
Computational Models on Pathological Redox Signalling Driven by Pregnancy: A Review

 ,
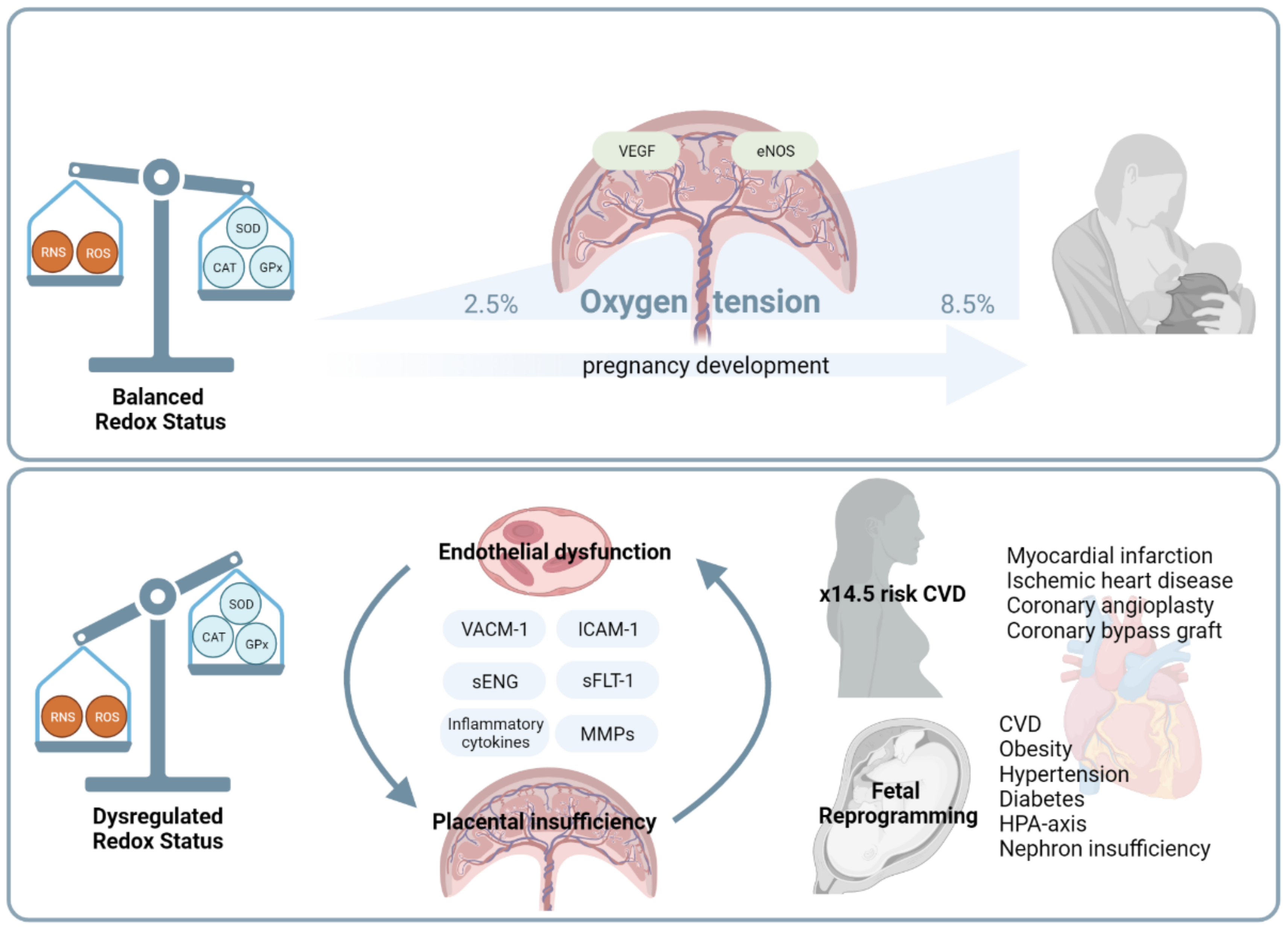
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Redox Signalling
1.2. Reactive Species
1.3. Antioxidant Cellular Systems
2. Redox Signalling in Pregnancy
2.1. Role of Oxidative Stress in Adverese Pregnancy Outcomes
2.2. Effects on Oxidative Damage to Foetal Health
2.3. Effects on Oxidative Damage in Pregnancy on Future Maternal Health
2.4. Oxidative Stress and Endothelial Dysfunction
2.5. Oxidative Stress, NOX, and Cardiovascular Disease
2.6. Therapeutic Interventions Targeting Oxidative Stress during Pregnancy
2.6.1. Vitamins
2.6.2. Selenium
2.6.3. Lifestyle Intervention
2.6.4. Sildenafil Citrate
2.6.5. Chronotherapy
2.6.6. Mitochondrial Antioxidant Therapy
2.7. Detection and Assessment of Oxidative Stress
2.7.1. Measurement of RONS
2.7.2. Assessment of Oxidative Damage
3. Current Computational Models—Enhanced Interpretation of Complexities of Oxidative Stress
Network-Based Redox Models
4. Future Perspectives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maritim, A.C.; Sanders, R.A.; Watkins, J.B., 3rd. Diabetes, oxidative stress, and antioxidants: A review. J. Biochem. Mol. Toxicol. 2003, 17, 24–38. [Google Scholar] [CrossRef]
- Mert, I.; Oruc, A.S.; Yuksel, S.; Cakar, E.S.; Buyukkagnıcı, U.; Karaer, A.; Danısman, N. Role of oxidative stress in preeclampsia and intrauterine growth restriction. J. Obstet. Gynaecol. Res. 2012, 38, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Rashid, C.; Bansal, A.; Simmons, R.A. Oxidative Stress, Intrauterine Growth Restriction, and Developmental Programming of Type 2 Diabetes. Physiology 2018, 33, 348–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyall, F.; Robson, S.C.; Bulmer, J.N. Spiral Artery Remodeling and Trophoblast Invasion in Preeclampsia and Fetal Growth Restriction: Relationship to Clinical Outcome. Hypertension 2013, 62, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ness, R.B.; Sibai, B.M. Shared and disparate components of the pathophysiologies of fetal growth restriction and preeclampsia. Am. J. Obstet. Gynecol. 2006, 195, 40–49. [Google Scholar] [CrossRef]
- Melchiorre, K.; Giorgione, V.; Thilaganathan, B. The placenta and preeclampsia: Villain or victim? Am. J. Obstet. Gynecol. 2022, 226, S954–S962. [Google Scholar] [CrossRef]
- Pillay, C.S.; Eagling, B.D.; Driscoll, S.R.; Rohwer, J. Quantitative measures for redox signaling. Free Radic. Biol. Med. 2016, 96, 290–303. [Google Scholar] [CrossRef]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, C.G.; Casalongué, C.A.; Simontacchi, M.; Garcia, B.M.; Foyer, C.H. Interactions between hormone and redox signalling pathways in the control of growth and cross tolerance to stress. Environ. Exp. Bot. 2013, 94, 73–88. [Google Scholar] [CrossRef]
- Brüne, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; Von Knethen, A.; Weigert, A. Redox Control of Inflammation in Macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarugi, P. From anchorage dependent proliferation to survival: Lessons from redox signalling. IUBMB Life 2008, 60, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Tabet, F.; Schiffrin, E.L. Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin. Exp. Pharmacol. Physiol. 2003, 30, 860–866. [Google Scholar] [CrossRef]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated Turnover of Nrf2 Is Determined by at Least Two Separate Protein Domains, the Redox-sensitive Neh2 Degron and the Redox-insensitive Neh6 Degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [Green Version]
- Haddad, J.J. Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell. Signal. 2002, 14, 879–897. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Zhao, R.-Z.; Jiang, S.; Zhang, L.; Yu, Z.-B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworakowski, R.; Anilkumar, N.; Zhang, M.; Shah, A. Redox signalling involving NADPH oxidase-derived reactive oxygen species. Biochem. Soc. Trans. 2006, 34, 960–964. [Google Scholar] [CrossRef] [PubMed]
- De Deken, X.; Wang, D.; Many, M.-C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of Two Human Thyroid cDNAs Encoding New Members of the NADPH Oxidase Family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzel, B.; John, M.; Klatt, P.; Böhme, E.; Mayer, B. Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem. J. 1992, 281, 627–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, C.; Fridovich, I. A mechanism for the production of ethylene from methional. The generation of the hydroxyl radical by xanthine oxidase. J. Biol. Chem. 1970, 245, 4641–4646. [Google Scholar] [CrossRef]
- Albertolle, M.E.; Guengerich, F.P. The relationships between cytochromes P450 and H2O2: Production, reaction, and inhibition. J. Inorg. Biochem. 2018, 186, 228–234. [Google Scholar] [CrossRef]
- Palmer, R.M.J.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [Green Version]
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussler, A.K.; Billiar, T.R. Inflammation, immunoregulation, and inducible nitric oxide synthase. J. Leukoc. Biol. 1993, 54, 171–178. [Google Scholar] [CrossRef]
- Bredt, D.S.; Hwang, P.M.; Snyder, S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 1990, 347, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Lei, H.; Qin, H.; Xia, Y. Molecular Mechanisms of Endothelial NO Synthase Uncoupling. Curr. Pharm. Des. 2014, 20, 3548–3553. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Are Reactive Sulfur Species the New Reactive Oxygen Species? Antioxid. Redox Signal. 2020, 33, 1125–1142. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Pérez, P.S.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.S.; Devalaraja, M.N.; Clair, D.K.S. Molecular Structure and Organization of the Human Manganese Superoxide Dismutase Gene. DNA Cell Biol. 1994, 13, 1127–1136. [Google Scholar] [CrossRef] [PubMed]
- Barra, D.; Schinina, M.E.; Simmaco, M.; Bannister, J.V.; Bannister, W.H.; Rotilio, G.; Bossa, F. The primary structure of human liver manganese superoxide dismutase. J. Biol. Chem. 1984, 259, 12595–12601. [Google Scholar] [CrossRef]
- Tainer, J.; Getzoff, E.D.; Richardson, J.; Richardson, D.C. Structure and mechanism of copper, zinc superoxide dismutase. Nature 1983, 306, 284–287. [Google Scholar] [CrossRef]
- Reid, T.J.; Murthy, M.R.; Sicignano, A.; Tanaka, N.; Musick, W.D.; Rossmann, M.G. Structure and heme environment of beef liver catalase at 2.5 A resolution. Proc. Natl. Acad. Sci. USA 1981, 78, 4767–4771. [Google Scholar] [CrossRef] [Green Version]
- Goyal, M.M.; Basak, A. Human catalase: Looking for complete identity. Protein Cell 2010, 1, 888–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, A. Thioredoxin structure and mechanism: Conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure 1995, 3, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Arnér, E.S.J.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. BioChem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staal, G.; Helleman, P.; De Wael, J.; Veeger, C. Purification and properties of an abrnomal glutathione reductace from human erythrocytes. Biochim. Biophys. Acta Enzym. 1969, 185, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; Van Goor, H.; Hillebrands, J.-L. Oxidative stress in placental pathology. Placenta 2018, 69, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannaerts, D.; Faes, E.; Cos, P.; Briedé, J.J.; Gyselaers, W.; Cornette, J.; Gorbanev, Y.; Bogaerts, A.; Spaanderman, M.; Van Craenenbroeck, E.; et al. Oxidative stress in healthy pregnancy and preeclampsia is linked to chronic inflammation, iron status and vascular function. PLoS ONE 2018, 13, e0202919. [Google Scholar] [CrossRef] [Green Version]
- Toboła-Wróbel, K.; Pietryga, M.; Dydowicz, P.; Napierała, M.; Brązert, J.; Florek, E. Association of Oxidative Stress on Pregnancy. Oxidative Med. Cell. Longev. 2020, 2020, 6398520. [Google Scholar] [CrossRef]
- Bolat, F.; Haberal, N.; Tunali, N.; Aslan, E.; Bal, N.; Tuncer, I. Expression of vascular endothelial growth factor (VEGF), hypoxia inducible factor 1 alpha (HIF-1α), and transforming growth factors β1 (TGFβ1) and β3 (TGFβ;3) in gestational trophoblastic disease. Pathol. Res. Pract. 2010, 206, 19–23. [Google Scholar] [CrossRef]
- Nanduri, J.; Vaddi, D.R.; Khan, S.A.; Wang, N.; Makarenko, V.; Semenza, G.L.; Prabhakar, N.R. HIF-1α Activation by Intermittent Hypoxia Requires NADPH Oxidase Stimulation by Xanthine Oxidase. PLoS ONE 2015, 10, e0119762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarello, D.I.; Abad, C.L.; Rojas, D.; Toledo, F.; Vazquez, C.; Mate, A.; Sobrevia, L.; Marín, R. Oxidative stress: Normal pregnancy versus preeclampsia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165354. [Google Scholar] [CrossRef] [PubMed]
- Sferruzzi-Perri, A.N.; Higgins, J.S.; Vaughan, O.R.; Murray, A.J.; Fowden, A.L. Placental mitochondria adapt developmentally and in response to hypoxia to support fetal growth. Proc. Natl. Acad. Sci. USA 2019, 116, 1621–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElwain, C.; Tuboly, E.; McCarthy, F.P.; McCarthy, C.M. Mechanisms of Endothelial Dysfunction in Pre-eclampsia and Gestational Diabetes Mellitus: Windows into Future Cardiometabolic Health? Front. Endocrinol. 2020, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Duhig, K.; Chappell, L.C.; Shennan, A.H. Oxidative stress in pregnancy and reproduction. Obstet. Med. 2016, 9, 113–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobrevia, L.; Ooi, L.; Ryan, S.; Steinert, J.R. Nitric Oxide: A Regulator of Cellular Function in Health and Disease. Oxidative Med. Cell. Longev. 2015, 2016, 9782346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visiedo, F.; Santos-Rosendo, C.; Mateos-Bernal, R.M.; Gil-Sánchez, M.D.M.; Bugatto, F.; Diosdado, M.A.; Segundo, C.; López-Tinoco, C. Characterization of NO-Induced Nitrosative Status in Human Placenta from Pregnant Women with Gestational Diabetes Mellitus. Oxidative Med. Cell. Longev. 2017, 2017, 5629341. [Google Scholar] [CrossRef] [PubMed]
- Sultana, Z.; Maiti, K.; Aitken, R.J.; Morris, J.; Dedman, L.; Smith, R. Oxidative stress, placental ageing-related pathologies and adverse pregnancy outcomes. Am. J. Reprod. Immunol. 2017, 77, e12653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manna, S.; McCarthy, C.; McCarthy, F. Placental Ageing in Adverse Pregnancy Outcomes: Telomere Shortening, Cell Senescence, and Mitochondrial Dysfunction. Oxidative Med. Cell. Longev. 2019, 2019, 3095383. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, P.; Cortijo, D.R.; Reyes-Hernández, C.G.; De Pablo, A.L.L.; González, M.C.; Arribas, S.M. Implication of Oxidative Stress in Fetal Programming of Cardiovascular Disease. Front. Physiol. 2018, 9, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J.P. Fetal origins of coronary heart disease. BMJ 1995, 311, 171–174. [Google Scholar] [CrossRef]
- Helle, E.; Priest, J.R. Maternal Obesity and Diabetes Mellitus as Risk Factors for Congenital Heart Disease in the Offspring. J. Am. Heart Assoc. 2020, 9, e011541. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli, M.; Chiefari, E.; Tocci, V.; Greco, E.; Foti, D.; Brunetti, A. Gestational diabetes: Implications for fetal growth, intervention timing, and treatment options. Curr. Opin. Pharmacol. 2021, 60, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Palinski, W. Effect of Maternal Cardiovascular Conditions and Risk Factors on Offspring Cardiovascular Disease. Circulation 2014, 129, 2066–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fall, C.H.D. Fetal Malnutrition and Long-Term Outcomes. In Nestlé Nutrition Workshop Series: Pediatric Program; S. Karger AG: Basel, Switzerland, 2013; Volume 74, pp. 11–25. [Google Scholar] [CrossRef] [Green Version]
- Evans, L.; Myatt, L. Sexual dimorphism in the effect of maternal obesity on antioxidant defense mechanisms in the human placenta. Placenta 2017, 51, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Ahn, Y.J.; Asmis, R. Sexual dimorphism in glutathione metabolism and glutathione-dependent responses. Redox Biol. 2020, 31, 101410. [Google Scholar] [CrossRef] [PubMed]
- Muralimanoharan, S.; Guo, C.; Myatt, L.; Maloyan, A. Sexual dimorphism in miR-210 expression and mitochondrial dysfunction in the placenta with maternal obesity. Int. J. Obes. 2015, 39, 1274–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orabona, R.; Sciatti, E.; Prefumo, F.; Vizzardi, E.; Bonadei, I.; Valcamonico, A.; Metra, M.; Frusca, T. Pre-eclampsia and heart failure: A close relationship. Ultrasound Obstet. Gynecol. 2018, 52, 297–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turbeville, H.R.; Sasser, J.M. Preeclampsia beyond pregnancy: Long-term consequences for mother and child. Am. J. Physiol. Physiol. 2020, 318, F1315–F1326. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Garcia, G.; Contag, S. Maternal Preeclampsia and Risk for Cardiovascular Disease in Offspring. Curr. Hypertens. Rep. 2014, 16, 475. [Google Scholar] [CrossRef]
- Ramsay, E.J.; Stewart, F.; Greer, I.A.; Sattar, N. Microvascular dysfunction: A link between pre-eclampsia and maternal coronary heart disease. BJOG 2003, 110, 1029–1031. [Google Scholar] [CrossRef] [PubMed]
- Tihtonen, K.M.; Kööbi, T.; Uotila, J.T. Arterial stiffness in preeclamptic and chronic hypertensive pregnancies. Eur. J. Obstet. Gynecol. Reprod. Biol. 2006, 128, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Free Radic. Biol. Med. 2017, 109, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y.; Satoh, A.; Chatterjee, S.; Toomre, D.K.; Chalouni, C.M.; Fulton, D.; Groszmann, R.J.; Shah, V.H.; Sessa, W.C. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc. Natl. Acad. Sci. USA 2006, 103, 19777–19782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hess, D.T.; Matsumoto, A.; Kim, S.-O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Erwin, P.A.; Lin, A.J.; Golan, D.E.; Michel, T. Receptor-regulated Dynamic S-Nitrosylation of Endothelial Nitric-oxide Synthase in Vascular Endothelial Cells. J. Biol. Chem. 2005, 280, 19888–19894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [Green Version]
- Endemann, D.H. Endothelial Dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef]
- Ma, S.; Ma, C.C.-H. Recent developments in the effects of nitric oxide-donating statins on cardiovascular disease through regulation of tetrahydrobiopterin and nitric oxide. Vasc. Pharmacol. 2014, 63, 63–70. [Google Scholar] [CrossRef]
- Durrant, J.R.; Seals, D.R.; Connell, M.L.; Russell, M.J.; Lawson, B.R.; Folian, B.J.; Donato, A.J.; Lesniewski, L.A. Voluntary wheel running restores endothelial function in conduit arteries of old mice: Direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down regulation of NADPH oxidase. J. Physiol. 2009, 587, 3271–3285. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.H.; Kader, K.N. Mechanisms of H2O2-Induced Oxidative Stress in Endothelial Cells Exposed to Physiologic Shear Stress. ASAIO J. 2007, 53, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Touyz, R.M. NADPH Oxidases, Reactive Oxygen Species, and Hypertension: Clinical implications and therapeutic possibilities. Diabetes Care 2008, 31 (Suppl. S2), S170–S180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, X.; Yu, L.; Liang, P.; Li, L.; Dai, X.; Zhou, B.; Wu, X.; Xu, H.; Fang, M.; Chen, Q.; et al. A crosstalk between chromatin remodeling and histone H3K4 methyltransferase complexes in endothelial cells regulates angiotensin II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2015, 82, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Serpillon, S.; Floyd, B.C.; Gupte, R.S.; George, S.; Kozicky, M.; Neito, V.; Recchia, F.; Stanley, W.; Wolin, M.S.; Gupte, S.A. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am. J. Physiol. Circ. Physiol. 2009, 297, H153–H162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortuño, A.; José, G.S.; Moreno, M.U.; Beloqui, O.; Díez, J.; Zalba, G. Phagocytic NADPH Oxidase Overactivity Underlies Oxidative Stress in Metabolic Syndrome. Diabetes 2006, 55, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassègue, B.; Clempus, R.E.; Szöcs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide Production and Expression of Nox Family Proteins in Human Atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Sen, S.; Iyer, C.; Meydani, S.N. Obesity during pregnancy alters maternal oxidant balance and micronutrient status. J. Perinatol. 2013, 34, 105–111. [Google Scholar] [CrossRef]
- Chappell, L.C.; Seed, P.T.; Briley, A.L.; Kelly, F.J.; Lee, R.; Hunt, B.J.; Parmar, K.; Bewley, S.; Shennan, A.H.; Steer, P.J.; et al. Effect of antioxidants on the occurrence of pre-eclampsia in women at increased risk: A randomised trial. Lancet 1999, 354, 810–816. [Google Scholar] [CrossRef]
- Hyppönen, E.; Cavadino, A.; Williams, D.; Fraser, A.; Vereczkey, A.; Fraser, W.D.; Bánhidy, F.; Lawlor, D.; Czeizel, A.E. Vitamin D and Pre-Eclampsia: Original Data, Systematic Review and Meta-Analysis. Ann. Nutr. Metab. 2013, 63, 331–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, R.D.; McCarthy, C.; McCarthy, F.; Kenny, L.C. Oxidative stress in pre-eclampsia; have we been looking in the wrong place? Pregnancy Hypertens. Int. J. Women Cardiovasc. Health 2017, 8, 1–5. [Google Scholar] [CrossRef]
- Duntas, L.H. Selenium and at-risk pregnancy: Challenges and controversies. Thyroid Res. 2020, 13, 16. [Google Scholar] [CrossRef]
- Hubalewska-Dydejczyk, A.; Duntas, L.; Gilis-Januszewska, A. Pregnancy, thyroid, and the potential use of selenium. Hormones 2019, 19, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasawara, K.T.; Nascimento, S.L.D.; Costa, M.L.; Surita, F.G.; Silva, J.L.P.E. Exercise and physical activity in the prevention of pre-eclampsia: Systematic review. Acta Obstet. Gynecol. Scand. 2012, 91, 1147–1157. [Google Scholar] [CrossRef]
- Dastjerdi, M.V.; Hosseini, S.; Bayani, L. Sildenafil citrate and uteroplacental perfusion in fetal growth restriction. J. Res. Med. Sci. 2012, 17, 632–636. [Google Scholar]
- Ferreira, R.D.D.S.; Negrini, R.; Bernardo, W.M.; Simões, R.; Piato, S. The effects of sildenafil in maternal and fetal outcomes in pregnancy: A systematic review and meta-analysis. PLoS ONE 2019, 14, e0219732. [Google Scholar] [CrossRef]
- Sharp, A.; Cornforth, C.; Jackson, R.; Harrold, J.; Turner, M.A.; Kenny, L.C.; Baker, P.N.; Johnstone, E.D.; Khalil, A.; Von Dadelszen, P.; et al. Maternal sildenafil for severe fetal growth restriction (STRIDER): A multicentre, randomised, placebo-controlled, double-blind trial. Lancet Child Adolesc. Health 2018, 2, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998, 280, 1564–1569. [Google Scholar] [CrossRef]
- Richards, J.; Diaz, A.N.; Gumz, M.L. Clock genes in hypertension. Blood Press. Monit. 2014, 19, 249–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala, D.E.; Ucieda, R.; Hermida, R.C. Chronotherapy with Low-Dose Aspirin for Prevention of Complications in Pregnancy. Chronobiol. Int. 2012, 30, 260–279. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, L.; D’Angelo, G.; Manti, S.; Reiter, R.J.; Gitto, E. Potential Utility of Melatonin in Preeclampsia, Intrauterine Fetal Growth Retardation, and Perinatal Asphyxia. Reprod. Sci. 2016, 23, 970–977. [Google Scholar] [CrossRef]
- Hobson, S.; Gurusinghe, S.; Lim, R.; Alers, N.O.; Miller, S.L.; Kingdom, J.C.; Wallace, E.M. Melatonin improves endothelial function in vitro and prolongs pregnancy in women with early-onset preeclampsia. J. Pineal Res. 2018, 65, e12508. [Google Scholar] [CrossRef] [PubMed]
- Adlam, V.J.; Harrison, J.; Porteous, C.M.; James, A.M.; Smith, R.A.J.; Murphy, M.; Sammut, I. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Cochemé, H.M.; Logan, A.; Prime, T.A.; Abakumova, I.; Quin, C.; McQuaker, S.J.; Patel, J.V.; Fearnley, I.M.; James, A.M.; Porteous, C.M.; et al. Using the mitochondria-targeted ratiometric mass spectrometry probe MitoB to measure H2O2 in living Drosophila. Nat. Protoc. 2012, 7, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Salin, K.; Auer, S.K.; Villasevil, E.M.; Anderson, G.J.; Cairns, A.; Mullen, W.; Hartley, R.; Metcalfe, N. Using the MitoB method to assess levels of reactive oxygen species in ecological studies of oxidative stress. Sci. Rep. 2017, 7, 41228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prime, T.A.; Forkink, M.; Logan, A.; Finichiu, P.G.; McLachlan, J.; Pun, P.B.L.; Koopman, W.J.; Larsen, L.; Latter, M.J.; Smith, R.A.; et al. A ratiometric fluorescent probe for assessing mitochondrial phospholipid peroxidation within living cells. Free Radic. Biol. Med. 2012, 53, 544–553. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.M.; Kenny, L.C. Therapeutically targeting mitochondrial redox signalling alleviates endothelial dysfunction in preeclampsia. Sci. Rep. 2016, 6, 32683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cochemé, H.M.; Murphy, M.P.; Dominiczak, A.F. Mitochondria-Targeted Antioxidant MitoQ10 Improves Endothelial Function and Attenuates Cardiac Hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Kerr, D.S. Treatment of mitochondrial electron transport chain disorders: A review of clinical trials over the past decade. Mol. Genet. Metab. 2010, 99, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Ronco, P.; Debiec, H. Molecular Pathogenesis of Membranous Nephropathy. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 287–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramaniam, S.; Yaplito-Lee, J. Riboflavin metabolism: Role in mitochondrial function. J. Transl. Genet. Genom. 2020, 4, 285–306. [Google Scholar] [CrossRef]
- Penn, A.; Lee, J.W.; Thuillier, P.; Wagner, M.; Maclure, K.M.; Menard, M.R.; Hall, L.D.; Kennaway, N.G. MELAS syndrome with mitochondrial tRNA Leu [UUR] mutation: Correlation of clinical state, nerve conduction, and muscle 31P magnetic resonance spectroscopy during treatment with nicotinamide and riboflavin. Neurology 1992, 42, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.M.; Abeling, N.G.G.M.; Ijlst, L.; Knoester, H.; Van Der Pol, W.L.; Stroomer, A.E.M.; Wanders, R.J.; Visser, G.; Wijburg, F.A.; Duran, M.; et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: A new inborn error of metabolism with potential treatment. J. Inherit. Metab. Dis. 2010, 34, 159–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischner, C.; Wenz, T. Keep the fire burning: Current avenues in the quest of treating mitochondrial disorders. Mitochondrion 2015, 24, 32–49. [Google Scholar] [CrossRef]
- Luo, K.; Yu, J.H.; Quan, Y.; Shin, Y.J.; Lee, K.E.; Kim, H.L.; Ko, E.J.; Chung, B.H.; Lim, S.W.; Yang, C.W. Therapeutic potential of coenzyme Q10 in mitochondrial dysfunction during tacrolimus-induced beta cell injury. Sci. Rep. 2019, 9, 7995. [Google Scholar] [CrossRef]
- Ostojic, S.M. Plasma creatine as a marker of mitochondrial dysfunction. Med. Hypotheses 2018, 113, 52–53. [Google Scholar] [CrossRef]
- Kazak, L.; Cohen, P. Creatine metabolism: Energy homeostasis, immunity and cancer biology. Nat. Rev. Endocrinol. 2020, 16, 421–436. [Google Scholar] [CrossRef]
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxidative Med. Cell. Longev. 2019, 2019, 8409329. [Google Scholar] [CrossRef] [Green Version]
- Di Nicola, S.; Proietti, S.; Cucina, A.; Bizzarri, M.; Fuso, A. Alpha-Lipoic Acid Downregulates IL-1β and IL-6 by DNA Hypermethylation in SK-N-BE Neuroblastoma Cells. Antioxidants 2017, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Morillon, A.-C.; Williamson, R.D.; Baker, P.N.; Kell, D.B.; Kenny, L.C.; English, J.A.; McCarthy, F.P.; McCarthy, C. Effect of L-Ergothioneine on the metabolic plasma profile of the RUPP rat model of pre-eclampsia. PLoS ONE 2020, 15, e0230977. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Ottosson, F.; Hellstrand, S.; Ericson, U.; Orho-Melander, M.; Fernandez, C.; Melander, O. Ergothioneine is associated with reduced mortality and decreased risk of cardiovascular disease. Heart 2019, 106, 691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, R.D.; McCarthy, F.P.; Manna, S.; Groarke, E.; Kell, D.B.; Kenny, L.C.; McCarthy, C.M. L-(+)-Ergothioneine Significantly Improves the Clinical Characteristics of Preeclampsia in the Reduced Uterine Perfusion Pressure Rat Model. Hypertension 2020, 75, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, A.M.; Camm, E.J.; Sferruzzi-Perri, A.; Ashmore, T.J.; Yung, H.-W.; Cindrova-Davies, T.; Spiroski, A.-M.; Sutherland, M.; Logan, A.; Austin-Williams, S.; et al. Placental Adaptation to Early-Onset Hypoxic Pregnancy and Mitochondria-Targeted Antioxidant Therapy in a Rodent Model. Am. J. Pathol. 2018, 188, 2704–2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aljunaidy, M.M.; Morton, J.S.; Kirschenman, R.; Phillips, T.; Case, C.P.; Cooke, C.-L.M.; Davidge, S.T. Maternal treatment with a placental-targeted antioxidant (MitoQ) impacts offspring cardiovascular function in a rat model of prenatal hypoxia. Pharmacol. Res. 2018, 134, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Miller, E.W.; Tulyanthan, O.; Isacoff, E.; Chang, C.J. Molecular imaging of hydrogen peroxide produced for cell signaling. Nat. Chem. Biol. 2007, 3, 263–267. [Google Scholar] [CrossRef]
- Itoh, Y.; Ma, F.H.; Hoshia, H.; Okaa, M.; Nodaa, K.; Ukaia, Y.; Kojimab, H.; Naganob, T.; Todaa, N. Determination and Bioimaging Method for Nitric Oxide in Biological Specimens by Diaminofluorescein Fluorometry. Anal. Biochem. 2000, 287, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Vasquez-Vivar, J.; Kalyanaraman, B. Detection of 2-hydroxyethidium in cellular systems: A unique marker product of superoxide and hydroethidine. Nat. Protoc. 2007, 3, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.; Touyz, R.M.; Zweier, J.L.; Dikalov, S.; Chilian, W.; Chen, Y.-R.; Harrison, D.G.; Bhatnagar, A. Measurement of Reactive Oxygen Species, Reactive Nitrogen Species, and Redox-Dependent Signaling in the Cardiovascular System. Circ. Res. 2016, 119, e39–e75. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Summers, F.A.; Mason, R.P. Photooxidation of Amplex red to resorufin: Implications of exposing the Amplex red assay to light. Free Radic. Biol. Med. 2012, 53, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paital, B. A Modified Fluorimetric Method for Determination of Hydrogen Peroxide Using Homovanillic Acid Oxidation Principle. BioMed Res. Int. 2014, 2014, 342958. [Google Scholar] [CrossRef]
- Chen, Y.; Zhong, Q.; Wang, Y.; Yuan, C.; Qin, X.; Xu, Y. Colorimetric detection of hydrogen peroxide and glucose by exploiting the peroxidase-like activity of papain. RSC Adv. 2019, 9, 16566–16570. [Google Scholar] [CrossRef] [Green Version]
- Tarpey, M.M.; Wink, D.A.; Grisham, M.B. Methods for detection of reactive metabolites of oxygen and nitrogen: In vitro and in vivo considerations. Am. J. Physiol. Integr. Comp. Physiol. 2004, 286, R431–R444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, C.; Sueyoshi, Y.; Ema, M.; Mori, Y.; Takaishi, K.; Hisatomi, H. Induction of oxidative stress by anticancer drugs in the presence and absence of cells. Oncol. Lett. 2017, 14, 6066–6070. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React. Oxyg. Species 2016, 2, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and Methods to Measure Oxidative Stress in Clinical Samples: Research Applications in the Cancer Field. Oxidative Med. Cell. Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef] [Green Version]
- Gryglewski, R.; Moncada, S.; Palmer, R. Bioassay of prostacyclin and endothelium-derived relaxing factor (EDRF) from porcine aortic endothelial cells. J. Cereb. Blood Flow Metab. 1986, 87, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Isbell, T.S.; Koenitzer, J.R.; Crawford, J.H.; White, C.; Kraus, D.W.; Patel, R.P. Assessing NO-Dependent Vasodilatation Using Vessel Bioassays at Defined Oxygen Tensions. Methods Enzymol. 2005, 396, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.; Davies, M.J.; Grune, T. Determination of protein carbonyls in plasma, cell extracts, tissue homogenates, isolated proteins: Focus on sample preparation and derivatization conditions. Redox Biol. 2015, 5, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Deng, Y.; Yang, X.; Xue, H.; Lang, Y. The Relationship Between Protein S-Nitrosylation and Human Diseases: A Review. Neurochem. Res. 2020, 45, 2815–2827. [Google Scholar] [CrossRef]
- Ichihara, K.; Fukubayashi, Y. Preparation of fatty acid methyl esters for gas-liquid chromatography. J. Lipid Res. 2010, 51, 635–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, H.; Smith, C.; Horning, E.; Mitchell, J. High-performance liquid chromatography and gas chromatography-mass spectrometry determination of specific lipid peroxidation products in vivo. Anal. Biochem. 1983, 130, 431–436. [Google Scholar] [CrossRef]
- Patton, G.M.; Fasulo, J.M.; Robins, S.J. Analysis of lipids by high performance liquid chromatography: Part I. J. Nutr. Biochem. 1990, 1, 493–500. [Google Scholar] [CrossRef]
- Ferreira, R.C.; Fragoso, M.B.T.; Tenório, M.C.D.S.; Martins, A.S.D.P.; Borbely, A.U.; Moura, F.A.; Goulart, M.O.F.; De Oliveira, A.C.M. Biomarkers of placental redox imbalance in pregnancies with preeclampsia and consequent perinatal outcomes. Arch. Biochem. Biophys. 2020, 691, 108464. [Google Scholar] [CrossRef] [PubMed]
- Karatas, F.; Karatepe, M.; Baysar, A. Determination of free malondialdehyde in human serum by high-performance liquid chromatography. Anal. Biochem. 2002, 311, 76–79. [Google Scholar] [CrossRef]
- Fisk, H.L.; West, A.L.; Childs, C.E.; Burdge, G.C.; Calder, P.C. The Use of Gas Chromatography to Analyze Compositional Changes of Fatty Acids in Rat Liver Tissue during Pregnancy. J. Vis. Exp. 2014, 85, e51445. [Google Scholar] [CrossRef] [Green Version]
- Enoiu, M.; Wellman, M.; Leroy, P.; Ziegler, J.-M.; Mitrea, N.; Siest, G. Gas and liquid chromatography-mass spectrometry of aldehydic products from lipid peroxidation. Analusis 2000, 28, 285–290. [Google Scholar] [CrossRef]
- Cháfer-Pericás, C.; Rahkonen, L.; Sánchez-Illana, A.; Kuligowski, J.; Torres-Cuevas, I.; Cernada, M.; Cubells, E.; Nuñez-Ramiro, A.; Andersson, S.; Vento, M.; et al. Ultra high performance liquid chromatography coupled to tandem mass spectrometry determination of lipid peroxidation biomarkers in newborn serum samples. Anal. Chim. Acta 2015, 886, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative Stress: An Essential Factor in the Pathogenesis of Gastrointestinal Mucosal Diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awooda, H.A. Pathophysiology of Cerebral Ischemia: Role of Oxidative/Nitrosative Stress. J. Biosci. Med. 2019, 7, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Figueroa-González, G.; Pérez-Plasencia, C. Strategies for the evaluation of DNA damage and repair mechanisms in cancer. Oncol. Lett. 2017, 13, 3982–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galson, S.; Austin, C.P.; Khandekar, E.; Hudson, L.D.; Di Masi, J.A.; Califf, R.; Wagner, J.A. The failure to fail smartly. Nat. Rev. Drug Discov. 2021, 20, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.J.; Carpenter, D.; Lauffenburger, J.; Wang, B.; Franklin, J.M.; Kesselheim, A. Failure of Investigational Drugs in Late-Stage Clinical Development and Publication of Trial Results. JAMA Intern. Med. 2016, 176, 1826–1833. [Google Scholar] [CrossRef] [PubMed]
- Kaye, D.K. The moral imperative to approve pregnant women’s participation in randomized clinical trials for pregnancy and newborn complications. Philos. Ethic Humanit. Med. 2019, 14, 11. [Google Scholar] [CrossRef] [Green Version]
- Tazhigulov, R.N.; Gurunathan, P.; Kim, Y.; Slipchenko, L.V.; Bravaya, K.B. Polarizable embedding for simulating redox potentials of biomolecules. Phys. Chem. Chem. Phys. 2019, 21, 11642–11650. [Google Scholar] [CrossRef]
- Brodland, G.W. How computational models can help unlock biological systems. Semin. Cell Dev. Biol. 2015, 47–48, 62–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemis, L.M. Computational probability applications. In Proceedings of the 2014 Winter Simulation Conference, Savannah, GA, USA, 7–10 December 2014; pp. 51–65. [Google Scholar]
- Guimera, A.M.; Shanley, D.P.; Proctor, C.J. Modelling the role of redox-related mechanisms in musculoskeletal ageing. Free Radic. Biol. Med. 2019, 132, 11–18. [Google Scholar] [CrossRef]
- Markevich, N.I.; Hoek, J.B. Computational modeling analysis of mitochondrial superoxide production under varying substrate conditions and upon inhibition of different segments of the electron transport chain. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 656–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.D.; Greenstein, J.L.; Cortassa, S.; O’Rourke, B.; Winslow, R.L. A Computational Model of Reactive Oxygen Species and Redox Balance in Cardiac Mitochondria. Biophys. J. 2013, 105, 1045–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haack, F.; Lemcke, H.; Ewald, R.; Rharass, T.; Uhrmacher, A.M. Spatio-temporal Model of Endogenous ROS and Raft-Dependent WNT/Beta-Catenin Signaling Driving Cell Fate Commitment in Human Neural Progenitor Cells. PLoS Comput. Biol. 2015, 11, e1004106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, G.; Gran, M.A.; Bagchi, P.; Kemp, M.L. Dynamic Redox Regulation of IL-4 Signaling. PLoS Comput. Biol. 2015, 11, e1004582. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, T.E.; Barnett, K.J.; Wallis, L.; Hanneman, W.H. A multimethod computational simulation approach for investigating mitochondrial dynamics and dysfunction in degenerative aging. Aging Cell 2017, 16, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Kembro, J.M.; Cortassa, S.; Lloyd, D.; Sollott, S.J.; Aon, M.A. Mitochondrial chaotic dynamics: Redox-energetic behavior at the edge of stability. Sci. Rep. 2018, 8, 15422. [Google Scholar] [CrossRef] [PubMed]
- Del Olmo, M.; Kramer, A.; Herzel, H. A Robust Model for Circadian Redox Oscillations. Int. J. Mol. Sci. 2019, 20, 2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Aon, M.A.; Almas, T.; Cortassa, S.; Winslow, R.L.; O’Rourke, B. A Reaction-Diffusion Model of ROS-Induced ROS Release in a Mitochondrial Network. PLoS Comput. Biol. 2010, 6, e1000657. [Google Scholar] [CrossRef] [Green Version]
- Sridharan, S.; Layek, R.; Datta, A.; Venkatraj, J. Boolean modeling and fault diagnosis in oxidative stress response. BMC Genom. 2012, 13 (Suppl. S6), S4. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.R.; Shanley, D.P. Computational modelling of the regulation of Insulin signalling by oxidative stress. BMC Syst. Biol. 2013, 7, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padayachee, L.; Rohwer, J.M.; Pillay, C.S. The thioredoxin redox potential and redox charge are surrogate measures for flux in the thioredoxin system. Arch. Biochem. Biophys. 2020, 680, 108231. [Google Scholar] [CrossRef] [PubMed]
- Gerken, M.; Kakorin, S.; Chibani, K.; Dietz, K.-J. Computational simulation of the reactive oxygen species and redox network in the regulation of chloroplast metabolism. PLoS Comput. Biol. 2020, 16, e1007102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, W.; Young, D.A.; Rowan, A.D.; Xu, X.; Cawston, T.E.; Proctor, C.J. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 2016, 75, 449–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazein, A.; Ostaszewski, M.; Kuperstein, I.; Watterson, S.; Le Novère, N.; Lefaudeux, D.; De Meulder, B.; Pellet, J.; Balaur, I.; Saqi, M.; et al. Systems medicine disease maps: Community-driven comprehensive representation of disease mechanisms. NPJ Syst. Biol. Appl. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2010, 12, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Gupta, S.K.; Perretti, M.; Godson, C.; Brennan, E.; Li, Y.; Soehnlein, O.; Shimizu, T.; Werz, O.; Chiurchiù, V.; et al. The Atlas of Inflammation Resolution (AIR). Mol. Asp. Med. 2020, 74, 100894. [Google Scholar] [CrossRef] [PubMed]
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish-Bowden, A. The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Correia, Y.; Scheel, J.; Gupta, S.; Wang, K. Placental mitochondrial function as a driver of angiogenesis and placental dysfunction. Biol. Chem. 2021, 402, 887–909. [Google Scholar] [CrossRef]
- Karlstaedt, A.; Khanna, R.; Thangam, M.; Taegtmeyer, H. Glucose 6-Phosphate Accumulates via Phosphoglucose Isomerase Inhibition in Heart Muscle. Circ. Res. 2020, 126, 60–74. [Google Scholar] [CrossRef] [PubMed]
- D’Annibale, V.; Nardi, A.N.; Amadei, A.; D’Abramo, M. Theoretical Characterization of the Reduction Potentials of Nucleic Acids in Solution. J. Chem. Theory Comput. 2021, 17, 1301–1307. [Google Scholar] [CrossRef]
- Yu, J.; Horsley, J.R.; Abell, A.D. Unravelling electron transfer in peptide-cation complexes: A model for mimicking redox centres in proteins. Phys. Chem. Chem. Phys. 2020, 22, 8409–8417. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, J.; Bras, N.; Fernandes, P.; Rangel, M.; Ramos, M.J. A computational study on the redox properties and binding affinities of iron complexes of hydroxypyridinones. J. Mol. Model. 2019, 25, 172. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.J.; Smolko, C.M.; Janes, K.A. Computational Models of Reactive Oxygen Species as Metabolic Byproducts and Signal-Transduction Modulators. Front. Pharmacol. 2016, 7, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touré, V.; Zobolas, J.; Kuiper, M.; Vercruysse, S. Causal Builder: Bringing the MI2CAST causal interaction annotation standard to the curator. Database 2021, 2021, baaa107. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manna, S.; Ruano, C.S.M.; Hegenbarth, J.-C.; Vaiman, D.; Gupta, S.; McCarthy, F.P.; Méhats, C.; McCarthy, C.; Apicella, C.; Scheel, J. Computational Models on Pathological Redox Signalling Driven by Pregnancy: A Review. Antioxidants 2022, 11, 585. https://doi.org/10.3390/antiox11030585
Manna S, Ruano CSM, Hegenbarth J-C, Vaiman D, Gupta S, McCarthy FP, Méhats C, McCarthy C, Apicella C, Scheel J. Computational Models on Pathological Redox Signalling Driven by Pregnancy: A Review. Antioxidants. 2022; 11(3):585. https://doi.org/10.3390/antiox11030585
Chicago/Turabian StyleManna, Samprikta, Camino S. M. Ruano, Jana-Charlotte Hegenbarth, Daniel Vaiman, Shailendra Gupta, Fergus P. McCarthy, Céline Méhats, Cathal McCarthy, Clara Apicella, and Julia Scheel. 2022. "Computational Models on Pathological Redox Signalling Driven by Pregnancy: A Review" Antioxidants 11, no. 3: 585. https://doi.org/10.3390/antiox11030585
APA StyleManna, S., Ruano, C. S. M., Hegenbarth, J.-C., Vaiman, D., Gupta, S., McCarthy, F. P., Méhats, C., McCarthy, C., Apicella, C., & Scheel, J. (2022). Computational Models on Pathological Redox Signalling Driven by Pregnancy: A Review. Antioxidants, 11(3), 585. https://doi.org/10.3390/antiox11030585