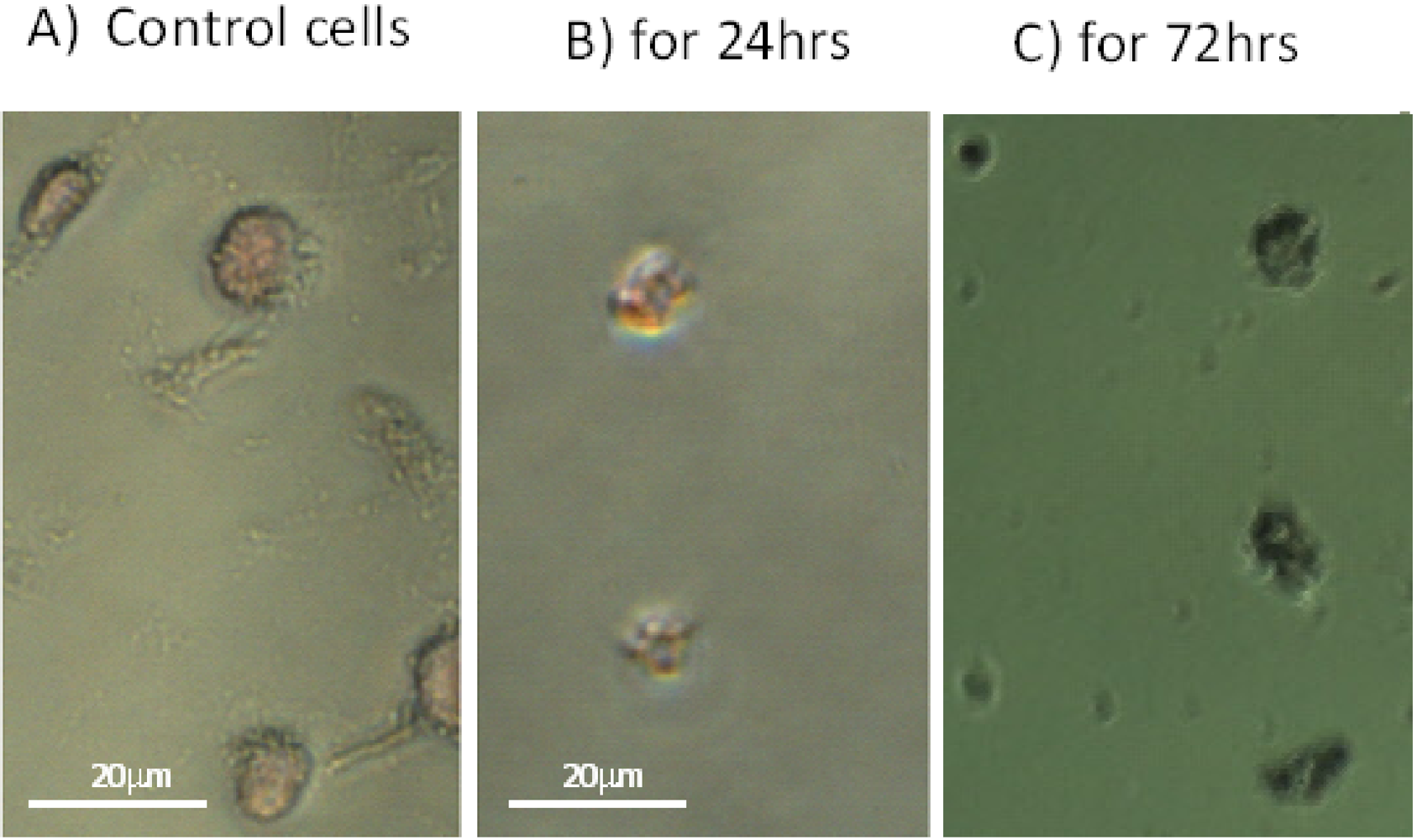
2.5. Effect of SD on the Total Protein Contents in Melanoma Cells
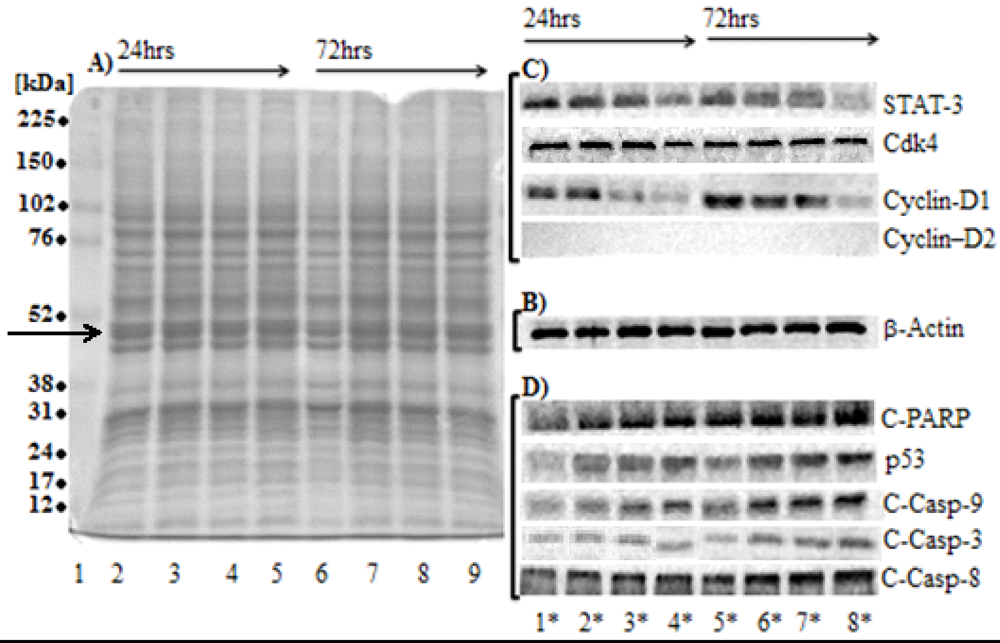
2.6. SD Inhibits the Levels of Protein Markers for Cell Proliferation
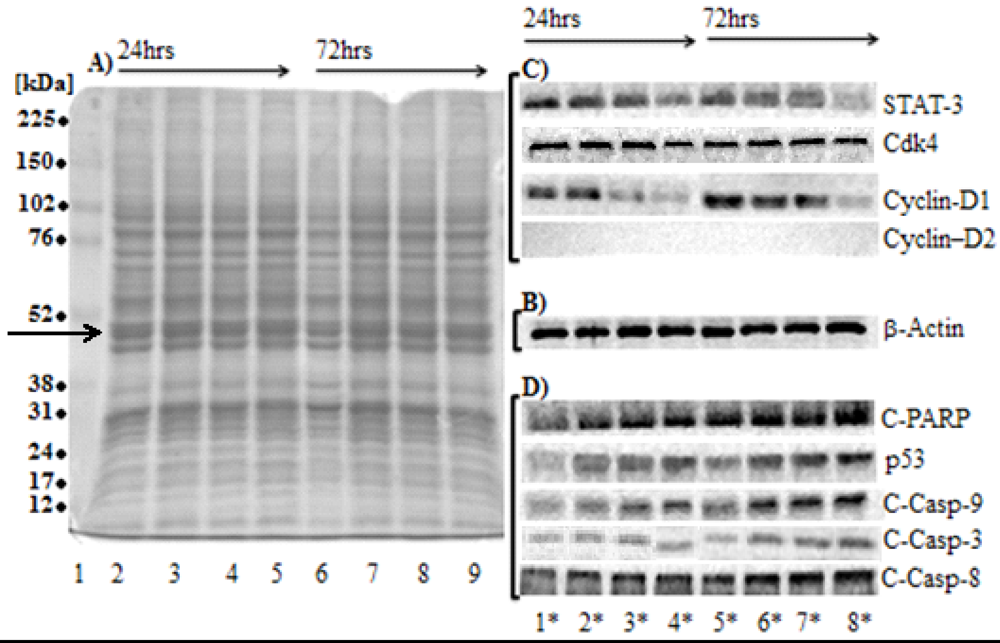

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sarcophine-diol concentration (µM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0 | 62.5 | 125 | 250 | 0 | 62.5 | 125 | 250 | ||
| 24 h treatment | 72 h treatment | ||||||||
| Proliferation markers | STAT-3 | 100% | 78.8 ± 9.5 n = 4 | 62.9 ± 10.4 n = 4 (*) | 36.0 ± 6.0 n = 4 (*) | 100% | 70.4 ± 18.2 n = 3 | 59.8 ± 8.4 n = 3 (*) | 41.8 ± 10.5 n = 3 (*) |
| Cdk4 | 100% | 105.3 ± 9.2 n = 3 | 92.8 ± 14.3 n = 3 | 101.8 ± 15.3 n = 3 | 100% | 112.5 ± 27.2 n = 3 | 97.4 ± 18.9 n = 3 | 118.7 ± 20.1 n = 3 | |
| Cyclin-D1 | 100% | 75.3 ± 10.5 n = 3 | 35.5 ± 7.2 n = 3 (*) | 7.7 ± 14.2 n = 3 (*) | 100% | 87.8 ± 12.6 n = 3 | 48.9 ± 9.8 n = 3 (*) | 4.9 ± 6.2 n = 3 (*) | |
| Cyclin-D2 | ND | ND | ND | ND | ND | ND | ND | ND | |
| Apoptosis markers | C-PARP | 100% | 123.2 ± 18.5 n = 3 | 132.1 ± 18.1 n = 3 (*) | 168.3 ± 24.1 n = 3 (*) | 100% | 189.3 ± 32.2 n = 3 (*) | 216.0 ± 42.7 n = 3 (*) | 276.0 ± 56.8 n = 3 (*) |
| p53 | 100% | 128.0 ± 23.9 n = 3 | 132.1 ± 19.6 n = 3 (*) | 132.7 ± 16.9 n = 3 (*) | 100% | 141.5 ± 33.3 n = 3 | 159.9 ± 22.6 n = 3 (*) | 177.6 ± 19.4 n = 3 (*) | |
| C-Casp-9 | 100% | 124.3 ± 27.7 n = 3 | 132.3 ± 20.5 n = 3 (*) | 147.4 ± 18.1 n = 3 (*) | 100% | 130.6 ± 23.5 n = 3 | 158.0 ± 31.6 n = 3 (*) | 179.1 ± 18.9 n = 3 (*) | |
| C-Casp-3 | 100% | 120.2 ± 14.3 n = 3 | 133.3 ± 9.9 n = 3 (*) | 140.3 ± 16.7 n = 3 (*) | 100% | 128.2 ± 25.5 n = 3 | 141.7 ± 22.5 n = 3 (*) | 174.2 ± 21.0 n = 3 (*) | |
| C-Casp-8 | 100% | 119.5 ± 22.6 n = 3 | 132.3 ± 25.3 n = 3 (*) | 160.3 ± 18.7 n = 3 (*) | 100% | 123.9 ± 25.1 n = 3 | 178.3 ± 33.6 n = 3 (*) | 213 ± 51.2 n = 3 (*) | |
2.7. SD Increases the Expression Levels of Protein Markers for Apoptosis
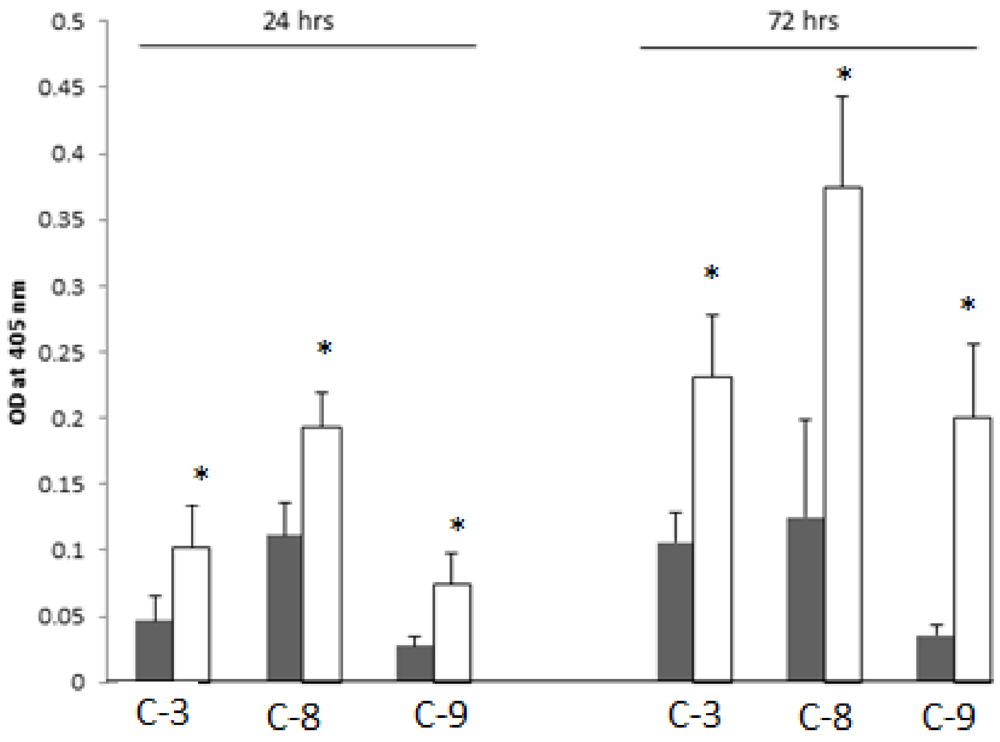
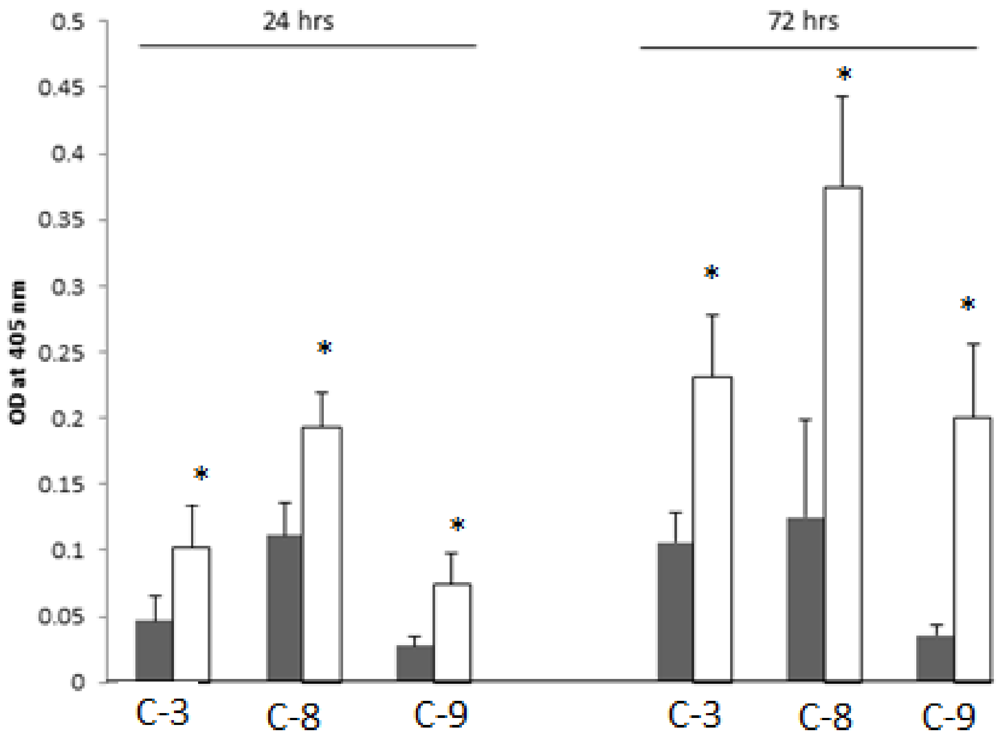
2.8. SD Increases Enzymatic Activities of the Cleaved-Caspases-3, -8 and -9
2.9. Discussion
3. Experimental Section
3.1. Materials and Reagents
3.2. Synthesis of SD
3.3. Cell Culture
3.4. Total Cellular Protein Extract
3.5. Determination of Protein Content
3.6. Western Blot Analyses of Protein Markers for Cell Proliferation and Apoptosis
3.7. Cell Viability Assays
3.7.1. Staining of the Cells with Trypan-Blue
3.7.2. MTT Assay
3.8. Determination of DNA Synthesis
3.9. Determination of DNA Fragmentation in Situ
3.10. Determination of Enzymatic Activities of the Cleaved-Caspases-3, -8 and -9
3.11. Statistical Analysis
Acknowledgements
- Samples Availability: Available from the authors.
References
- Zhang, X.; Kundoor, V.; Khalifa, S.; Zeman, D.; Fahmy, H.; Dwivedi, C. Chemopreventive effects of sarcophine-diol on skin tumor development in CD-1 mice. Cancer Lett. 2007, 253, 53–59. [Google Scholar]
- Zang, X.; Bommareddy, A.; Chen, W.; Hildreth, M.B.; Kaushik, R.S.; Zeman, D.; Khalifa, S.; Fahmy, H.; Dwivedi, C. Chemopreventive effects of sarcophine-diol on ultraviolet B-induced skin tumor development in SKH-1 hairless mice. Mar. Drugs 2009, 7, 153–165. [Google Scholar]
- Zhang, X.; Bommareddy, A.; Chen, W.; Khalifa, S.; Kaushik, R.S.; Fahmy, H.; Dwivedi, C. Sarcophine-diol, a chemopreventive agent of skin cancer, inhibits apoptosis through extrinsic pathway in human epidermoid carcinoma A431 Cells. Trans. Oncol. 2009, 2, 21–30. [Google Scholar]
- Rigel, D.S.; Friedman, R.J.; Kopf, A.W. Lifetime risk for development of skin cancer in US population: Current estimate is now 1 in 5. J. Am. Acad. Dermatol. 1996, 35, 1012–1013. [Google Scholar]
- Sarasin, A. The molecular pathways of ultraviolet-induced carcinogenesis. Mutat. Res. 1999, 428, 5–10. [Google Scholar]
- Jarnat, A.F.; Johnson, J.T.; Sheridan, C.D.; Caffrey, T.J. Early detection and treatment of skin cancer. Am. Fam. Physician 2000, 62, 357-368, 375-376, 381-382. [Google Scholar]
- Urteaga, O.; Pack, G. On the antiquity of melanoma. Cancer 1966, 19, 607–610. [Google Scholar]
- National Cancer Institute. Skin Cancer Facts. Available online: http://www.skincancer.org (accessed on 3 October 2005).
- Haefner, B. Chemoprevention of cancer. Cancer Res. 2003, 45, 1–8. [Google Scholar]
- Fahmy, H.; Khalifa, S.; Konoshima, T.; Zjawiony, J.K. An improved synthesis of 7,8-epoxy-1,3,11-cembratriene-15R(α), 16-diol, a cembranoid of marine origin with a potent cancer chemopreventive activity. Mar. Drugs 2004, 2, 1–7. [Google Scholar]
- Fahmy, H.; Zjawiony, J.K.; Konoshima, T.; Tokuda, H.; Khan, S.; Khalifa, S. Potent skin cancer chemopreventing activity of some novel semi-synthesis cembranoids from marine sources. Mar. Drugs 2006, 4, 1–9. [Google Scholar]
- Kundoor, V.; Zhang, X.; Khalifa, S.; Fahmy, H.; Dwivedi, C. A possible mechanism of action of the chemopreventive effects of sarcotriol on skin tumor development in CD-1 mice. Mar. Drugs 2006, 4, 274–285. [Google Scholar]
- Fujiki, H.; Suganuma, M.; Takagi, K. Sarcophytol A and Its Analogs. In Cancer Preventive Activity in Phenolic Compounds in Food and Their Effects on Health II, Antioxidants and Cancer Prevention; Huang, M.T., Ho, C.T., Lee, C.Y., Eds.; American Chemical Society: Washington, DC, USA, 1992; pp. 380–387. [Google Scholar]
- Katsuyama, I.; Fahmy, H.; Zjawiony, J.K.; Khalifa, S.; Kilada, R.W.; Konoshima, T.; Takasaki, M.; Tokuda, H. Semisynthesis of new sarcophine derivatives with chemopreventive activity. J. Nat. Prod. 2002, 65, 1809–1814. [Google Scholar]
- Sawant, S.; Youssef, D.; Mayer, A.; Sylvester, P.; Wali, V.; Arant, M.; Sayed, K.E. Anticancer and anti-inflammatory sulfur-containing semisynthetic derivatives of sarcophine. Chem. Pharm. Bull. 2006, 54, 1119–1123. [Google Scholar]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar]
- Darnell, J.E. Validating STAT3 in cancer therapy. Nat. Med. 2005, 11, 595–596. [Google Scholar]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian caspases: Structure, activation, substrates and functions during apoptosis. Annu. Rev. Biochem. 1999, 68, 383–424. [Google Scholar]
- Thornberry, N.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316. [Google Scholar]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar]
- Bate, S.; Vousden, K.H. p53 in signaling checkpoint arrest in apoptosis. Curr. Opin. Genet. Dev. 1996, 6, 12–18. [Google Scholar]
- Ziemer, M.A.; Mason, A.; Carlson, D.M. Cell-free translations of proline-rich protein mRNAs. J. Biol. Chem. 1982, 257, 11176–11180. [Google Scholar]
- Black, M.M.; Speer, F.D. Effects of cancer chemotherapeutic agents on dehydrogenase activity of human cancer tissue in vitro. Am. J. Clin. Pathol. 1953, 23, 218–227. [Google Scholar]
- Altman, F.P. Tetrazolium salts and formazans. Prog. Histochem. 1977, 9, 1–56. [Google Scholar]
- Benchimol, S. p53-dependent pathways of apoptosis. Cell Death Diff. 2001, 8, 1049–1051. [Google Scholar]
- Hoffman, W.H.; Biade, S.; Zilfou, J.T.; Chen, J.; Murphy, M. Transcriptional repression of the anti-apoptotic surviving gene by wild type p53. J. Biol. Chem. 2002, 277, 3247–3257. [Google Scholar]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar]
- Bennett, M.; Macdonald, K.; Chan, S.W.; Luzio, J.P.; Simari, R.; Weissberg, P. Cell surface trafficking of Fas: A rapid mechanism of p53-mediated apoptosis. Science 1998, 282, 290–293. [Google Scholar]
- Boni, R.; Burg, G.; Doguoglu, A.; Ilg, E.C.; Schafer, B.W.; Muller, B.; Heizmann, C.W. Immunihistochemical localization of Ca2+ binding S100 proteins in normal human skin and melanocytic lesions. Br. J. Dermatol. 1977, 137, 39–43. [Google Scholar]
- Delphin, C.; Ronjat, M.; Deloulme, J.C.; Garin, G.; Debussche, L.; Highasimoto, Y.; Sakaguchi, K.; Baudier, J. Calcium-dependent interaction of S100B with the C-terminal domain of the tumor suppressor p53. J. Biol. Chem. 1999, 274, 10539–10544. [Google Scholar]
- Lin, J.; Yang, Q.; Yan, Z.; Markowitz, J.; Wilder, P.T.; Carrier, F.; Weber, D.J. Inhibiting S100B restores p53 levels in primary malignant melanoma cancer cells. J. Biol. Chem. 2004, 279, 34071–34077. [Google Scholar]
- Lin, J.; Blake, M.; Tang, C.; Zimmer, D.; Rustandi, R.R.; Weber, D.J.; Carrier, F. Inhibition of p53 transcriptional activity by the S100B calcium-binding protein. J. Biol. Chem. 2001, 276, 35037–35041. [Google Scholar]
- Lin, J.; Yang, Q.; Wilder, P.T.; Carrier, F.; Weber, D.J. The calcium-binding protein S100B down-regulates p53 and apoptosis in malignant melanoma. J. Biol. Chem. 2010, 285, 27487–27498. [Google Scholar]
- Ihle, J.N. STAT’s signal transducers and activators of transcription. Cell 1996, 84, 331–334. [Google Scholar]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. STAT3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar]
- Horvath, C.M.; Darnell, J.E. The state of the STAT’s: recent developments in the study of signal transduction to the nucleus. Curr. Opin. Cell Biol. 1997, 9, 233–239. [Google Scholar]
- Brivanlou, A.H.; Darnell, J.E., Jr. Signal transduction and the control gene expression. Science 2002, 295, 813–818. [Google Scholar]
- Coffer, P.J.; Koenderman, L.; de Groot, R.P. The role of STATs in myeloid differentiation and laukemia. Oncogene 2000, 19, 2511–2522. [Google Scholar]
- Quintanilla-Martinez, L.; Kremer, M.; Specht, K.; Calzada-Wack, J.; Nathrath, M.; Schaich, R.; Hofler, H.; Freud, F. Analysis of signal transducer and transcription 3 (STAT3) pathway in multiple myelonoma: STAT3 activation and cyclin D1 dysregulation are mutually exclusive events. Am. J. Pathol. 2003, 162, 1449–1461. [Google Scholar]
- Mora, L.B.; Buettner, R.; Seigne, J.; Diaz, J.; Ahmad, N.; Bowman, T.; Falcone, R.; Fairelough, R.; Cantor, A.; Moro-Cacho, C.; et al. Constitutive activation of STAT3 in human prostate tumors and cell lines: direct inhibition of STAT3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002, 62, 6659–6666. [Google Scholar] [PubMed]
- Norbury, C.; Nurse, P. Animal cell cycles and their control. Annu. Rev. Biochem. 1992, 61, 441–470. [Google Scholar]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks, and microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar]
- Kato, Y.T. Control of G1 progression by D-type cyclins; key event for cell proliferation. Leukemia 1997, 11, S347–S351. [Google Scholar]
- Sherr, C.J. Growth factor-regulated G1 cyclins. Stem Cells 1994, 12, S47–S55. [Google Scholar]
- Taules, M.; Rius, E.; Talaya, D.; Lopez-Garcia, A.; Bachs, O.; Agell, N. Calmodulin is essential for cyclin-dependent kinase 4 (Cdk4) activity and nuclear accumulation of cyclin D1-Cdk4 during G1. J. Biol. Chem. 1998, 273, 33279–33286. [Google Scholar]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar]
- Lazebnik, Y.A.; Kaufman, S.H.; Desnoyers, S.; Poirier, G.G.; Earnshaw, W.C. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties of ICE. Nature 1994, 371, 346–347. [Google Scholar]
- Cohen, G.M. Caspases: Executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar]
- Oliver, F.J.; dela Rubia, G.; Rolli, V.; Ruiz-Ruiz, M.C.; de Murcia, G.; Murcia, J.M. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis: lesson from an uncleavable mutant. J. Biol. Chem. 1998, 273, 33533–33539. [Google Scholar]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian caspases: structure, substrates and functions during apoptosis. Annu. Rev. Biochem. 1999, 68, 383–424. [Google Scholar]
- Chinnaiyan, A.M.; Dixit, V.M. The cell-death machine. Curr. Biol. 1996, 6, 555–562. [Google Scholar]
- Salvesen, G.S.; Dixit, V.M. Caspases: Intracellular signaling by proteolysis. Cell 1997, 91, 443–446. [Google Scholar]
- Green, D.; Kroemer, G. The central executioners of apoptosis: Caspases or mitochondria? Trends Cell Biol. 1998, 8, 267–271. [Google Scholar]
- Nicholson, D.W.; Thornberry, N.A. Caspases: Killer proteases. Trends Biochem. Sci. 1997, 22, 299–306. [Google Scholar]
- Debatin, K. Apoptosis pathway in cancer and cancer therapy. Cancer Immunol. Immunother. 2004, 53, 153–159. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Szymanski, P.T.; Kuppast, B.; Ahmed, S.A.; Khalifa, S.; Fahmy, H. Sarcophine-Diol, a Skin Cancer Chemopreventive Agent, Inhibits Proliferation and Stimulates Apoptosis in Mouse Melanoma B16F10 Cell Line. Mar. Drugs 2012, 10, 1-19. https://doi.org/10.3390/md10010001
Szymanski PT, Kuppast B, Ahmed SA, Khalifa S, Fahmy H. Sarcophine-Diol, a Skin Cancer Chemopreventive Agent, Inhibits Proliferation and Stimulates Apoptosis in Mouse Melanoma B16F10 Cell Line. Marine Drugs. 2012; 10(1):1-19. https://doi.org/10.3390/md10010001
Chicago/Turabian StyleSzymanski, Pawel T., Bhimanna Kuppast, Safwat A. Ahmed, Sherief Khalifa, and Hesham Fahmy. 2012. "Sarcophine-Diol, a Skin Cancer Chemopreventive Agent, Inhibits Proliferation and Stimulates Apoptosis in Mouse Melanoma B16F10 Cell Line" Marine Drugs 10, no. 1: 1-19. https://doi.org/10.3390/md10010001
APA StyleSzymanski, P. T., Kuppast, B., Ahmed, S. A., Khalifa, S., & Fahmy, H. (2012). Sarcophine-Diol, a Skin Cancer Chemopreventive Agent, Inhibits Proliferation and Stimulates Apoptosis in Mouse Melanoma B16F10 Cell Line. Marine Drugs, 10(1), 1-19. https://doi.org/10.3390/md10010001