
A Multi-Model Machine Learning Framework for Identifying Raloxifene as a Novel RNA Polymerase Inhibitor from FDA-Approved Drugs



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Traditional Machine Learning
2.2. Convolutional Neural Network (CNN) Deep Machine Learning
2.3. FDA-Approved Drug Library
2.4. RxNav Drug Classification
2.5. Molecular Docking
2.6. Molecular Dynamics Simulation
3. Results
3.1. Performance Evaluation of Machine Learning Classifiers for RNA Polymerase Inhibitor Prediction
3.2. Evaluation and Performance Analysis of Convolutional Neural Network Deep Learning Model for RNA Polymerase Inhibitor Prediction
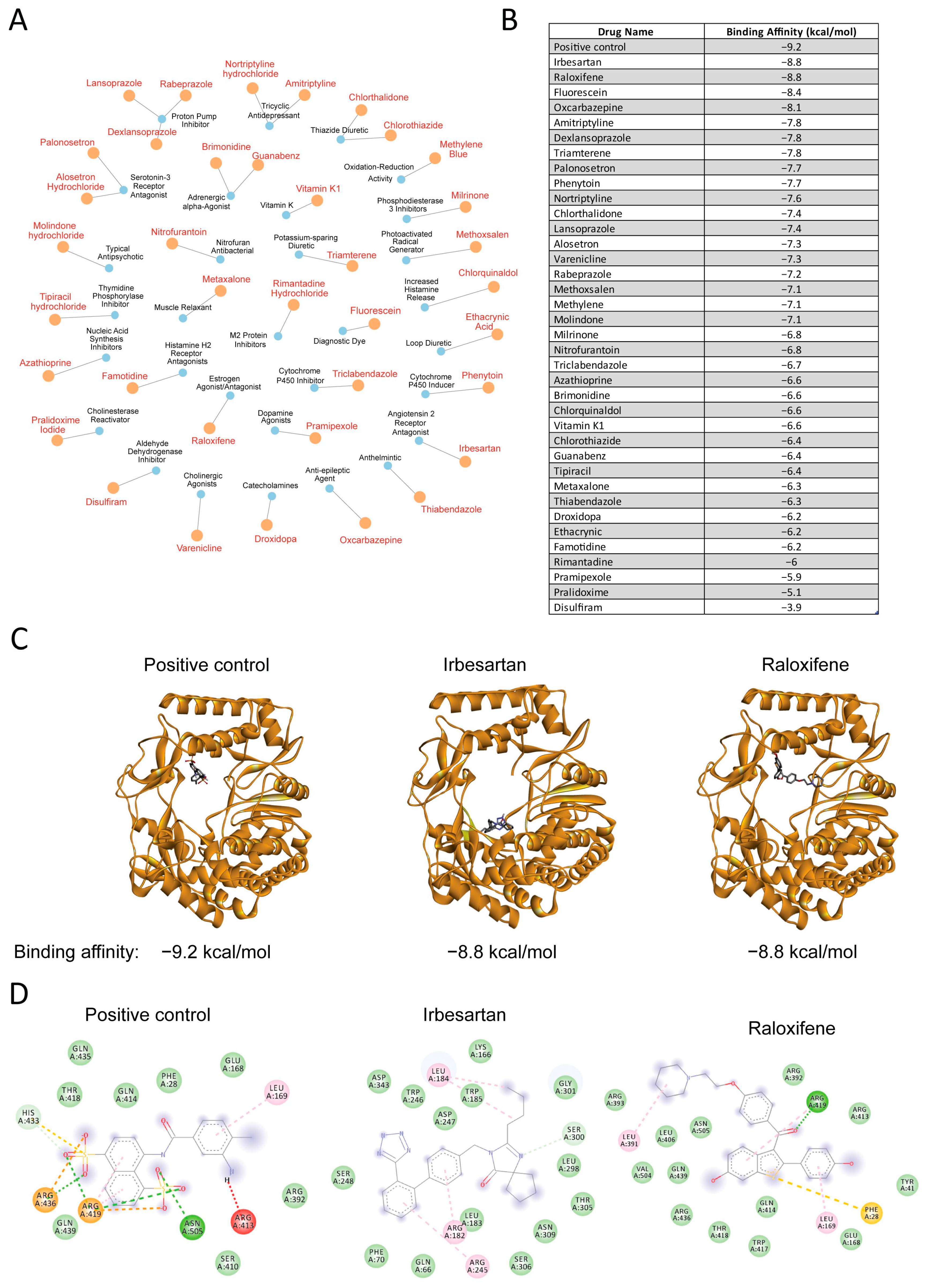
3.3. Drug-Target Network Analysis and Molecular Docking Results
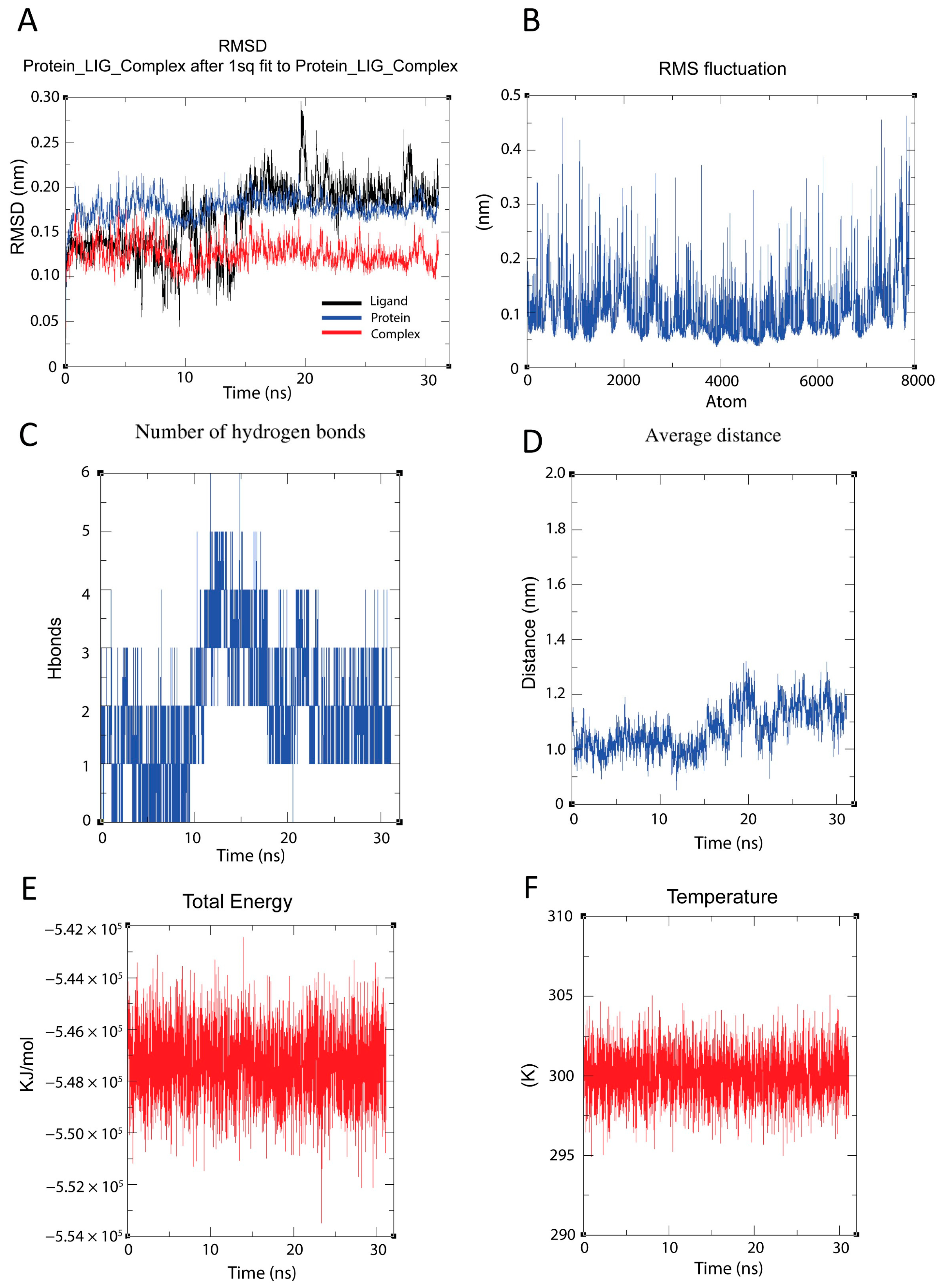
3.4. Molecular Dynamics Simulation Analysis Reveals Stable Binding Between RdRP and Raloxifene
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pathania, S.; Rawal, R.K.; Singh, P.K. RdRp (RNA-dependent RNA polymerase): A key target providing anti-virals for the management of various viral diseases. J. Mol. Struct. 2022, 1250, 131756. [Google Scholar] [CrossRef] [PubMed]
- Ivashkina, N.; Wölk, B.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Penin, F.; Moradpour, D. The hepatitis C virus RNA-dependent RNA polymerase membrane insertion sequence is a transmembrane segment. J. Virol. 2002, 76, 13088–13093. [Google Scholar] [CrossRef] [PubMed]
- Deval, J.; Jin, Z.; Chuang, Y.-C.; Kao, C.C. Structure(s), function(s), and inhibition of the RNA-dependent RNA polymerase of noroviruses. Virus Res. 2017, 234, 21–33. [Google Scholar] [CrossRef]
- Tian, L.; Qiang, T.; Liang, C.; Ren, X.; Jia, M.; Zhang, J.; Li, J.; Wan, M.; YuWen, X.; Li, H.; et al. RNA-dependent RNA polymerase (RdRp) inhibitors: The current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021, 213, 113201. [Google Scholar] [CrossRef]
- Venkataraman, S.; Prasad, B.; Selvarajan, R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef]
- Malone, B.F.; Perry, J.K.; Olinares, P.D.B.; Lee, H.W.; Chen, J.; Appleby, T.C.; Feng, J.Y.; Bilello, J.P.; Ng, H.; Sotiris, J.; et al. Structural basis for substrate selection by the SARS-CoV-2 replicase. Nature 2023, 614, 781–787. [Google Scholar] [CrossRef]
- Nie, Z.; Zhai, F.; Zhang, H.; Zheng, H.; Pei, J. The multiple roles of viral 3D(pol) protein in picornavirus infections. Virulence 2024, 15, 2333562. [Google Scholar] [CrossRef]
- Rohayem, J.; Jäger, K.; Robel, I.; Scheffler, U.; Temme, A.; Rudolph, W. Characterization of norovirus 3Dpol RNA-dependent RNA polymerase activity and initiation of RNA synthesis. J. Gen. Virol. 2006, 87, 2621–2630. [Google Scholar] [CrossRef] [PubMed]
- “Raloxifene Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 26 February 2025. Available online: https://www.drugs.com/ (accessed on 26 February 2025).
- National Center for Biotechnology Information. PubChem Bioassay Record for AID 588519, A Screen for Compounds That Inhibit Viral RNA Polymerase Binding and Polymerization Activities, Source: ICCB-Longwood Screening Facility, Harvard Medical School. Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/588519 (accessed on 21 February 2025).
- Campagnola, G.; Gong, P.; Peersen, O.B. High-throughput screening identification of poliovirus RNA-dependent RNA polymerase inhibitors. Antivir. Res. 2011, 91, 241–251. [Google Scholar] [CrossRef]
- RDKit: Open-Source Cheminformatics. Available online: https://www.rdkit.org (accessed on 20 October 2024).
- Muhamad Iqbal Januadi, P.; Vincent, A. Comparison of Machine Learning Land Use-Land Cover Supervised Classifiers Performance on Satellite Imagery Sentinel 2 using Lazy Predict Library. Indones. J. Data Sci. 2023, 4, 183–189. [Google Scholar] [CrossRef]
- Li, W.; Ma, W.; Yang, M.; Tang, X. Drug repurposing based on the DTD-GNN graph neural network: Revealing the relationships among drugs, targets and diseases. BMC Genom. 2024, 25, 584. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Yun, J.; Coban Akdemir, Z.H.; Jiang, X.; Wu, E.; Huang, J.H.; Sahni, N.; Yi, S.S. AI-DrugNet: A network-based deep learning model for drug repurposing and combination therapy in neurological disorders. Comput. Struct. Biotechnol. J. 2023, 21, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Amiri, R.; Razmara, J.; Parvizpour, S.; Izadkhah, H. A novel efficient drug repurposing framework through drug-disease association data integration using convolutional neural networks. BMC Bioinform. 2023, 24, 442. [Google Scholar] [CrossRef] [PubMed]
- Croci, R.; Pezzullo, M.; Tarantino, D.; Milani, M.; Tsay, S.C.; Sureshbabu, R.; Tsai, Y.J.; Mastrangelo, E.; Rohayem, J.; Bolognesi, M.; et al. Structural bases of norovirus RNA dependent RNA polymerase inhibition by novel suramin-related compounds. PLoS ONE 2014, 9, e91765. [Google Scholar] [CrossRef] [PubMed]
- Cousins, K.R. Computer Review of ChemDraw Ultra 12.0. J. Am. Chem. Soc. 2011, 133, 8388. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef]
- Kim, G.; Van, N.T.H.; Nam, J.H.; Lee, W. Unraveling the Molecular Reason of Opposing Effects of α-Mangostin and Norfluoxetine on TREK-2 at the Same Binding Site. ChemMedChem 2024, 19, e202400409. [Google Scholar] [CrossRef]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System, Open Source PyMOL. 2010. Available online: https://www.pymol.org/ (accessed on 20 November 2024).
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Lee, G.J.; Kim, B.; Kim, H.J.; Tak, J.; Park, M.K.; Kim, E.J.; Kang, G.J.; Rho, S.B.; Lee, H.; et al. Penfluridol suppresses MYC-driven ANLN expression and liver cancer progression by disrupting the KEAP1–NRF2 interaction. Pharmacol. Res. 2024, 210, 107512. [Google Scholar] [CrossRef]
- Tuan, N.M.; Lee, C.H. Penfluridol as a Candidate of Drug Repurposing for Anticancer Agent. Molecules 2019, 24, 3659. [Google Scholar] [CrossRef]
- D’Amelio, P.; Isaia, G.C. The use of raloxifene in osteoporosis treatment. Expert. Opin. Pharmacother. 2013, 14, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Vogel, V.G. Update on raloxifene: Role in reducing the risk of invasive breast cancer in postmenopausal women. Breast Cancer Targets Ther. 2011, 3, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Chang, J.; Jeong, K.; Lee, W. Raloxifene as a treatment option for viral infections. J. Microbiol. 2021, 59, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Iaconis, D.; Bordi, L.; Matusali, G.; Talarico, C.; Manelfi, C.; Cesta, M.C.; Zippoli, M.; Caccuri, F.; Bugatti, A.; Zani, A.; et al. Characterization of raloxifene as a potential pharmacological agent against SARS-CoV-2 and its variants. Cell Death Dis. 2022, 13, 498. [Google Scholar] [CrossRef]
- Allegretti, M.; Cesta, M.C.; Zippoli, M.; Beccari, A.; Talarico, C.; Mantelli, F.; Bucci, E.M.; Scorzolini, L.; Nicastri, E. Repurposing the estrogen receptor modulator raloxifene to treat SARS-CoV-2 infection. Cell Death Differ. 2022, 29, 156–166. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Ben Ouirane, K.; Boulard, Y.; Bressanelli, S. The hepatitis C virus RNA-dependent RNA polymerase directs incoming nucleotides to its active site through magnesium-dependent dynamics within its F motif. J. Biol. Chem. 2019, 294, 7573–7587. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, Q.; Shao, Y.; Ding, S.; Guo, J.; Gao, G.F.; Deng, T. Influenza A virus NS2 protein acts on vRNA-resident polymerase to drive the transcription to replication switch. Nucleic Acids Res. 2025, 53, gkaf027. [Google Scholar] [CrossRef]
- Tchesnokov, E.P.; Raeisimakiani, P.; Ngure, M.; Marchant, D.; Götte, M. Recombinant RNA-Dependent RNA Polymerase Complex of Ebola Virus. Sci. Rep. 2018, 8, 3970. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van, N.T.H.; Nguyen, M.T. A Multi-Model Machine Learning Framework for Identifying Raloxifene as a Novel RNA Polymerase Inhibitor from FDA-Approved Drugs. Curr. Issues Mol. Biol. 2025, 47, 315. https://doi.org/10.3390/cimb47050315
Van NTH, Nguyen MT. A Multi-Model Machine Learning Framework for Identifying Raloxifene as a Novel RNA Polymerase Inhibitor from FDA-Approved Drugs. Current Issues in Molecular Biology. 2025; 47(5):315. https://doi.org/10.3390/cimb47050315
Chicago/Turabian StyleVan, Nhung Thi Hong, and Minh Tuan Nguyen. 2025. "A Multi-Model Machine Learning Framework for Identifying Raloxifene as a Novel RNA Polymerase Inhibitor from FDA-Approved Drugs" Current Issues in Molecular Biology 47, no. 5: 315. https://doi.org/10.3390/cimb47050315
APA StyleVan, N. T. H., & Nguyen, M. T. (2025). A Multi-Model Machine Learning Framework for Identifying Raloxifene as a Novel RNA Polymerase Inhibitor from FDA-Approved Drugs. Current Issues in Molecular Biology, 47(5), 315. https://doi.org/10.3390/cimb47050315