
Retinal Dystrophies Associated with Mutations in the RP1 Gene: Genotype–Phenotype Correlations

 , , , ,
, , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
Molecular Genetic Analysis
3. Results
3.1. Phenotype–Genotype Correlation in RP1-Associated RP
3.2. Genetic Characteristics
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.T.; Moekotte, L.; Plomp, A.S.; Bergen, A.A.; van Genderen, M.M.; Boon, C.J.F. Retinitis pigmentosa: Current clinical management and emerging therapies. Int. J. Mol. Sci. 2023, 24, 7481. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Ramprasad, V.L.; Soumittra, N.; Mohamed, M.D.; Jafri, H.; Rashid, Y.; Danciger, M.; McKibbin, M.; Kumaramanickavel, G.; Inglehearn, C.F. A missense mutation in the nuclear localization signal sequence of CERKL (p.R106S) causes autosomal recessive retinal degeneration. Mol. Vis. 2008, 14, 1960–1964. [Google Scholar]
- García Bohórquez, B.; Aller, E.; Rodríguez Muñoz, A.; Jaijo, T.; García García, G.; Millán, J.M. Updating the genetic landscape of inherited retinal dystrophies. Front. Cell Dev. Biol. 2021, 9, 645600. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.; Bartmann, L.; Ramesar, R.; Greenberg, J. Novel variants in the hotspot region of RP1 in South African patients with retinitis pigmentosa. Mol. Vis. 2006, 12, 177–183. [Google Scholar]
- Pierce, E.A.; Quinn, T.; Meehan, T.; McGee, T.L.; Berson, E.L.; Dryja, T.P. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat. Genet. 1999, 22, 248–254. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V.; Iannaccone, A.; Weleber, R.G.; Fishman, G.A.; Maguire, A.M.; Affatigato, L.M.; Bennett, J.; Pierce, E.A.; Danciger, M.; et al. Disease expression of RP1 mutations causing autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1898–1908. [Google Scholar]
- Berson, E.L.; Grimsby, J.L.; Adams, S.M.; McGee, T.L.; Sweklo, E.; Pierce, E.A.; Sandberg, M.A.; Dryja, T.P. Clinical features and mutations in patients with dominant retinitis pigmentosa-1 (RP1). Investig. Ophthalmol. Vis. Sci. 2001, 42, 2217–2224. [Google Scholar]
- Audo, I.; Lancelot, M.E.; Mohand-Saïd, S.; Antonio, A.; Germain, A.; Sahel, J.A.; Bhattacharya, S.S.; Zeitz, C. Novel C2orf71 mutations account for ∼1% of cases in a large French arRP cohort. Hum. Mutat. 2011, 32, E2091–E2103. [Google Scholar] [CrossRef]
- Khaliq, S.; Abid, A.; Ismail, M.; Hameed, A.; Mohyuddin, A.; Lall, P.; Aziz, A.; Anwar, K.; Mehdi, S.Q. Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. J. Med. Genet. 2005, 42, 436–438. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.S.; Salles, M.V.; Motta, F.L.; Sallum, J.M.F. Retinitis pigmentosa due to Rp1 biallelic variants. Sci. Rep. 2020, 10, 1603. [Google Scholar]
- Colombo, L.; Maltese, P.E.; Castori, M.; El Shamieh, S.; Zeitz, C.; Audo, I.; Zulian, A.; Marinelli, C.; Benedetti, S.; Costantini, A.; et al. Molecular epidemiology in 591 italian probands with nonsyndromic retinitis pigmentosa and usher syndrome. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar]
- El Shamieh, S.; Boulanger-Scemama, E.; Lancelot, M.E.; Antonio, A.; Démontant, V.; Condroyer, C.; Letexier, M.; Saraiva, J.P.; Mohand-Saïd, S.; Sahel, J.A.; et al. Targeted next generation sequencing identifies novel mutations in RP1 as a relatively common cause of autosomal recessive rod-cone dystrophy. BioMed Res. Int. 2015, 2015, 485624. [Google Scholar]
- Avila-Fernandez, A.; Corton, M.; Nishiguchi, K.M.; Muñoz-Sanz, N.; Benavides-Mori, B.; Blanco-Kelly, F.; Riveiro-Alvarez, R.; Garcia-Sandoval, B.; Rivolta, C.; Ayuso, C. Identification of an RP1 prevalent founder mutation and related phenotype in spanish patients with early-onset autosomal recessive retinitis. Ophthalmology 2012, 119, 2616–2621. [Google Scholar]
- D’Esposito, F.; Randazzo, V.; Vega, M.I.; Esposito, G.; Maltese, P.E.; Torregrossa, S.; Scibetta, P.; Listì, F.; Gagliano, C.; Scalia, L.; et al. RP1 dominant p.Ser740* pathogenic variant in 20 knowingly unrelated families affected by rod-cone dystrophy: Potential founder effect in western sicily. Medicina 2024, 60, 254. [Google Scholar] [CrossRef]
- Verbakel, S.K.; van Huet, R.A.C.; den Hollander, A.I.; Geerlings, M.J.; Kersten, E.; Klevering, B.J.; Klaver, C.C.W.; Plomp, A.S.; Wesseling, N.L.; Bergen, A.A.B.; et al. Macular dystrophy and cone-rod dystrophy caused by mutations in the RP1 gene: Extending the RP1 disease spectrum. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1192–1203. [Google Scholar]
- Riera, M.; Abad-Morales, V.; Navarro, R.; Ruiz-Nogales, S.; Méndez-Vendrell, P.; Corcostegui, B.; Pomares, E. Expanding the retinal phenotype of RP1: From retinitis pigmentosa to a novel and singular macular dystrophy. Br. J. Ophthalmol. 2020, 104, 173–181. [Google Scholar] [CrossRef]
- Payne, A.; Vithana, E.; Khaliq, S.; Hameed, A.; Deller, J.; Abu-Safieh, L.; Kermani, S.; Leroy, B.P.; Mehdi, S.Q.; Moore, A.T.; et al. RP1 protein truncating mutations predominate at the RP1 adRP locus. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4069–4073. [Google Scholar]
- Schwartz, S.B.; Aleman, T.S.; Cideciyan, A.V.; Swaroop, A.; Jacobson, S.G.; Stone, E.M. De novo mutation in the RP1 gene (Arg677ter) associated with retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3593–3597. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.J.; Law, J.P.; Lai, T.Y.; Chiang, S.W.; Tam, P.O.; Chu, K.Y.; Wang, N.; Zhang, M.; Pang, C.P. Differential pattern of RP1 mutations in retinitis pigmentosa. Mol. Vis. 2010, 16, 1353–1360. [Google Scholar] [PubMed]
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Self, J.E.; Dunn, M.J.; Erichsen, J.T.; Gottlob, I.; Griffiths, H.J.; Harris, C.; Lee, H.; Owen, J.; Sanders, J.; Shawkat, F.; et al. Nystagmus UK Eye research group (NUKE). Management of nystagmus in children: A review of the literature and current practice in UK specialist services. Eye 2020, 34, 1515–1534. [Google Scholar] [CrossRef] [PubMed]
- Karali, M.; Testa, F.; Di Iorio, V.; Torella, A.; Zeuli, R.; Scarpato, M.; Romano, F.; Onore, M.E.; Pizzo, M.; Melillo, P.; et al. Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy. Sci. Rep. 2022, 12, 20815. [Google Scholar] [CrossRef]
- Méndez-Vidal, C.; Bravo-Gil, N.; González-Del Pozo, M.; Vela-Boza, A.; Dopazo, J.; Borrego, S.; Antiñolo, G. Novel RP1 mutations and a recurrent BBS1 variant explain the co-existence of two distinct retinal phenotypes in the same pedigree. BMC Genet. 2014, 15, 143. [Google Scholar] [CrossRef]
- Bowne, S.J.; Daiger, S.P.; Hims, M.M.; Sohocki, M.M.; Malone, K.A.; McKie, A.B.; Heckenlively, J.R.; Birch, D.G.; Inglehearn, C.F.; Bhattacharya, S.S.; et al. Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 1999, 8, 2121–2128. [Google Scholar] [CrossRef]
- Chiang, S.W.; Wang, D.Y.; Chan, W.M.; Tam, P.O.; Chong, K.K.; Lam, D.S.; Pang, C.P. A novel missense RP1 mutation in retinitis pigmentosa. Eye 2006, 20, 602–605. [Google Scholar] [CrossRef]
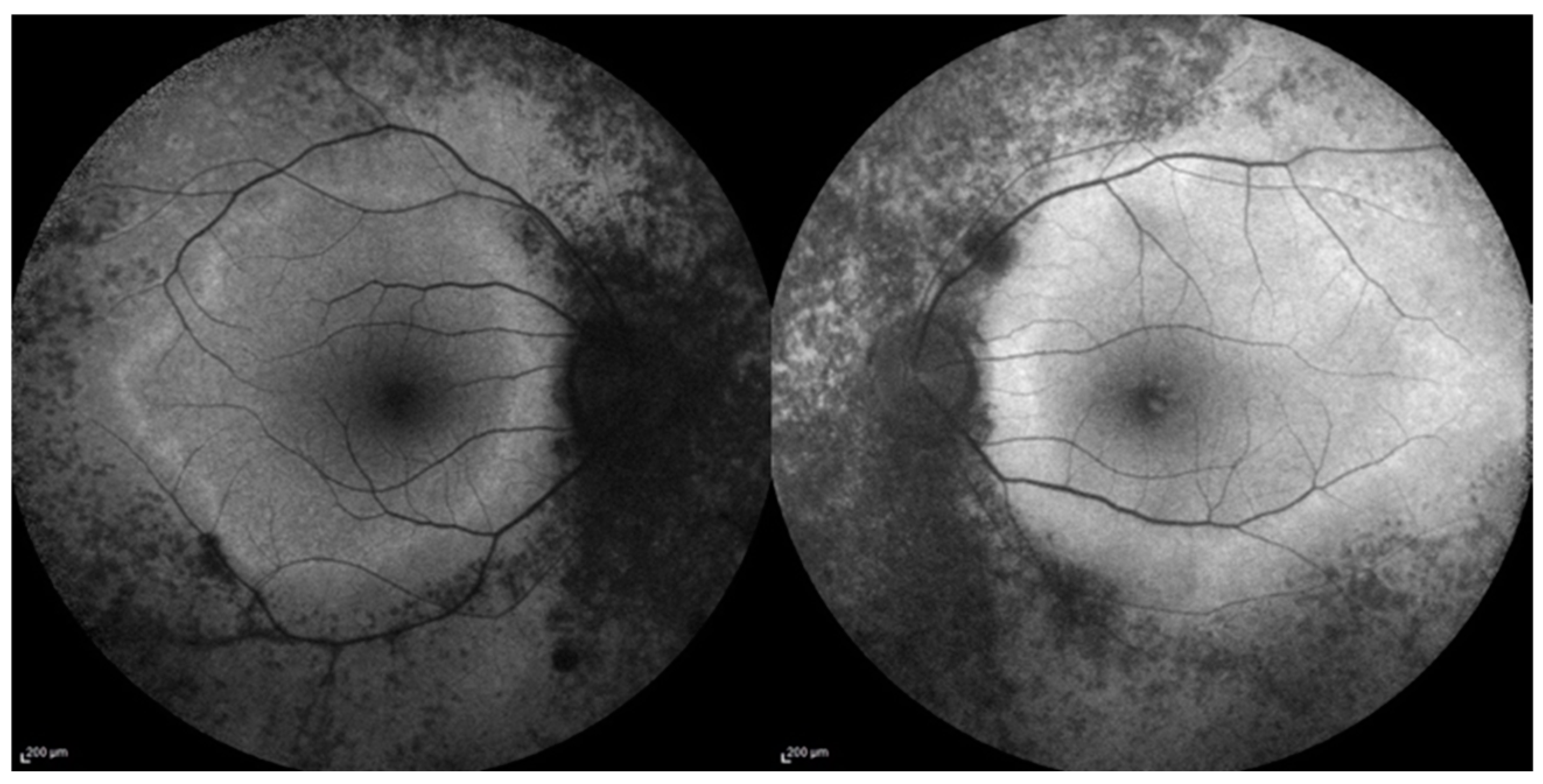
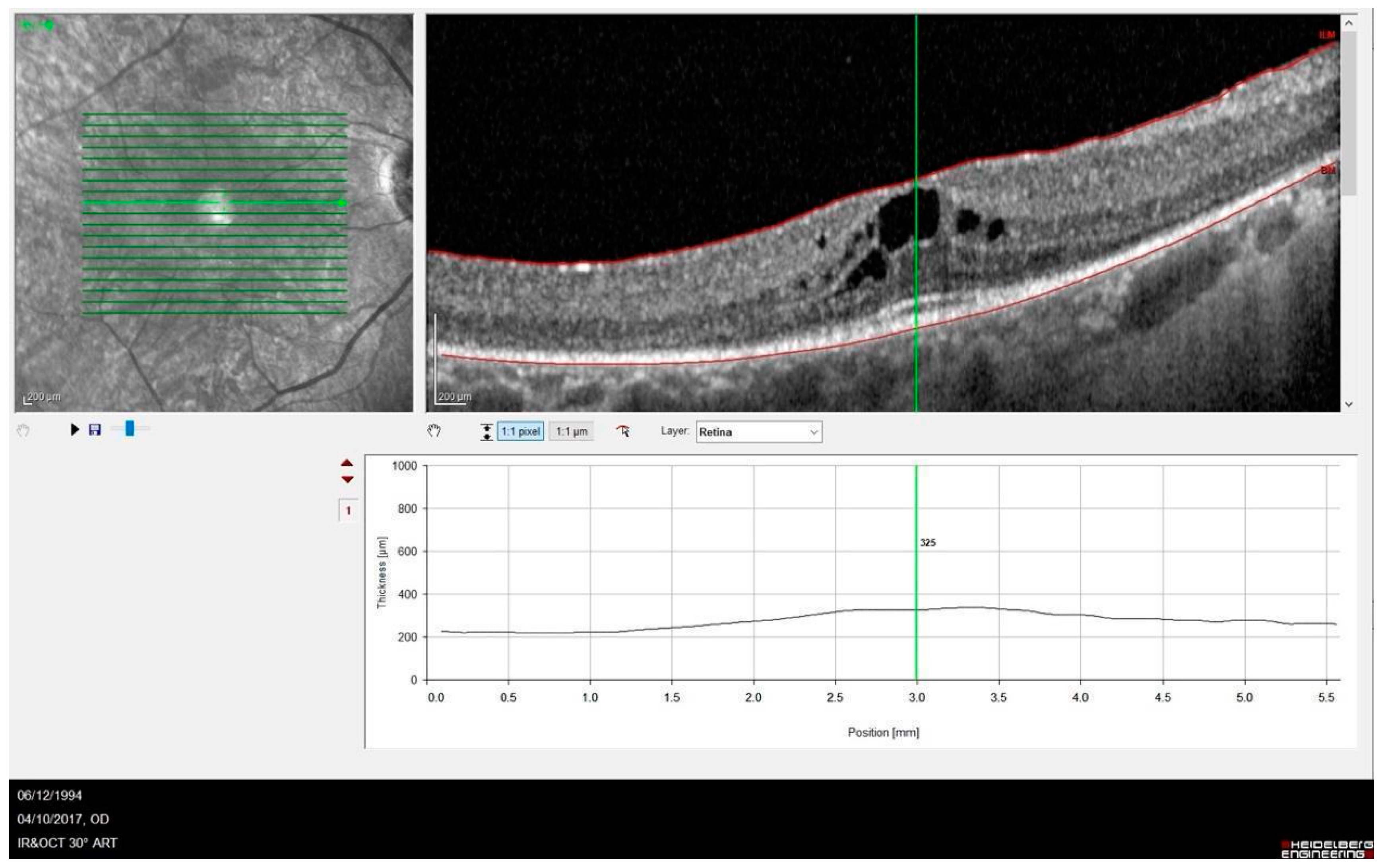


{kind=link}
{kind=link}
{kind=link}
| Patient | Gender | Age | Symptoms at Time of Diagnosis | Age of Symptom Onset | BCVA logMAR (RE; LE) | Genetic Variants |
|---|---|---|---|---|---|---|
| 1 | F | 66 | Reduced visual field | 55 | 0.1; 0.1 | c.2029C>T p.(Arg677*)/WT |
| 2 | F | 58 | Hemeralopia | 4 | HM; HM | c.5962dup p.(Ile1988Asnfs*3)/c.5962dup p.(Ile1988Asnfs*3) |
| 3 | F | 32 | Nystagmus | 2 | HM; 0.7 | c.2019C>T p.Arg677*)/WT |
| 4 | M | 60 | Hemeralopia | 43 | 0.2; 0.5 | c.2019C>T p.Arg677*)/WT |
| 5 | F | 77 | Hemeralopia | 47 | LP; LP | c.2029C>T p.(Arg677*)/WT |
| 6 | M | 52 | Reduced visual field | 48 | 0.1; 0.1 | c.2029C>T p.(Arg677*)/WT |
| 7 | M | 32 | Hemeralopia | 12 | HM; HM | c.1234dup (p.Met412Asnfs*7)/c.1234dup (p.Met412Asnfs*7) |
| 8 | F | 79 | Reduced visual field | 74 | LP; no LP | c.2029C>T p.(Arg677*)/WT |
| 9 | F | 77 | Hemeralopia | 30 | 0.8; 1.0 | c.2029C>T p.(Arg677*)/WT |
| 10 | M | 57 | Reduced visual field | 6 | LP; LP | c.5962dup p.(Ile1988Asnfs*3)/c.5962dup p.(Ile1988Asnfs*3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spagnuolo, V.; Piergentili, M.; Passerini, I.; Murro, V.; Mucciolo, D.P.; Giorgio, D.; Maccari, M.; Pelo, E.; Biagini, I.; Giansanti, F.; et al. Retinal Dystrophies Associated with Mutations in the RP1 Gene: Genotype–Phenotype Correlations. Curr. Issues Mol. Biol. 2025, 47, 212. https://doi.org/10.3390/cimb47030212
Spagnuolo V, Piergentili M, Passerini I, Murro V, Mucciolo DP, Giorgio D, Maccari M, Pelo E, Biagini I, Giansanti F, et al. Retinal Dystrophies Associated with Mutations in the RP1 Gene: Genotype–Phenotype Correlations. Current Issues in Molecular Biology. 2025; 47(3):212. https://doi.org/10.3390/cimb47030212
Chicago/Turabian StyleSpagnuolo, Vito, Marco Piergentili, Ilaria Passerini, Vittoria Murro, Dario Pasquale Mucciolo, Dario Giorgio, Martina Maccari, Elisabetta Pelo, Ilaria Biagini, Fabrizio Giansanti, and et al. 2025. "Retinal Dystrophies Associated with Mutations in the RP1 Gene: Genotype–Phenotype Correlations" Current Issues in Molecular Biology 47, no. 3: 212. https://doi.org/10.3390/cimb47030212
APA StyleSpagnuolo, V., Piergentili, M., Passerini, I., Murro, V., Mucciolo, D. P., Giorgio, D., Maccari, M., Pelo, E., Biagini, I., Giansanti, F., Virgili, G., & Sodi, A. (2025). Retinal Dystrophies Associated with Mutations in the RP1 Gene: Genotype–Phenotype Correlations. Current Issues in Molecular Biology, 47(3), 212. https://doi.org/10.3390/cimb47030212