


Exploring the Molecular Mechanism of 1,25(OH)2D3 Reversal of Sorafenib Resistance in Hepatocellular Carcinoma Based on Network Pharmacology and Experimental Validation

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. GEO Microarray and Acquisition of Sorafenib-Resistant Targets in Hepatocellular Carcinoma
2.2. Target Prediction for 1,25(OH)2D3 and Its Derivatives
2.3. Prediction of Potential Targets for 1,25(OH)2D3 Chemosensitization
2.4. Biofunctional Analysis
2.5. Construction of “Compound–Target” and “Compound–Target–Pathway” Network Diagrams
2.6. Molecular Docking
2.7. Molecular Dynamics Simulations
2.8. Cell Culture
2.9. CCK8 Assay and Combination Index Calculations
2.10. Cell Clone Formation Experiments
2.11. Apoptosis and Cell Cycle Analysis
2.12. Monodansylcadaverine (MDC) Staining
2.13. Transmission Electron Microscopy
2.14. Western Blot
2.15. Statistical Methods
3. Results
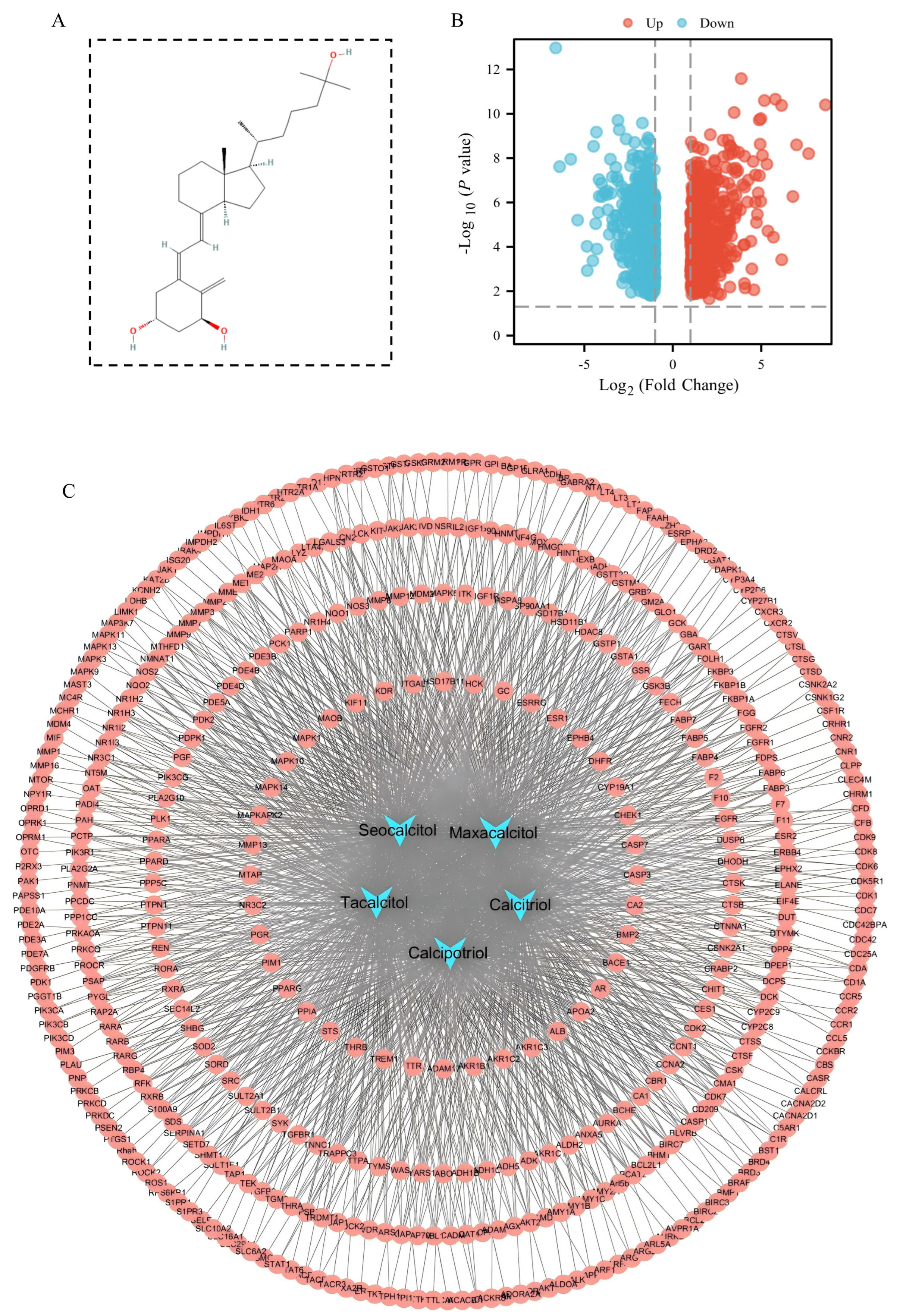
3.1. 1,25(OH)2D3 Target Prediction and Screening of Sorafenib Resistance Genes in Hepatocellular Carcinoma
3.2. Identification of Potential Targets for Sorafenib Resistance in 1,25(OH)2D3-Treated HCC
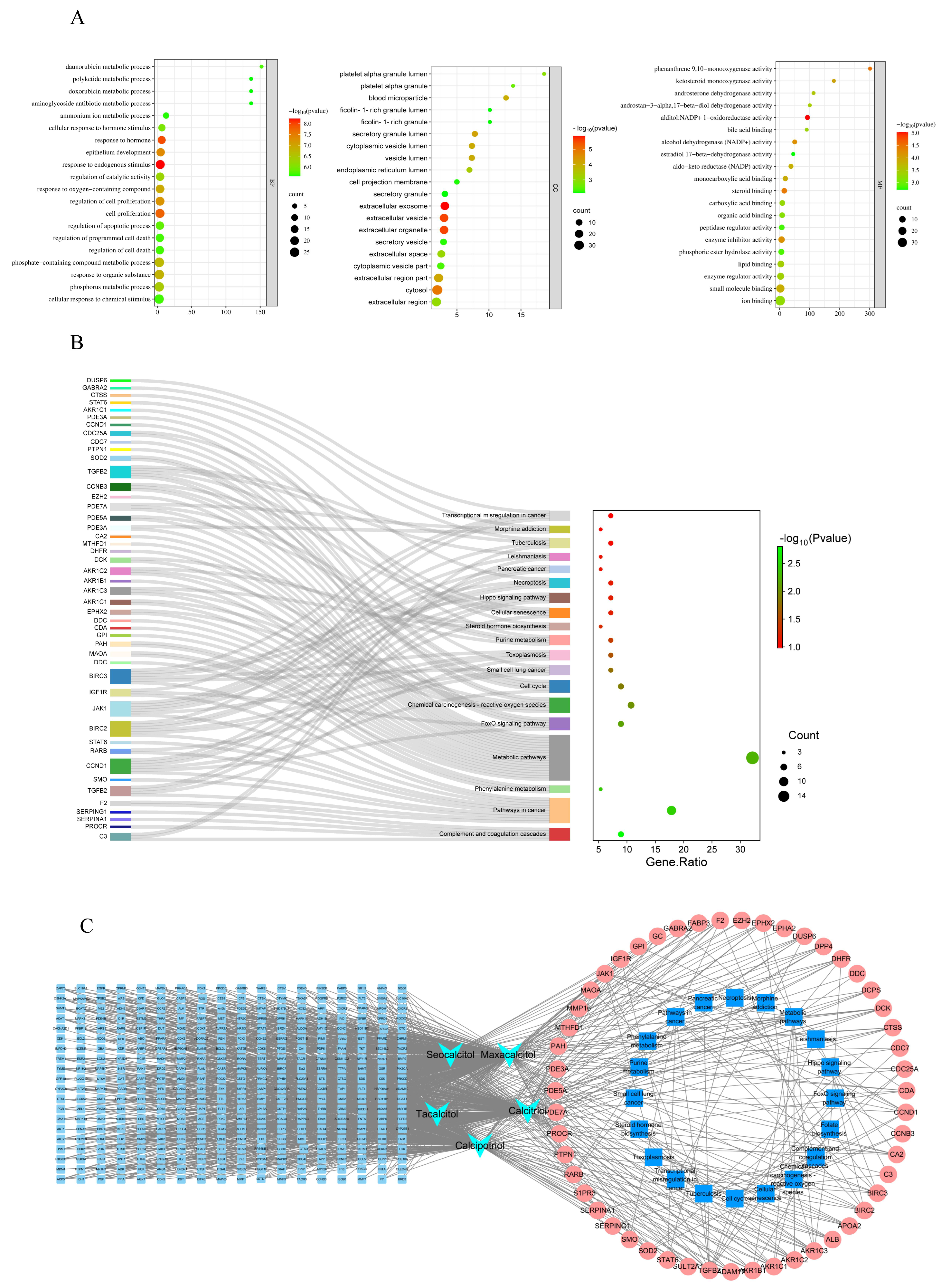
3.3. GO and KEGG Analysis
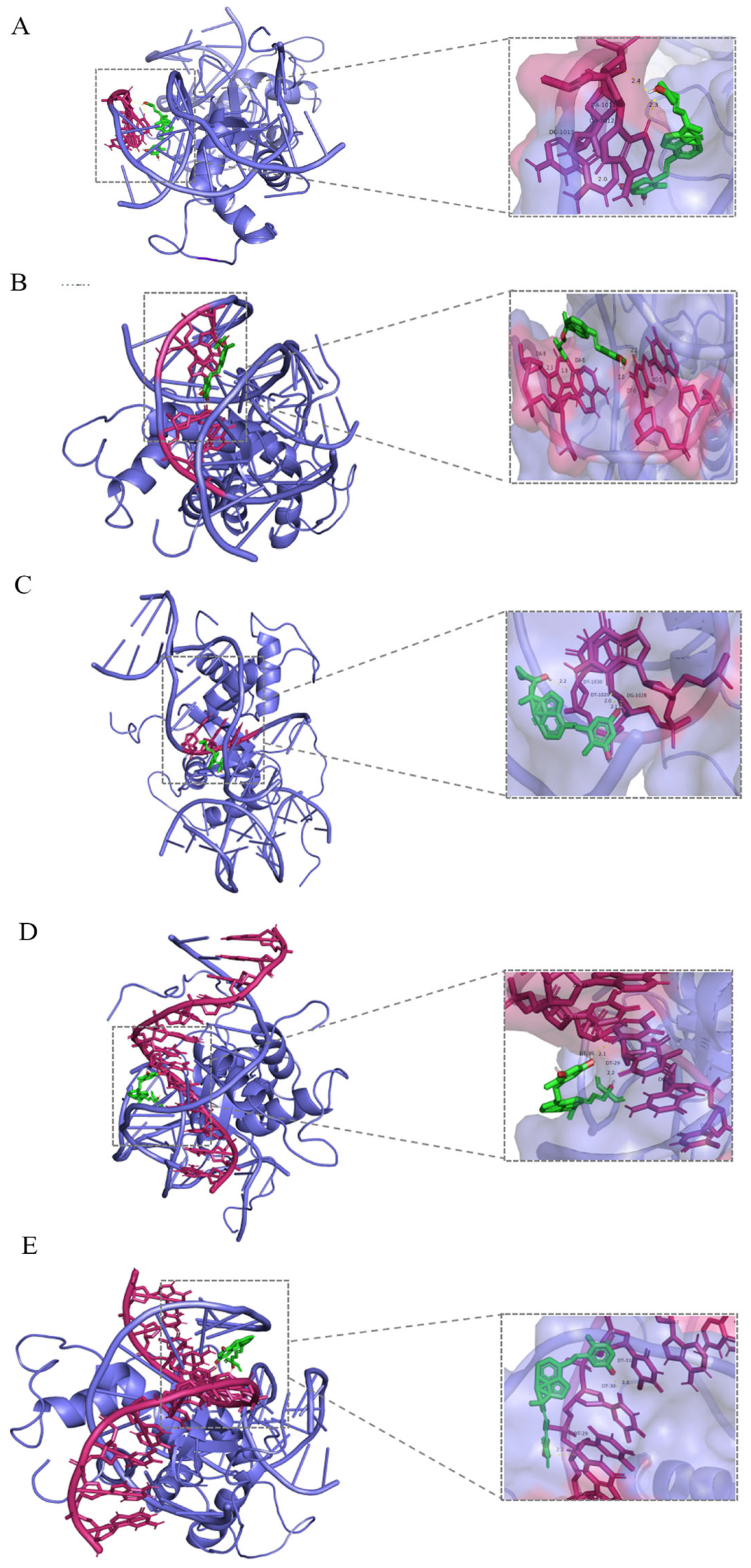
3.4. Molecular Docking of Small Molecule Models with FOXO3A
3.5. Molecular Dynamics Simulations
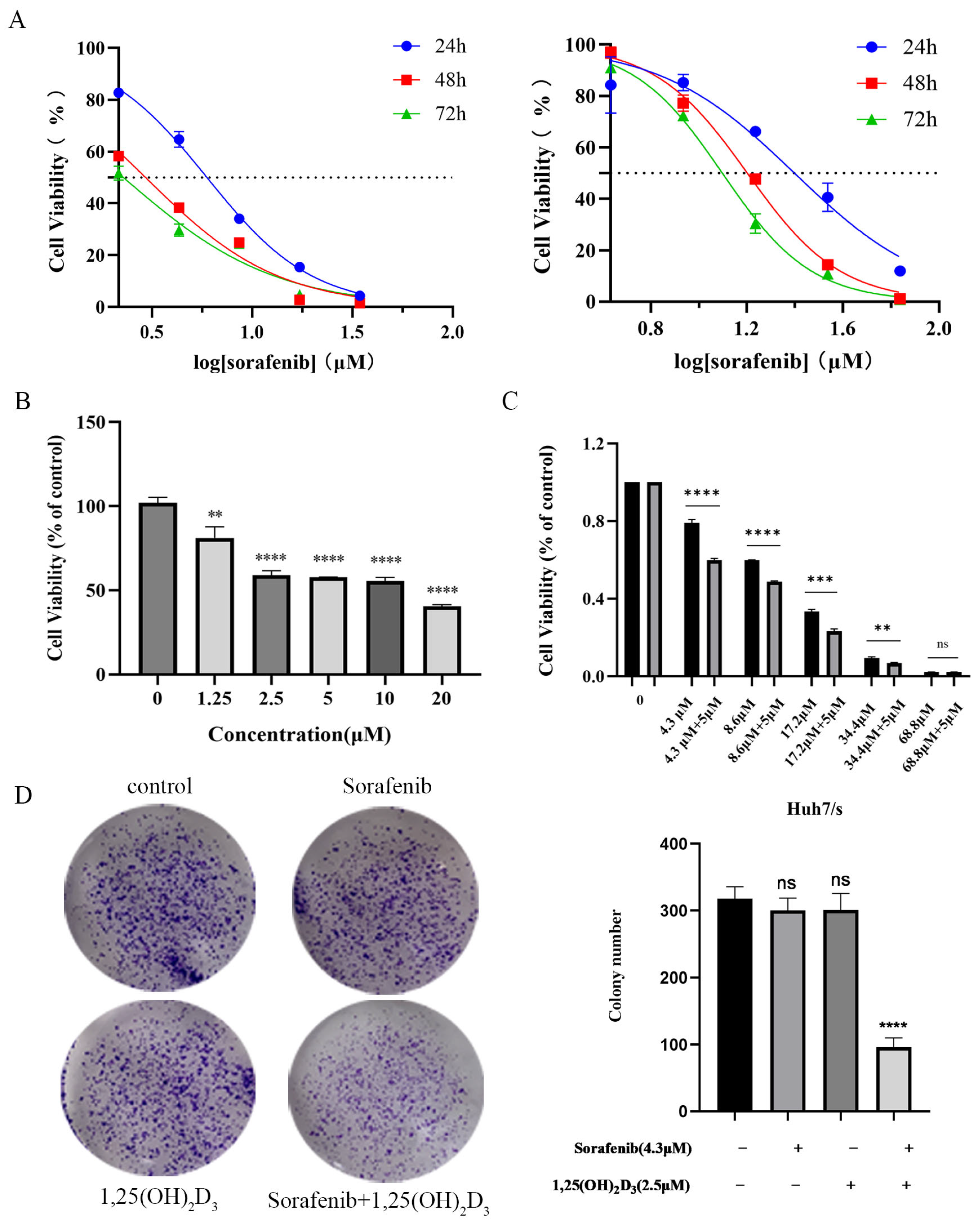
3.6. 1,25(OH)2D3 in Combination with Sorafenib Inhibits the Proliferation of Huh7/s Cells
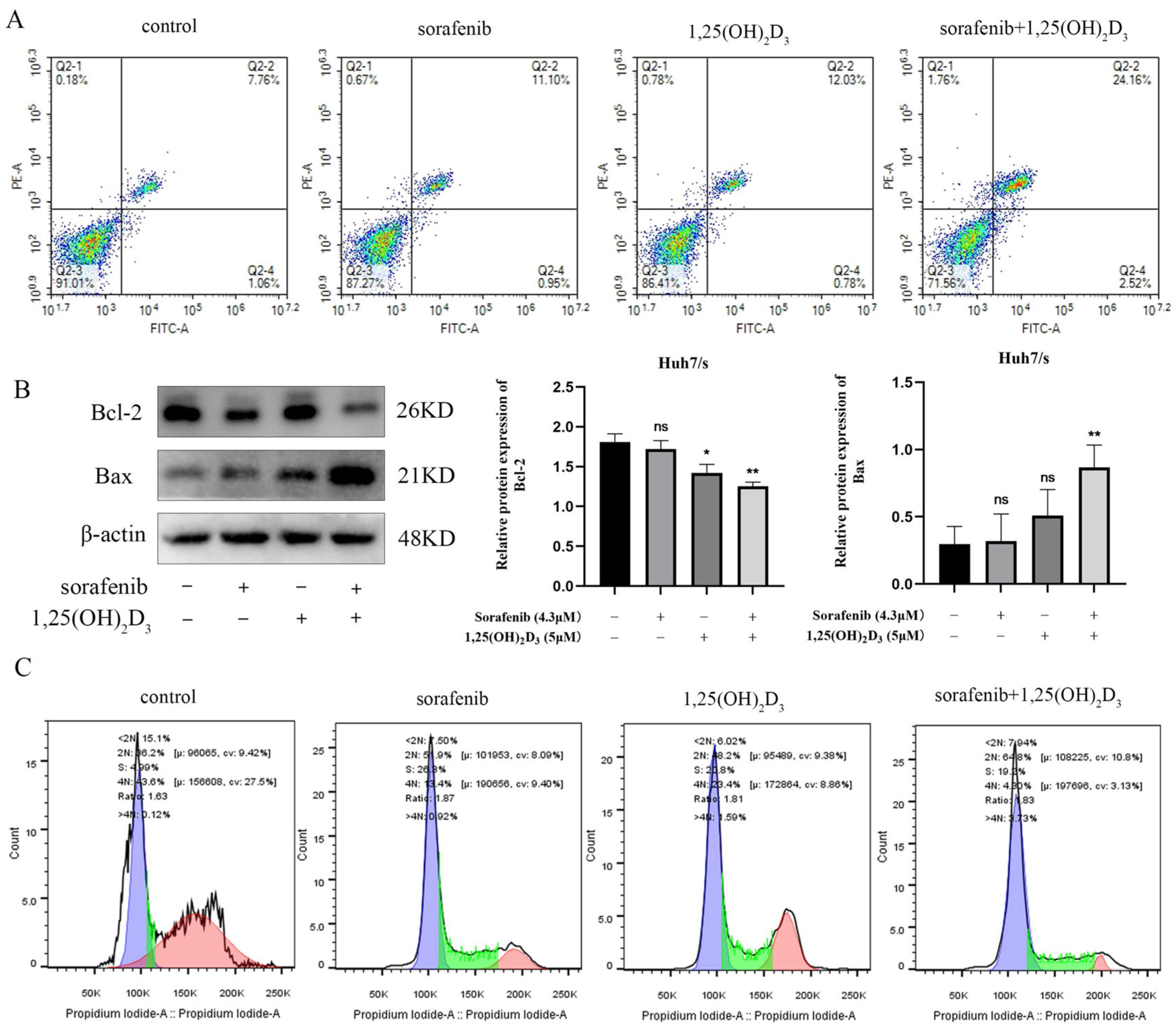
3.7. 1,25(OH)2D3 in Combination with Sorafenib Promotes Apoptosis and Cycle Blockade in Huh7/s Cells
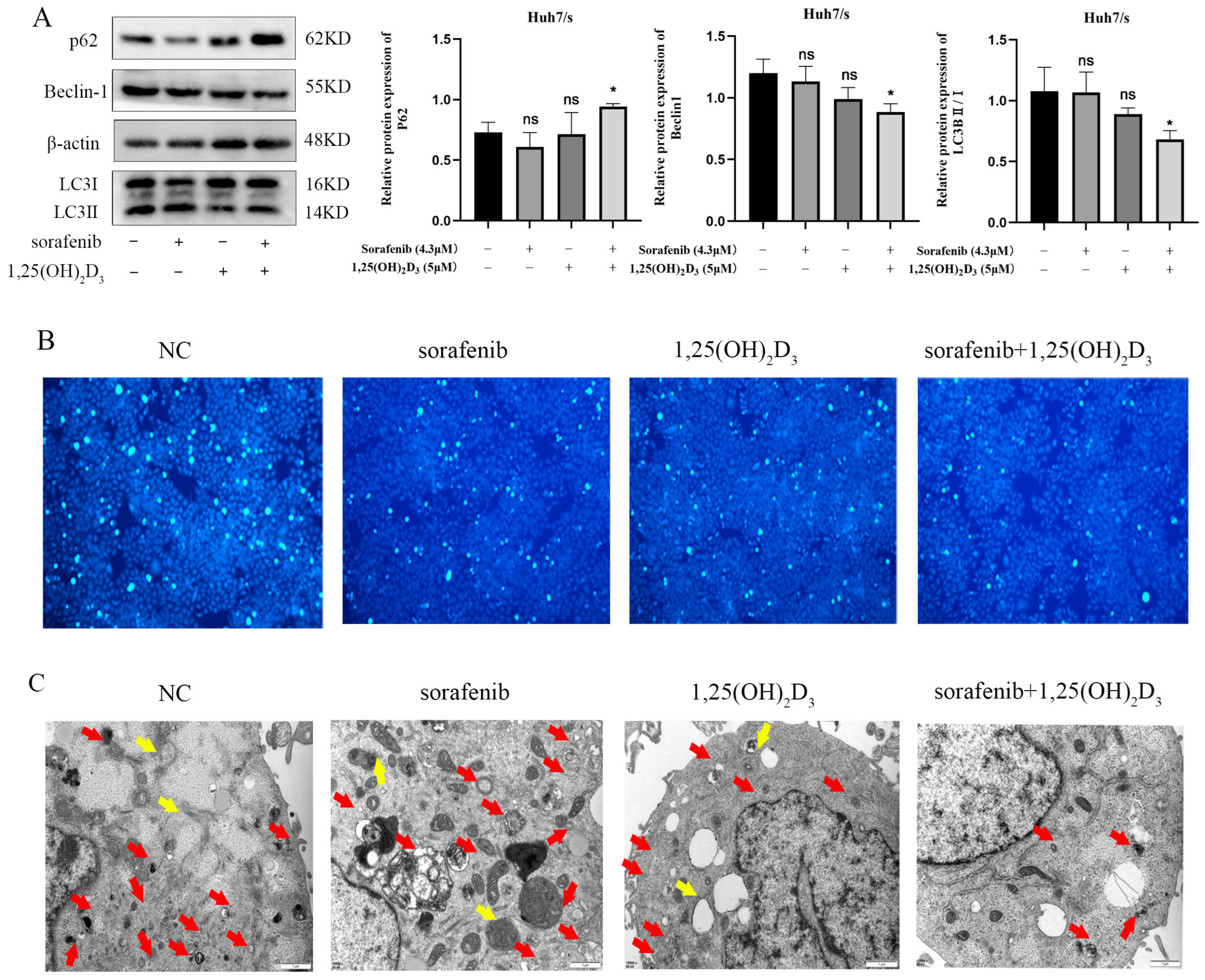
3.8. 1,25(OH)2D3 Combined with Sorafenib Inhibits Autophagy in Huh7/s Cells
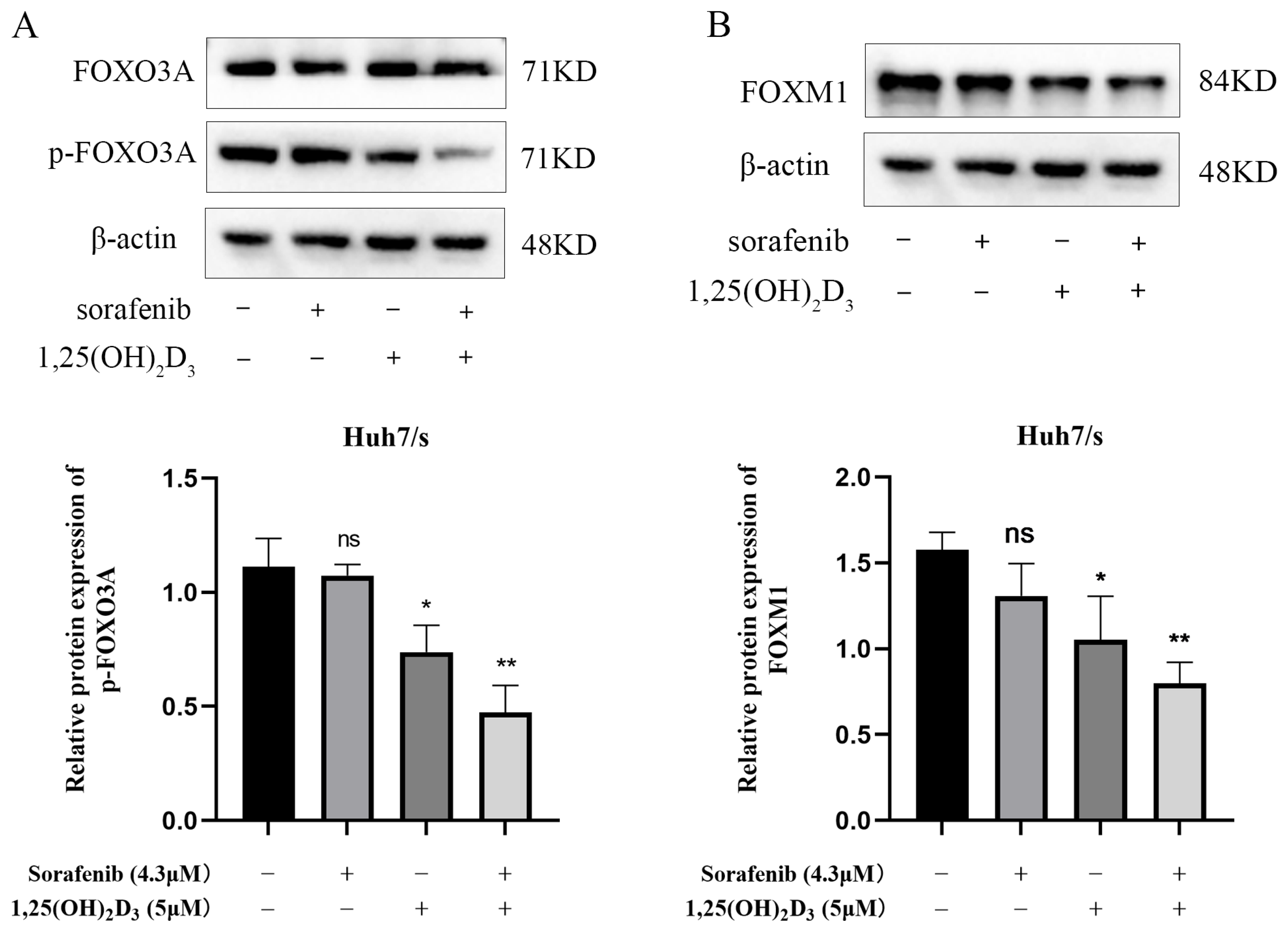
3.9. Effect of 1,25(OH)2D3 Combined with Sorafenib on FOXO3A/FOXM1 Axis
4. Discussion
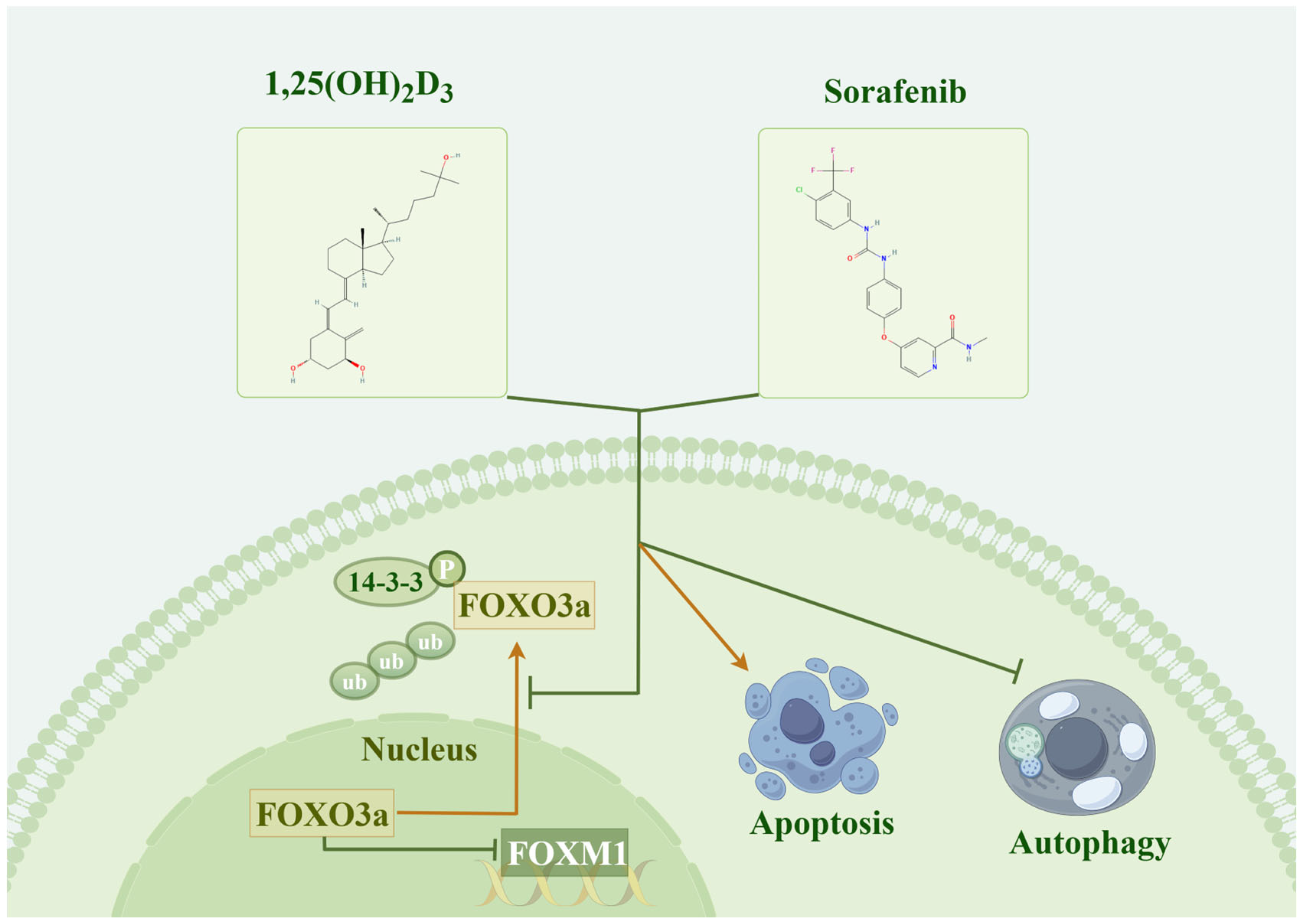
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Laube, R.; Sabih, A.H.; Strasser, S.I.; Lim, L.; Cigolini, M.; Liu, K. Palliative care in hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2021, 36, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Sun, X.Y. Mechanisms of resistance to sorafenib and the corresponding strategies in hepatocellular carcinoma. World J. Hepatol. 2013, 5, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ma, Z.; Li, W.; Ma, Q.; Guo, J.; Hu, A.; Li, R.; Wang, F.; Han, S. The mechanism of calcitriol in cancer prevention and treatment. Curr. Med. Chem. 2013, 20, 4121–4130. [Google Scholar] [CrossRef]
- Vanoirbeek, E.; Krishnan, A.; Eelen, G.; Verlinden, L.; Bouillon, R.; Feldman, D.; Verstuyf, A. The anti-cancer and anti-inflammatory actions of 1,25(OH)2D3. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 593–604. [Google Scholar] [CrossRef]
- Deeb, K.K.; Trump, D.L.; Johnson, C.S. Vitamin D signalling pathways in cancer: Potential for anticancer therapeutics. Nat. Rev. Cancer 2007, 7, 684–700. [Google Scholar] [CrossRef]
- Abu El Maaty, M.A.; Wölfl, S. Effects of 1,25(OH)2D3 on Cancer Cells and Potential Applications in Combination with Established and Putative Anti-Cancer Agents. Nutrients 2017, 9, 87. [Google Scholar] [CrossRef]
- Ma, Y.; Yu, W.D.; Hershberger, P.A.; Flynn, G.; Kong, R.X.; Trump, D.L.; Johnson, C.S. 1alpha,25-Dihydroxyvitamin D3 potentiates cisplatin antitumor activity by p73 induction in a squamous cell carcinoma model. Mol. Cancer Ther. 2008, 7, 3047–3055. [Google Scholar] [CrossRef]
- Hershberger, P.A.; Yu, W.D.; Modzelewski, R.A.; Rueger, R.M.; Johnson, C.S.; Trump, D.L. Calcitriol (1,25-dihydroxycholecalciferol) enhances paclitaxel antitumor activity in vitro and in vivo and accelerates paclitaxel-induced apoptosis. Clin. Cancer Res. 2001, 7, 1043–1051. [Google Scholar]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Yokoyama, A. Sunitinib, an orally available receptor tyrosine kinase inhibitor, induces monocytic differentiation of acute myelogenous leukemia cells that is enhanced by 1,25-dihydroxyvitamin D3. Leukemia 2009, 23, 2171–2173. [Google Scholar] [CrossRef]
- Luo, T.T.; Lu, Y.; Yan, S.K.; Xiao, X.; Rong, X.L.; Guo, J. Network Pharmacology in Research of Chinese Medicine Formula: Methodology, Application and Prospective. Chin. J. Integr. Med. 2020, 26, 72–80. [Google Scholar] [CrossRef]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical role of FOXO3a in carcinogenesis. Mol. Cancer 2018, 17, 104. [Google Scholar] [CrossRef]
- Chen, Y.F.; Pandey, S.; Day, C.H.; Chen, Y.F.; Jiang, A.Z.; Ho, T.J.; Chen, R.J.; Padma, V.V.; Kuo, W.W.; Huang, C.Y. Synergistic effect of HIF-1α and FoxO3a trigger cardiomyocyte apoptosis under hyperglycemic ischemia condition. J. Cell Physiol. 2018, 233, 3660–3671. [Google Scholar] [CrossRef] [PubMed]
- McClelland Descalzo, D.L.; Satoorian, T.S.; Walker, L.M.; Sparks, N.R.; Pulyanina, P.Y.; Zur Nieden, N.I. Glucose-Induced Oxidative Stress Reduces Proliferation in Embryonic Stem Cells via FOXO3A/β-Catenin-Dependent Transcription of p21cip1. Stem Cell Rep. 2016, 7, 55–68. [Google Scholar] [CrossRef]
- McGowan, S.E.; McCoy, D.M. Platelet-derived growth factor-A regulates lung fibroblast S-phase entry through p27kip1 and FoxO3a. Respir. Res. 2013, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Kim, S. Getting the most out of PubChem for virtual screening. Expert Opin. Drug Discov. 2016, 11, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Murmu, S.; Aravinthkumar, A.; Singh, M.K.; Sharma, S.; Das, R.; Jha, G.K.; Prakash, G.; Rana, V.S.; Kaushik, P.; Farooqi, M.S. Identification of potent phytochemicals against Magnaporthe oryzae through machine learning aided-virtual screening and molecular dynamics simulation approach. Comput. Biol. Med. 2025, 188, 109862. [Google Scholar] [CrossRef] [PubMed]
- Muhetaer, H.; Li, H.; Wang, B.; Cai, X.; Zhang, Y.; Li, Y.; Li, C.; Wu, B. Exploring the Effects and Mechanisms of Valerian Volatile Oil in Treating Insomnia Using Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation-Based Approaches. Int. J. Mol. Sci. 2025, 26, 1726. [Google Scholar] [CrossRef]
- Li, Y.; Mu, Y.; Chen, X.; Zhao, Y.; Ji, C.; Xu, R.; Jiang, R.; Liu, F.; Wang, M.; Sun, L. Deoxyshikonin from Arnebiae Radix promotes hair growth by targeting the Wnt/β-catenin signaling pathway. Phytomedicine 2025, 140, 156590. [Google Scholar] [CrossRef]
- Jin, L.; Guan, Y.; Li, X.; Wang, M.; Shen, Y.; Wang, N.; He, Z. Combining Network Pharmacology, Molecular Docking and Experimental Validation to Explore the Effects and Mechanisms of Indirubin on Acute Lymphoblastic Leukemia. Drug Des. Devel. Ther. 2025, 19, 1083–1103. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Cai, L.; Qin, X.; Xu, Z.; Song, Y.; Jiang, H.; Wu, Y.; Ruan, H.; Chen, J. Comparison of Cytotoxicity Evaluation of Anticancer Drugs between Real-Time Cell Analysis and CCK-8 Method. ACS Omega J. 2019, 4, 12036–12042. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yan, J.; Huang, J.; Wu, X.; Yuan, Y.; Yuan, Y.; Zhang, S.; Mo, F. Exploring the mechanism by which quercetin re-sensitizes breast cancer to paclitaxel: Network pharmacology, molecular docking, and experimental verification. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 3045–3059. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dong, M.; Qin, H.; An, G.; Cen, L.; Deng, L.; Cui, H. Mulberrin suppresses gastric cancer progression and enhances chemosensitivity to oxaliplatin through HSP90AA1/PI3K/AKT axis. Phytomedicine 2025, 139, 156441. [Google Scholar] [CrossRef]
- Li, Y.; Su, Y.; Zhao, Y.; Hu, X.; Zhao, G.; He, J.; Wan, S.; Lü, M.; Cui, H. Demethylzeylasteral inhibits proliferation, migration, and invasion through FBXW7/c-Myc axis in gastric cancer. MedComm 2021, 2, 467–480. [Google Scholar] [CrossRef]
- Ji, H.; Zhang, K.; Pan, G.; Li, C.; Li, C.; Hu, X.; Yang, L.; Cui, H. Deoxyelephantopin Induces Apoptosis and Enhances Chemosensitivity of Colon Cancer via miR-205/Bcl2 Axis. Int. J. Mol. Sci. 2022, 23, 5051. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef]
- Abu El Maaty, M.A.; Wölfl, S. Vitamin D as a Novel Regulator of Tumor Metabolism: Insights on Potential Mechanisms and Implications for Anti-Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2184. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Banach-Petrosky, W.; Ouyang, X.; Gao, H.; Nader, K.; Ji, Y.; Suh, N.; DiPaola, R.S.; Abate-Shen, C. Vitamin D inhibits the formation of prostatic intraepithelial neoplasia in Nkx3.1;Pten mutant mice. Clin. Cancer Res. 2006, 12, 5895–5901. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Feldman, D. Mechanisms of the anti-cancer and anti-inflammatory actions of vitamin D. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 311–336. [Google Scholar] [CrossRef]
- Krishnan, A.V.; Trump, D.L.; Johnson, C.S.; Feldman, D. The role of vitamin D in cancer prevention and treatment. Endocrinol. Metab. Clin. N. Am. 2010, 39, 401–418, table of contents. [Google Scholar] [CrossRef]
- Li, Y.; Cook, K.L.; Yu, W.; Jin, L.; Bouker, K.B.; Clarke, R.; Hilakivi-Clarke, L. Inhibition of Antiestrogen-Promoted Pro-Survival Autophagy and Tamoxifen Resistance in Breast Cancer through Vitamin D Receptor. Nutrients 2021, 13, 1715. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Zhang, Y.; Yan, A.; Wang, M.; Han, Q.; Wang, K.; Wang, J.; Qiao, C.; Pan, Z.; Chen, C.; et al. 1,25-dihydroxyvitamin D3 signaling-induced decreases in IRX4 inhibits NANOG-mediated cancer stem-like properties and gefitinib resistance in NSCLC cells. Cell Death Dis. 2020, 11, 670. [Google Scholar] [CrossRef]
- Davoust, N.; Wion, D.; Chevalier, G.; Garabedian, M.; Brachet, P.; Couez, D. Vitamin D receptor stable transfection restores the susceptibility to 1,25-dihydroxyvitamin D3 cytotoxicity in a rat glioma resistant clone. J. Neurosci. Res. 1998, 52, 210–219. [Google Scholar] [CrossRef]
- Zigmont, V.; Garrett, A.; Peng, J.; Seweryn, M.; Rempala, G.A.; Harris, R.; Holloman, C.; Gundersen, T.E.; Ahlbom, A.; Feychting, M.; et al. Association Between Prediagnostic Serum 25-Hydroxyvitamin D Concentration and Glioma. Nutr. Cancer 2015, 67, 1120–1130. [Google Scholar] [CrossRef]
- Bak, D.H.; Kang, S.H.; Choi, D.R.; Gil, M.N.; Yu, K.S.; Jeong, J.H.; Lee, N.S.; Lee, J.H.; Jeong, Y.G.; Kim, D.K.; et al. Autophagy enhancement contributes to the synergistic effect of vitamin D in temozolomide-based glioblastoma chemotherapy. Exp. Ther. Med. 2016, 11, 2153–2162. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.S.; Kiang, K.M.; Leung, G.K. Anti-tumor effects of vitamin D in glioblastoma: Mechanism and therapeutic implications. Lab. Investig. 2022, 102, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Woloszynska-Read, A.; Johnson, C.S.; Trump, D.L. Vitamin D and cancer: Clinical aspects. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 605–615. [Google Scholar] [CrossRef]
- Leyssens, C.; Verlinden, L.; Verstuyf, A. The future of vitamin D analogs. Front. Physiol. 2014, 5, 122. [Google Scholar] [CrossRef]
- Yee, Y.K.; Chintalacharuvu, S.R.; Lu, J.; Nagpal, S. Vitamin D receptor modulators for inflammation and cancer. Mini-Rev. Med. Chem. 2005, 5, 761–778. [Google Scholar] [CrossRef] [PubMed]
- Prudencio, J.; Akutsu, N.; Benlimame, N.; Wang, T.; Bastien, Y.; Lin, R.; Black, M.J.; Alaoui-Jamali, M.A.; White, J.H. Action of low calcemic 1alpha,25-dihydroxyvitamin D3 analogue EB1089 in head and neck squamous cell carcinoma. J. Natl. Cancer Inst. 2001, 93, 745–753. [Google Scholar] [CrossRef]
- Verlinden, L.; Verstuyf, A.; Van Camp, M.; Marcelis, S.; Sabbe, K.; Zhao, X.Y.; De Clercq, P.; Vandewalle, M.; Bouillon, R. Two novel 14-Epi-analogues of 1,25-dihydroxyvitamin D3 inhibit the growth of human breast cancer cells in vitro and in vivo. Cancer Res. 2000, 60, 2673–2679. [Google Scholar]
- Ma, Y.; Yu, W.D.; Hidalgo, A.A.; Luo, W.; Delansorne, R.; Johnson, C.S.; Trump, D.L. Inecalcitol, an analog of 1,25D3, displays enhanced antitumor activity through the induction of apoptosis in a squamous cell carcinoma model system. Cell Cycle 2013, 12, 743–752. [Google Scholar] [CrossRef]
- Okamoto, R.; Delansorne, R.; Wakimoto, N.; Doan, N.B.; Akagi, T.; Shen, M.; Ho, Q.H.; Said, J.W.; Koeffler, H.P. Inecalcitol, an analog of 1α,25(OH)2 D3, induces growth arrest of androgen-dependent prostate cancer cells. Int. J. Cancer 2012, 130, 2464–2473. [Google Scholar] [CrossRef]
- Farhan, M.; Wang, H.; Gaur, U.; Little, P.J.; Xu, J.; Zheng, W. FOXO Signaling Pathways as Therapeutic Targets in Cancer. Int. J. Biol. Sci. 2017, 13, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Cao, Q.; Wang, F.; Huang, L.Y.; Sang, T.T.; Liu, F.; Chen, S.Y. SIRT1 Protects Against Oxidative Stress-Induced Endothelial Progenitor Cells Apoptosis by Inhibiting FOXO3a via FOXO3a Ubiquitination and Degradation. J. Cell. Physiol. 2015, 230, 2098–2107. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Hu, J.; Li, X.; Gao, H.; Kong, D.; Jin, M. Glutamine transporter inhibitor enhances the sensitivity of NSCLC to trametinib through GSDME-dependent pyroptosis. Biochem. Pharmacol. 2025, 233, 116796. [Google Scholar] [CrossRef]
- Tang, Q.; Li, J.; Zhang, L.; Zeng, S.; Bao, Q.; Hu, W.; He, L.; Huang, G.; Wang, L.; Liu, Y.; et al. Orlistat facilitates immunotherapy via AKT-FOXO3a-FOXM1-mediated PD-L1 suppression. J. Immunother. Cancer 2025, 13, e008923. [Google Scholar] [CrossRef]
- Zhong, M.; Fang, Z.; Zou, J.; Chen, X.; Qiu, Z.; Zhou, L.; Le, Y.; Chen, Z.; Liao, Y.; Nie, F.; et al. SPIN1 accelerates tumorigenesis and confers radioresistance in non-small cell lung cancer by orchestrating the FOXO3a/FOXM1 axis. Cell Death Dis. 2024, 15, 832. [Google Scholar] [CrossRef] [PubMed]
- Nestal de Moraes, G.; Bella, L.; Zona, S.; Burton, M.J.; Lam, E.W. Insights into a Critical Role of the FOXO3a-FOXM1 Axis in DNA Damage Response and Genotoxic Drug Resistance. Curr. Drug Targets 2016, 17, 164–177. [Google Scholar] [CrossRef]
- Yao, S.; Fan, L.Y.; Lam, E.W. The FOXO3-FOXM1 axis: A key cancer drug target and a modulator of cancer drug resistance. Semin. Cancer. Biol. 2018, 50, 77–89. [Google Scholar] [CrossRef]
- Li, J.; Chen, P.; Wu, Q.; Guo, L.; Leong, K.W.; Chan, K.I.; Kwok, H.F. A novel combination treatment of antiADAM17 antibody and erlotinib to overcome acquired drug resistance in non-small cell lung cancer through the FOXO3a/FOXM1 axis. Cell. Mol. Life Sci. 2022, 79, 614. [Google Scholar] [CrossRef]
- Liu, H.; Song, Y.; Qiu, H.; Liu, Y.; Luo, K.; Yi, Y.; Jiang, G.; Lu, M.; Zhang, Z.; Yin, J.; et al. Downregulation of FOXO3a by DNMT1 promotes breast cancer stem cell properties and tumorigenesis. Cell Death Differ. 2020, 27, 966–983. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Li, X.; Jia, Y.; Wang, J.; Ao, X. FOXO3a in cancer drug resistance. Cancer Lett. 2022, 540, 215724. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y. Mechanisms of sorafenib resistance in hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2024, 48, 102434. [Google Scholar] [CrossRef]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef] [PubMed]
- Tutusaus, A.; Stefanovic, M.; Boix, L.; Cucarull, B.; Zamora, A.; Blasco, L.; de Frutos, P.G.; Reig, M.; Fernandez-Checa, J.C.; Marí, M.; et al. Antiapoptotic BCL-2 proteins determine sorafenib/regorafenib resistance and BH3-mimetic efficacy in hepatocellular carcinoma. Oncotarget 2018, 9, 16701–16717. [Google Scholar] [CrossRef]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2017, 1868, 564–570. [Google Scholar] [CrossRef] [PubMed]
- de Melo Silva, A.J.; de Melo Gama, J.E.; de Oliveira, S.A. The Role of Bcl-2 Family Proteins and Sorafenib Resistance in Hepatocellular Carcinoma. Int. J. Cell Biol. 2024, 2024, 4972523. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, X.; Li, X.; Li, C.; Zhao, L.; Zhou, Y.; Hou, H. Butein sensitizes HeLa cells to cisplatin through the AKT and ERK/p38 MAPK pathways by targeting FoxO3a. Int. J. Mol. Med. 2015, 36, 957–966. [Google Scholar] [CrossRef]
- Ding, Q.; Chen, Y.; Zhang, Q.; Guo, Y.; Huang, Z.; Dai, L.; Cao, S. 8-bromo-7-methoxychrysin induces apoptosis by regulating Akt/FOXO3a pathway in cisplatin-sensitive and resistant ovarian cancer cells. Mol. Med. Rep. 2015, 12, 5100–5108. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, J.; Tang, J.; Liu, X.; Zhong, Q.; Wang, F.; Hu, W.; Yuan, Z.; Nie, C.; Wei, Y. JNK- and Akt-mediated Puma expression in the apoptosis of cisplatin-resistant ovarian cancer cells. Biochem. J. 2012, 444, 291–301. [Google Scholar] [CrossRef]
- Shoeb, M.; Ramana, K.V.; Srivastava, S.K. Aldose reductase inhibition enhances TRAIL-induced human colon cancer cell apoptosis through AKT/FOXO3a-dependent upregulation of death receptors. Free Radic. Biol. Med. 2013, 63, 280–290. [Google Scholar] [CrossRef]
- Dong, Z.B.; Wu, H.M.; He, Y.C.; Huang, Z.T.; Weng, Y.H.; Li, H.; Liang, C.; Yu, W.M.; Chen, W. MiRNA-124-3p.1 sensitizes hepatocellular carcinoma cells to sorafenib by regulating FOXO3a by targeting AKT2 and SIRT1. Cell Death Dis. 2022, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.H.; Ding, Z.B.; Zhou, J.; Hui, B.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; Dai, Z.; Peng, Y.F.; Gu, C.Y.; et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef]
- Yuan, H.; Li, A.J.; Ma, S.L.; Cui, L.J.; Wu, B.; Yin, L.; Wu, M.C. Inhibition of autophagy significantly enhances combination therapy with sorafenib and HDAC inhibitors for human hepatoma cells. World J. Gastroenterol. 2014, 20, 4953–4962. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Fu, J.; Xu, A.; Su, B.; Ren, Y.; Li, N.; Zhu, J.; Zhao, X.; Dai, R.; Cao, J.; et al. PSMD10/gankyrin induces autophagy to promote tumor progression through cytoplasmic interaction with ATG7 and nuclear transactivation of ATG7 expression. Autophagy 2016, 12, 1355–1371. [Google Scholar] [CrossRef]
- Ling, S.; Song, L.; Fan, N.; Feng, T.; Liu, L.; Yang, X.; Wang, M.; Li, Y.; Tian, Y.; Zhao, F.; et al. Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int. J. Oncol. 2017, 50, 297–309. [Google Scholar] [CrossRef]
- Liang, C.; Dong, Z.; Cai, X.; Shen, J.; Xu, Y.; Zhang, M.; Li, H.; Yu, W.; Chen, W. Hypoxia induces sorafenib resistance mediated by autophagy via activating FOXO3a in hepatocellular carcinoma. Cell Death Dis. 2020, 11, 1017. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Niu, Y.; Wan, A.; Chen, D.; Liang, H.; Chen, X.; Sun, L.; Zhan, S.; Chen, L.; Cheng, C.; et al. RNA m6A methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. 2020, 39, e103181. [Google Scholar] [CrossRef]
- Xi, H.; Wang, S.; Wang, B.; Hong, X.; Liu, X.; Li, M.; Shen, R.; Dong, Q. The role of interaction between autophagy and apoptosis in tumorigenesis (Review). Oncol. Rep. 2022, 48, 208. [Google Scholar] [CrossRef]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Lin, Y.; Gao, S.; Wang, Y.; Pan, H.; Wang, Z.; Pozzolini, M.; Yang, F.; Zhang, H.; Yang, Y.; et al. Luteolin inhibits triple-negative breast cancer by inducing apoptosis and autophagy through SGK1-FOXO3a-BNIP3 signaling. Front. Pharmacol. 2023, 14, 1200843. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Pan, P.; Zhao, Q.; Liu, W.; Sun, Y.; Wang, J.; Liu, C.; Wang, C. Overcoming Basal Autophagy, Kangai Injection Enhances Cisplatin Cytotoxicity by Regulating FOXO3a-Dependent Autophagic Cell Death and Apoptosis in Human Lung Adenocarcinoma A549/DDP Cells. BioMed Res. Int. 2022, 2022, 6022981. [Google Scholar] [CrossRef]
- Lee, I.H.; Kawai, Y.; Fergusson, M.M.; Rovira, I.I., II; Bishop, A.J.; Motoyama, N.; Cao, L.; Finkel, T. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science 2012, 336, 225–228. [Google Scholar] [CrossRef]
- Lim, S.M.; Mohamad Hanif, E.A.; Chin, S.F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Key Target | PDB ID | Binding Energy (−kcal/mol) |
|---|---|---|---|
| 1,25(OH)2D3 | FOXO3A | 2UZK | −8.62 |
| Maxacalcitol | FOXO3A | 2UZK | −8.47 |
| Calcipotriol | FOXO3A | 2UZK | −7.99 |
| Seocalcitol | FOXO3A | 2UZK | −8.54 |
| Tacalcitol | FOXO3A | 2UZK | −8.57 |
| Group | Joint Concentration | q-Value |
|---|---|---|
| Sorafenib + 1,25(OH)2D3 | 4.3 + 5 | 2.496671581 |
| Sorafenib + 1,25(OH)2D3 | 8.6 + 5 | 1.719277168 |
| Sorafenib + 1,25(OH)2D3 | 17.2 + 5 | 1.401718285 |
| Sorafenib + 1,25(OH)2D3 | 34.4 + 5 | 1.099641231 |
| Sorafenib + 1,25(OH)2D3 | 68.8 + 5 | 1.015836282 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, Z.; Wu, X.; Luo, T.; Chen, X.; Huang, J.; Zhang, S. Exploring the Molecular Mechanism of 1,25(OH)2D3 Reversal of Sorafenib Resistance in Hepatocellular Carcinoma Based on Network Pharmacology and Experimental Validation. Curr. Issues Mol. Biol. 2025, 47, 319. https://doi.org/10.3390/cimb47050319
Long Z, Wu X, Luo T, Chen X, Huang J, Zhang S. Exploring the Molecular Mechanism of 1,25(OH)2D3 Reversal of Sorafenib Resistance in Hepatocellular Carcinoma Based on Network Pharmacology and Experimental Validation. Current Issues in Molecular Biology. 2025; 47(5):319. https://doi.org/10.3390/cimb47050319
Chicago/Turabian StyleLong, Zhiyan, Xiangyi Wu, Tianxin Luo, Xiaomei Chen, Jian Huang, and Shu Zhang. 2025. "Exploring the Molecular Mechanism of 1,25(OH)2D3 Reversal of Sorafenib Resistance in Hepatocellular Carcinoma Based on Network Pharmacology and Experimental Validation" Current Issues in Molecular Biology 47, no. 5: 319. https://doi.org/10.3390/cimb47050319
APA StyleLong, Z., Wu, X., Luo, T., Chen, X., Huang, J., & Zhang, S. (2025). Exploring the Molecular Mechanism of 1,25(OH)2D3 Reversal of Sorafenib Resistance in Hepatocellular Carcinoma Based on Network Pharmacology and Experimental Validation. Current Issues in Molecular Biology, 47(5), 319. https://doi.org/10.3390/cimb47050319