_Kim.png)
Total Flavones of Rhododendron Protect Against Ischemic Cerebral Injury by Regulating the Phosphorylation of the RhoA-ROCK2 Pathway via Endothelial-Derived H2S

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Drugs
2.2. Animals
2.3. Isolation and Cultivation of ECs
2.4. H/R Injury Model
2.5. Cultivation of Hippocampal Neurons
2.6. Co-Culture System
2.7. Determination of Cell Viability and Biochemical Measurement
2.8. Western Blotting
2.9. Vessel Experiment
2.10. Cerebral I/R Model
2.11. Determination of Blood Flow in Brain Tissue
2.12. Morris Water Maze Assay
2.13. Open Field Test
2.14. Randomized Block Design
2.15. Statistical Analysis
3. Results
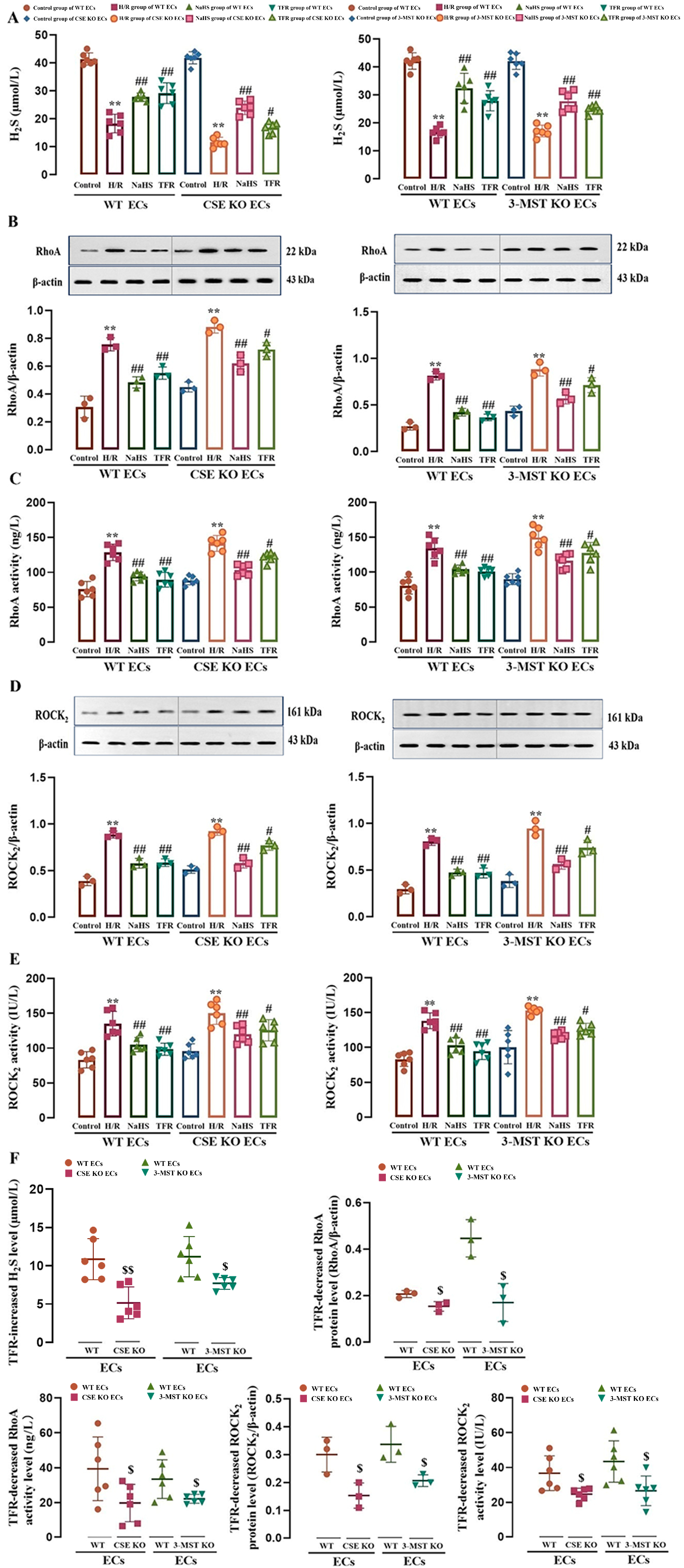
3.1. ROCK2-Mediated Inhibition in CSE and 3-MST Expressions and H2S Production in Mouse Cerebrovascular ECs
3.2. Effect of CSE or 3-MST KO on TFR Promoting H2S Production and Inhibiting the RhoA-ROCK2 Pathway in Mouse or Rat Cerebrovascular ECs Subjected to H/R Injury
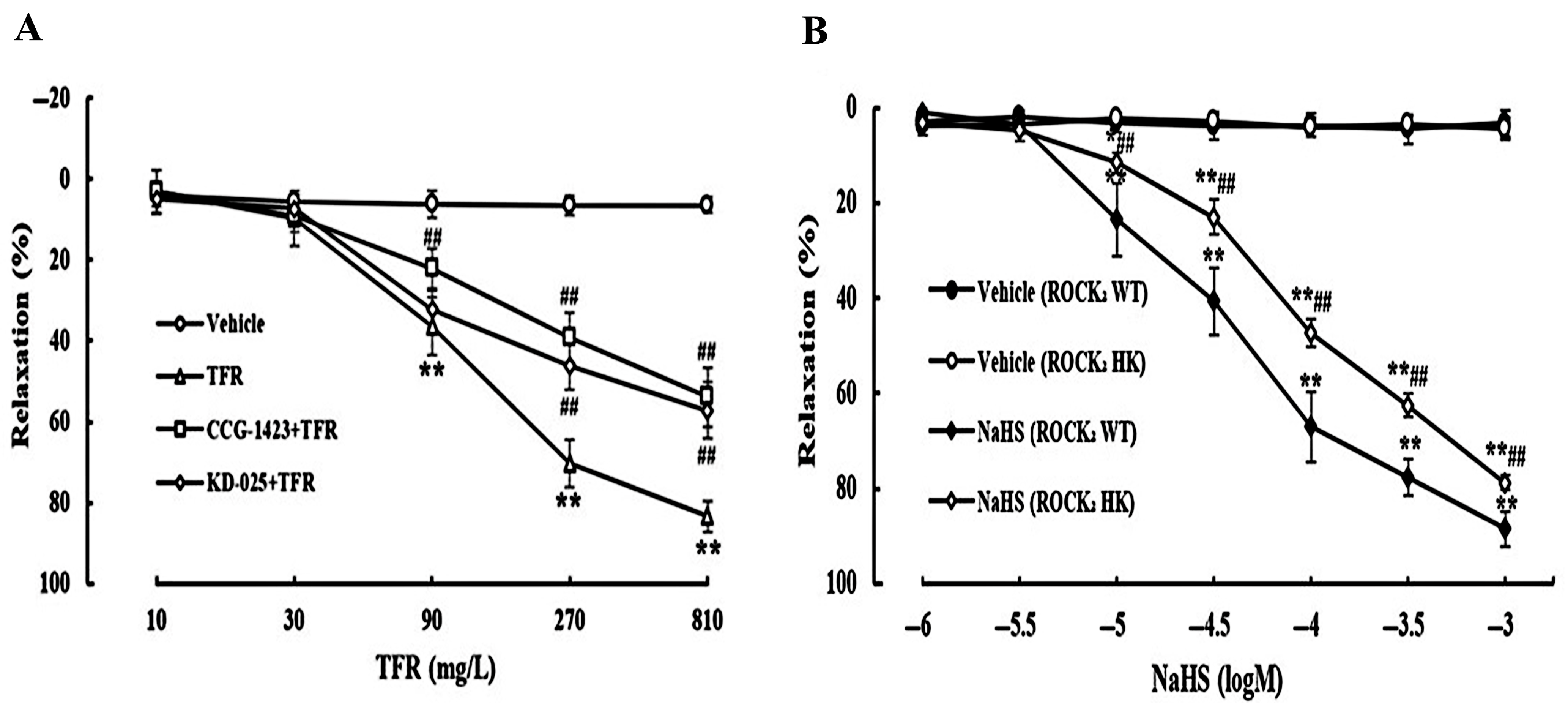
3.3. Effects of CCG-1423 and KD-025 on the Cerebral Vasodilation of TFR
3.4. Protective Effects of TFR Against Cerebral I/R Injury in WT and ROCK2 HK Mice
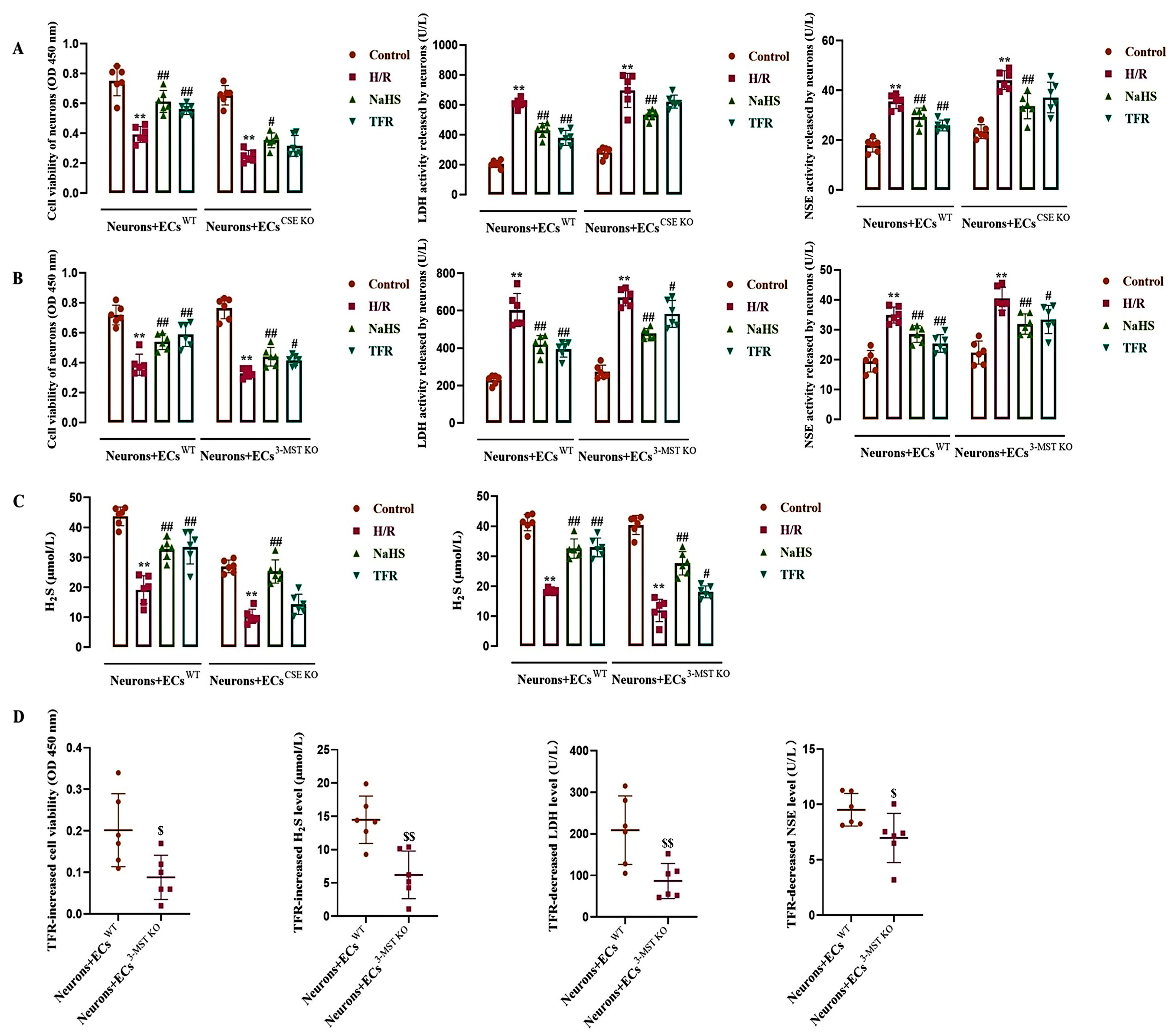
3.5. Effect of TFR on H/R Injury in the ECs-Co-Cultured Neurons and H2S Level in the Co-Cultured Medium
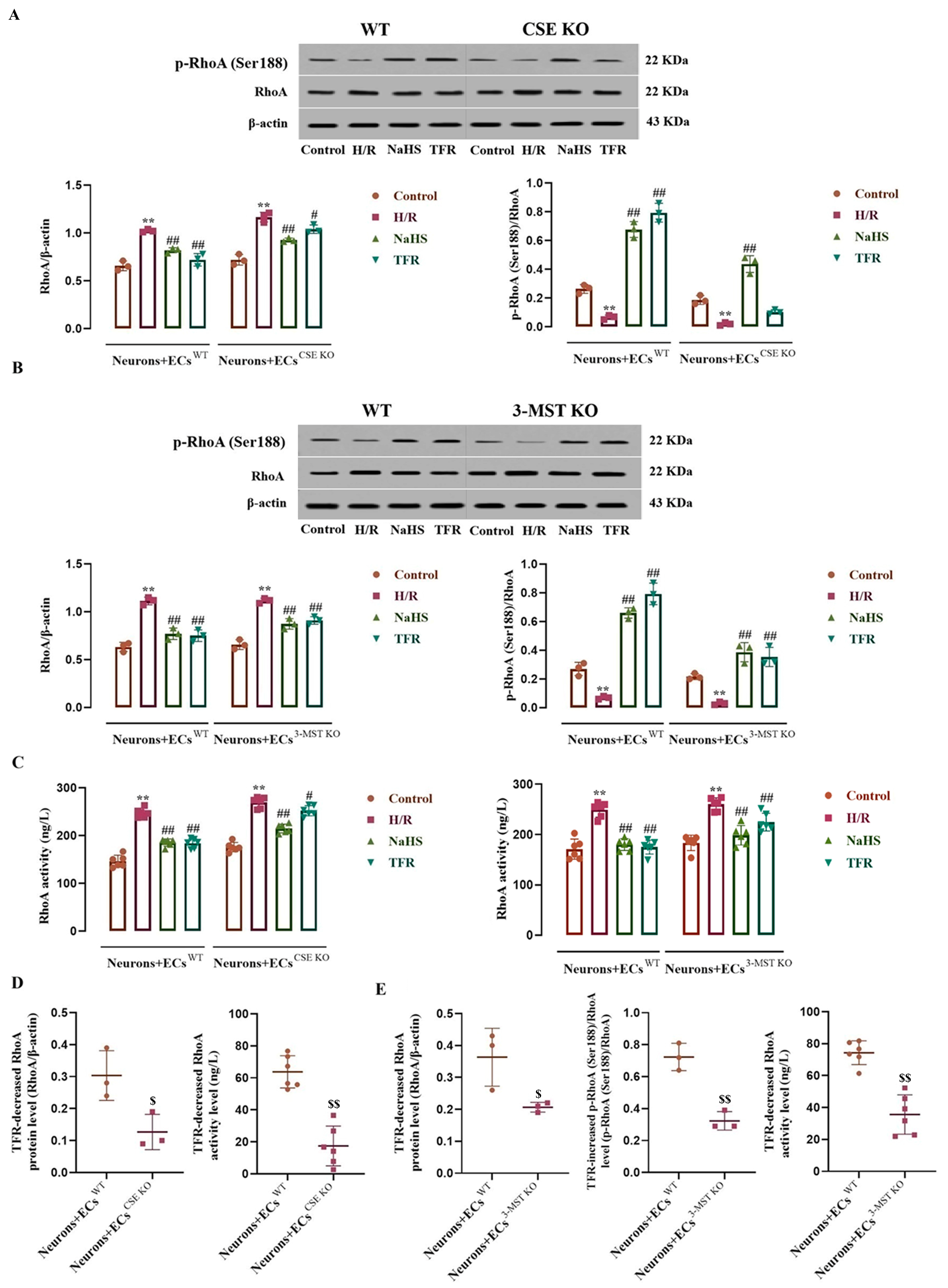
3.6. Impact of CSE or 3-MST KO in the ECs on the Effects of TFR on RhoA Expression, Activity, and Phosphorylation in the Co-Cultured Neurons Subjected to H/R Injury
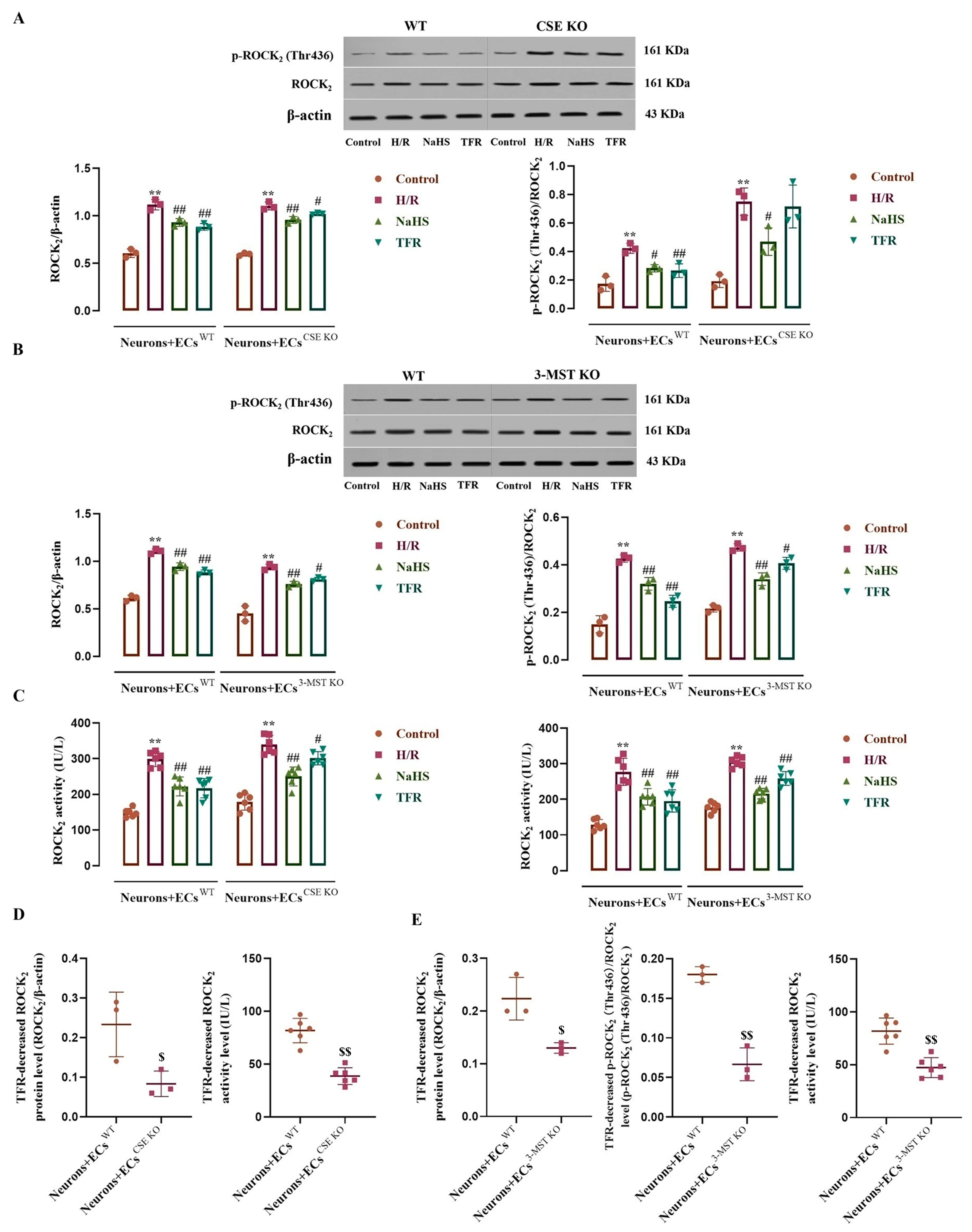
3.7. Effects of CSE or 3-MST KO in the ECs on TFR Inhibiting the H/R Injury-Increased ROCK2 Expression, Activity, and Phosphorylation in the Co-Cultured Neurons
3.8. Effects of Endothelial-Derived H2S on H/R Injury and the RhoA-ROCK2 Pathway in the Co-Cultured Neurons
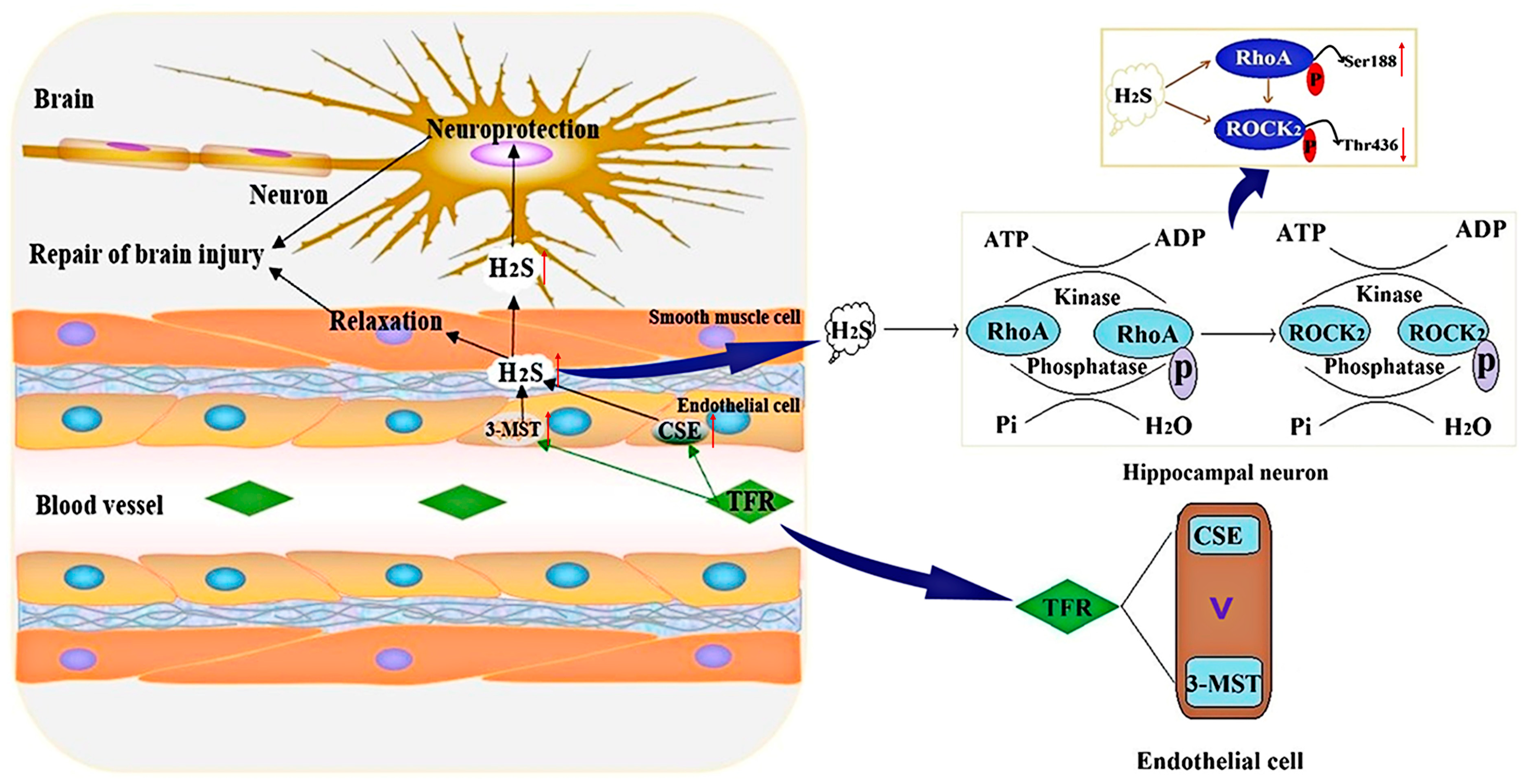
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3-MST | 3-mercaptopyruvate sulfurtransferase |
| CBF | cerebral blood flow |
| CBS | cystathionine-β-synthase |
| CSE | cystathionine-γ-lyase |
| ECs | endothelial cells |
| H/R | hypoxia/reoxygenation |
| H2S | hydrogen sulfide |
| HK | heterozygote knockout |
| I/R | ischemia/reperfusion |
| KO | knockout |
| LDH | lactate dehydrogenase |
| NSE | neuron-specific enolase |
| ROCK | rho kinase |
| TFR | total flavones of Rhododendron |
| WT | wild-type |
Appendix A. In Vivo Animal Experimental Methods
Appendix A.1. Experimental Animals
Appendix A.2. Experimental Design
Appendix A.3. Inclusion and Exclusion Criteria
Appendix A.4. Blinding
Appendix A.5. Outcome Measures
References
- Stocchetti, N.; Zanier, E.R. Chronic impact of traumatic brain injury on outcome and quality of life: A narrative review. Crit. Care 2016, 20, 148. [Google Scholar] [CrossRef]
- Chen, B.; Jin, W. A comprehensive review of stroke-related signaling pathways and treatment in western medicine and traditional Chinese medicine. Front. Neurosci. 2023, 17, 1200061. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chen, J.; Shen, J. Herbal medicines for ischemic stroke: Combating inflammation as therapeutic targets. J. Neuroimmune Pharmacol. 2014, 9, 313–339. [Google Scholar] [CrossRef]
- Pan, S.Y.; Zhou, S.F.; Gao, S.H.; Yu, Z.L.; Zhang, S.F.; Tang, M.K.; Sun, J.N.; Ma, D.L.; Han, Y.F.; Fong, W.F.; et al. New Perspectives on How to Discover Drugs from Herbal Medicines: CAM's Outstanding Contribution to Modern Therapeutics. Evid. Based Complement. Altern. Med. 2013, 2013, 627375. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Xu, H.H.; Chen, X.L.; Hu, H.R.; Hu, K.M.; Chen, Z.W.; He, G.W. Total Flavone of Rhododendron Improves Cerebral Ischemia Injury by Activating Vascular TRPV4 to Induce Endothelium-Derived Hyperpolarizing Factor-Mediated Responses. Evid. Based Complement. Altern. Med. 2018, 2018, 8919867. [Google Scholar] [CrossRef]
- Han, J.; He, G.W.; Chen, Z.W. Protective Effect and Mechanism of Total Flavones from Rhododendron simsii Planch on Endothelium-Dependent Dilatation and Hyperpolarization in Cerebral Ischemia-Reperfusion and Correlation to Hydrogen Sulphide Release in Rats. Evid. Based Complement. Altern. Med. 2014, 2014, 904019. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, J.H.; Hu, Y.Y.; Hu, D.H.; Gao, S.S.; Fan, Y.F.; Wang, Y.L.; Jiao, Y.; Chen, Z.W. Total Flavones of Rhododendron simsii Planch Flower Protect against Cerebral Ischemia-Reperfusion Injury via the Mechanism of Cystathionine-gamma-Lyase-Produced H(2)S. Evid. Based Complement. Altern. Med. 2018, 2018, 8903849. [Google Scholar] [CrossRef]
- Yin, X.; Liu, B.; Ding, Y.; Li, X.; Sheng, J.; Guo, Y.; Chen, Z.; Wen, J. Total flavones of Rhododendron induce the transformation of A1/A2 astrocytes via promoting the release of CBS-produced H(2)S. Phytomedicine 2023, 111, 154666. [Google Scholar] [CrossRef]
- Powell, C.R.; Dillon, K.M.; Matson, J.B. A review of hydrogen sulfide (H(2)S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef]
- Xin, D.; Chu, X.; Bai, X.; Ma, W.; Yuan, H.; Qiu, J.; Liu, C.; Li, T.; Zhou, X.; Chen, W.; et al. l-Cysteine suppresses hypoxia-ischemia injury in neonatal mice by reducing glial activation, promoting autophagic flux and mediating synaptic modification via H(2)S formation. Brain Behav. Immun. 2018, 73, 222–234. [Google Scholar] [CrossRef]
- Dyson, R.M.; Palliser, H.K.; Wilding, N.; Kelly, M.A.; Chwatko, G.; Glowacki, R.; Berry, M.J.; Ni, X.; Wright, I.M.R. Microvascular circulatory dysregulation driven in part by cystathionine gamma-lyase: A new paradigm for cardiovascular compromise in the preterm newborn. Microcirculation 2019, 26, e12507. [Google Scholar] [CrossRef]
- Rao, S.P.; Dobariya, P.; Bellamkonda, H.; More, S.S. Role of 3-Mercaptopyruvate Sulfurtransferase (3-MST) in Physiology and Disease. Antioxidants 2023, 12, 603. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.Y.; Gao, S.S.; Chen, F.L.; Chen, S.; Wang, M.; Chen, Z.W. Role of CSE-Produced H(2)S on Cerebrovascular Relaxation via RhoA-ROCK Inhibition and Cerebral Ischemia-Reperfusion Injury in Mice. ACS Chem. Neurosci. 2019, 10, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Wen, J. H(2)S-mediated inhibition of RhoA/ROCK pathway and noncoding RNAs in ischemic stroke. Metab. Brain Dis. 2023, 38, 163–176. [Google Scholar] [CrossRef]
- Lu, W.; Wen, J. H(2)S-RhoA/ROCK Pathway and Glial Cells in Axonal Remyelination After Ischemic Stroke. Mol. Neurobiol. 2023, 60, 5493–5504. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magid, A.F. Rho kinase inhibitors: Potentially versatile therapy for the treatment of cardiovascular diseases and more. ACS Med. Chem. Lett. 2015, 6, 371–372. [Google Scholar] [CrossRef]
- Lu, W.; Chen, Z.; Wen, J. RhoA/ROCK signaling pathway and astrocytes in ischemic stroke. Metab. Brain Dis. 2021, 36, 1101–1108. [Google Scholar] [CrossRef]
- Schmidt, S.I.; Blaabjerg, M.; Freude, K.; Meyer, M. RhoA Signaling in Neurodegenerative Diseases. Cells 2022, 11, 1520. [Google Scholar] [CrossRef]
- Cai, R.; Wang, Y.; Huang, Z.; Zou, Q.; Pu, Y.; Yu, C.; Cai, Z. Role of RhoA/ROCK signaling in Alzheimer’s disease. Behav. Brain Res. 2021, 414, 113481. [Google Scholar] [CrossRef]
- Fujita, Y.; Yamashita, T. Axon growth inhibition by RhoA/ROCK in the central nervous system. Front. Neurosci. 2014, 8, 338. [Google Scholar] [CrossRef]
- Lai, A.Y.; McLaurin, J. Rho-associated protein kinases as therapeutic targets for both vascular and parenchymal pathologies in Alzheimer’s disease. J. Neurochem. 2018, 144, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.C.; Tonges, L.; Barski, E.; Michel, U.; Bahr, M.; Lingor, P. ROCK2 is a major regulator of axonal degeneration, neuronal death and axonal regeneration in the CNS. Cell Death Dis. 2014, 5, e1225. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Chen, Y.; Wen, J.; Chen, Z. H2S protects hippocampal neurons against hypoxia-reoxygenation injury by promoting RhoA phosphorylation at Ser188. Cell Death Discov. 2021, 7, 132. [Google Scholar] [CrossRef]
- Chuang, H.H.; Liang, S.W.; Chang, Z.F.; Lee, H.H. Ser1333 phosphorylation indicates ROCKI activation. J. Biomed. Sci. 2013, 20, 83. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Fang, F.; Sheng, J.; Guo, Y.; Wen, J.; Chen, Z. Protection of H2S against Hypoxia/Reoxygenation Injury in Rat Hippocampal Neurons through Inhibiting Phosphorylation of ROCK2 at Thr436 and Ser575. Pharmaceuticals 2023, 16, 218. [Google Scholar] [CrossRef]
- Xia, H.; Li, Z.; Sharp, T.E., 3rd; Polhemus, D.J.; Carnal, J.; Moles, K.H.; Tao, Y.X.; Elrod, J.; Pfeilschifter, J.; Beck, K.F.; et al. Endothelial Cell Cystathionine gamma-Lyase Expression Level Modulates Exercise Capacity, Vascular Function, and Myocardial Ischemia Reperfusion Injury. J. Am. Heart Assoc. 2020, 9, e017544. [Google Scholar] [CrossRef]
- Groger, M.; Hogg, M.; Abdelsalam, E.; Kress, S.; Hoffmann, A.; Stahl, B.; Calzia, E.; Wachter, U.; Vogt, J.A.; Wang, R.; et al. Effects of Sodium Thiosulfate During Resuscitation From Trauma-and-Hemorrhage in Cystathionine-gamma-Lyase Knockout Mice With Diabetes Type 1. Front. Med. 2022, 9, 878823. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, S.; Wen, J.Y.; Chen, Z.W. 3-Mercaptopyruvate sulfurtransferase/hydrogen sulfide protects cerebral endothelial cells against oxygen-glucose deprivation/reoxygenation-induced injury via mitoprotection and inhibition of the RhoA/ROCK pathway. Am. J. Physiol. Cell Physiol. 2020, 319, C720–C733. [Google Scholar] [CrossRef]
- Chatterjee, S.; Patra, D.; Ghosh, P.; Banerjee, S.; Chowdhury, K.D.; Chakraborty, P.; Basu, A.; Sadhukhan, G.C. Activity of ROCKII not ROCKI promotes pulmonary metastasis of melanoma cells via modulating Smad2/3-MMP9 and FAK-Src-VEGF signalling. Cell. Signal. 2022, 97, 110389. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.Y.; Zhang, J.; Chen, S.; Chen, Y.; Zhang, Y.; Ma, Z.Y.; Zhang, F.; Xie, W.M.; Fan, Y.F.; Duan, J.S.; et al. Endothelium-derived hydrogen sulfide acts as a hyperpolarizing factor and exerts neuroprotective effects via activation of large-conductance Ca(2+) -activated K(+) channels. Br. J. Pharmacol. 2021, 178, 4155–4175. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.Y.; Wang, M.; Li, Y.N.; Jiang, H.H.; Sun, X.J.; Chen, Z.W. Vascular Protection of Hydrogen Sulfide on Cerebral Ischemia/Reperfusion Injury in Rats. Front. Neurol. 2018, 9, 779. [Google Scholar] [CrossRef]
- Chen, S.; Guo, F.; Liu, X.; Xi, J.; Xue, M.; Guo, Y.; Wen, J.; Dong, L.; Chen, Z. Roles of the RhoA-ROCK Signaling Pathway in the Endothelial H(2)S Production and Vasodilation in Rat Cerebral Arteries. ACS Omega 2022, 7, 18498–18508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, K.; Wang, X.; Ding, Y.; Ren, Z.; Fang, J.; Sun, T.; Guo, Y.; Chen, Z.; Wen, J. CSE-Derived H(2)S Inhibits Reactive Astrocytes Proliferation and Promotes Neural Functional Recovery after Cerebral Ischemia/Reperfusion Injury in Mice Via Inhibition of RhoA/ROCK(2) Pathway. ACS Chem. Neurosci. 2021, 12, 2580–2590. [Google Scholar] [CrossRef]
- Liu, D.; Tang, H.; Li, X.Y.; Deng, M.F.; Wei, N.; Wang, X.; Zhou, Y.F.; Wang, D.Q.; Fu, P.; Wang, J.Z.; et al. Targeting the HDAC2/HNF-4A/miR-101b/AMPK Pathway Rescues Tauopathy and Dendritic Abnormalities in Alzheimer's Disease. Mol. Ther. 2017, 25, 752–764. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, B.; Zhang, Y.; Fang, F.; Li, X.; Wang, S.; Wen, J. Hydrogen sulphide protects mice against the mutual aggravation of cerebral ischaemia/reperfusion injury and colitis. Eur. J. Pharmacol. 2022, 914, 174682. [Google Scholar] [CrossRef]
- Aggarwal, S.; Randhawa, P.K.; Singh, N.; Jaggi, A.S. Preconditioning at a distance: Involvement of endothelial vasoactive substances in cardioprotection against ischemia-reperfusion injury. Life Sci. 2016, 151, 250–258. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, C.; Jiang, J.; Xin, M.; Hao, J. Effect of TDP43-CTFs35 on Brain Endothelial Cell Functions in Cerebral Ischemic Injury. Mol. Neurobiol. 2022, 59, 4593–4611. [Google Scholar] [CrossRef]
- Abdulkadir, R.R.; Alwjwaj, M.; Othman, O.A.; Rakkar, K.; Bayraktutan, U. Outgrowth endothelial cells form a functional cerebral barrier and restore its integrity after damage. Neural Regen. Res. 2020, 15, 1071–1078. [Google Scholar] [CrossRef]
- Qi, M.; Hang, C.; Zhu, L.; Shi, J. Involvement of endothelial-derived relaxing factors in the regulation of cerebral blood flow. Neurol. Sci. 2011, 32, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Tierradentro-Garcia, L.O.; Saade-Lemus, S.; Freeman, C.; Kirschen, M.; Huang, H.; Vossough, A.; Hwang, M. Cerebral Blood Flow of the Neonatal Brain after Hypoxic-Ischemic Injury. Am. J. Perinatol. 2023, 40, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Mo, D.; Tian, W.; Liu, X.X.; Zhou, Y.G.; Sun, Y.; Feng, Y.D.; Xiao, X.; Hao, X.W.; Zhang, H.N.; et al. Inhibition of RhoA/ROCK signaling pathway ameliorates hypoxic pulmonary hypertension via HIF-1alpha-dependent functional TRPC channels. Toxicol. Appl. Pharmacol. 2019, 369, 60–72. [Google Scholar] [CrossRef]
- Liu, C.; Han, S.; Zheng, J.; Wang, H.; Li, S.; Li, J. EphA4 regulates white matter remyelination after ischemic stroke through Ephexin-1/RhoA/ROCK signaling pathway. Glia 2022, 70, 1971–1991. [Google Scholar] [CrossRef]
- Dokumacioglu, E.; Duzcan, I.; Iskender, H.; Sahin, A. RhoA/ROCK-1 Signaling Pathway and Oxidative Stress in Coronary Artery Disease Patients. Braz. J. Cardiovasc. Surg. 2022, 37, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, X.M.; Chen, S.; Wen, J.Y.; Chen, Z.W. Total flavones of Rhododendron simsii Planch flower protect rat hippocampal neuron from hypoxia-reoxygenation injury via activation of BK(Ca) channel. J. Pharm. Pharmacol. 2020, 72, 111–120. [Google Scholar] [CrossRef]
- Chen, J.; Sun, L.; Ding, G.B.; Chen, L.; Jiang, L.; Wang, J.; Wu, J. Oxygen-Glucose Deprivation/Reoxygenation Induces Human Brain Microvascular Endothelial Cell Hyperpermeability Via VE-Cadherin Internalization: Roles of RhoA/ROCK2. J. Mol. Neurosci. 2019, 69, 49–59. [Google Scholar] [CrossRef]
- Sycheva, M.; Sustarich, J.; Zhang, Y.; Selvaraju, V.; Geetha, T.; Gearing, M.; Babu, J.R. Pro-Nerve Growth Factor Induces Activation of RhoA Kinase and Neuronal Cell Death. Brain Sci. 2019, 9, 204. [Google Scholar] [CrossRef]
- Liu, M.; Wang, W.; Zhang, Y.; Xu, Z. Effects of combined electroacupuncture and medication therapy on the RhoA/ROCK-2 signaling pathway in the striatal region of rats afflicted by cerebral ischemia. Brain Res. Bull. 2023, 205, 110828. [Google Scholar] [CrossRef]
- Xue, M.; Chen, S.; Xi, J.; Guan, Q.; Chen, W.; Guo, Y.; Chen, Z. Protection against Hypoxia-Reoxygenation Injury of Hippocampal Neurons by H2S via Promoting Phosphorylation of ROCK2 at Tyr722 in Rat Model. Molecules 2022, 27, 4567. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Zhang, X.; Li, Y.; Wen, J.; Chen, Z.; Chen, S. Total Flavones of Rhododendron Protect Against Ischemic Cerebral Injury by Regulating the Phosphorylation of the RhoA-ROCK2 Pathway via Endothelial-Derived H2S. Curr. Issues Mol. Biol. 2025, 47, 513. https://doi.org/10.3390/cimb47070513
Sun X, Zhang X, Li Y, Wen J, Chen Z, Chen S. Total Flavones of Rhododendron Protect Against Ischemic Cerebral Injury by Regulating the Phosphorylation of the RhoA-ROCK2 Pathway via Endothelial-Derived H2S. Current Issues in Molecular Biology. 2025; 47(7):513. https://doi.org/10.3390/cimb47070513
Chicago/Turabian StyleSun, Xiaoqing, Xingyu Zhang, Yuwen Li, Jiyue Wen, Zhiwu Chen, and Shuo Chen. 2025. "Total Flavones of Rhododendron Protect Against Ischemic Cerebral Injury by Regulating the Phosphorylation of the RhoA-ROCK2 Pathway via Endothelial-Derived H2S" Current Issues in Molecular Biology 47, no. 7: 513. https://doi.org/10.3390/cimb47070513
APA StyleSun, X., Zhang, X., Li, Y., Wen, J., Chen, Z., & Chen, S. (2025). Total Flavones of Rhododendron Protect Against Ischemic Cerebral Injury by Regulating the Phosphorylation of the RhoA-ROCK2 Pathway via Endothelial-Derived H2S. Current Issues in Molecular Biology, 47(7), 513. https://doi.org/10.3390/cimb47070513