
Detection of Helicobacter pylori and the Genotypes of Resistance to Clarithromycin, Fluoroquinolones, and Metronidazole in Gastric Biopsies: An In Silico Analysis to Help Understand Antibiotic Resistance

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. DNA Extraction
2.3. Helicobacter pylori Detection
2.4. Molecular Characterization of Genes Encoding Antibiotic Resistance
2.5. In Silico Mutational Analysis
- 23S rRNA Modeling and Analysis
- DNA Gyrase Subunit A Homology Modeling
- Oxygen-Insensitive NADPH Nitroreductase
3. Results
3.1. Molecular Analysis of Genes Involved in H. pylori Antibiotic Resistance
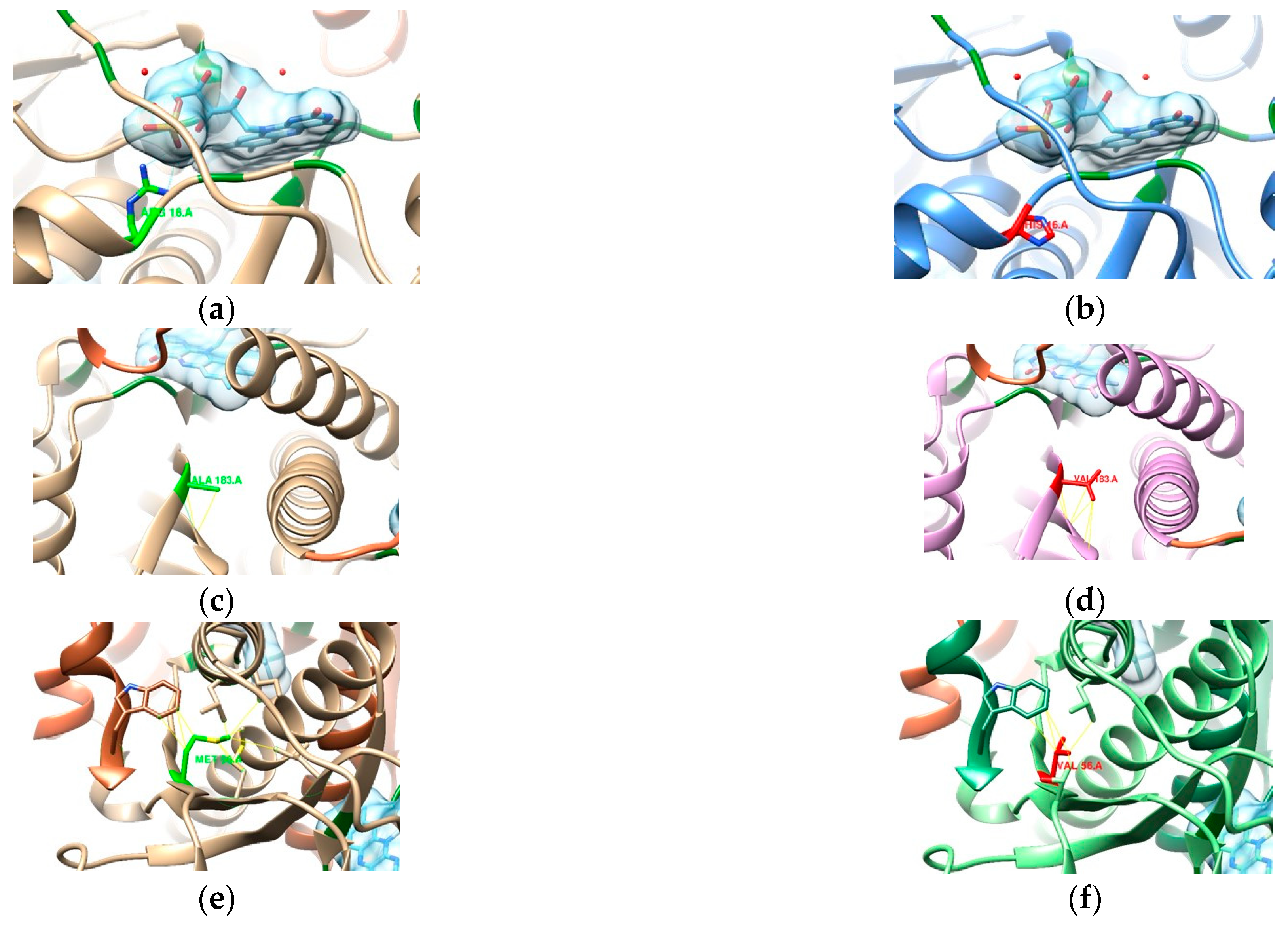
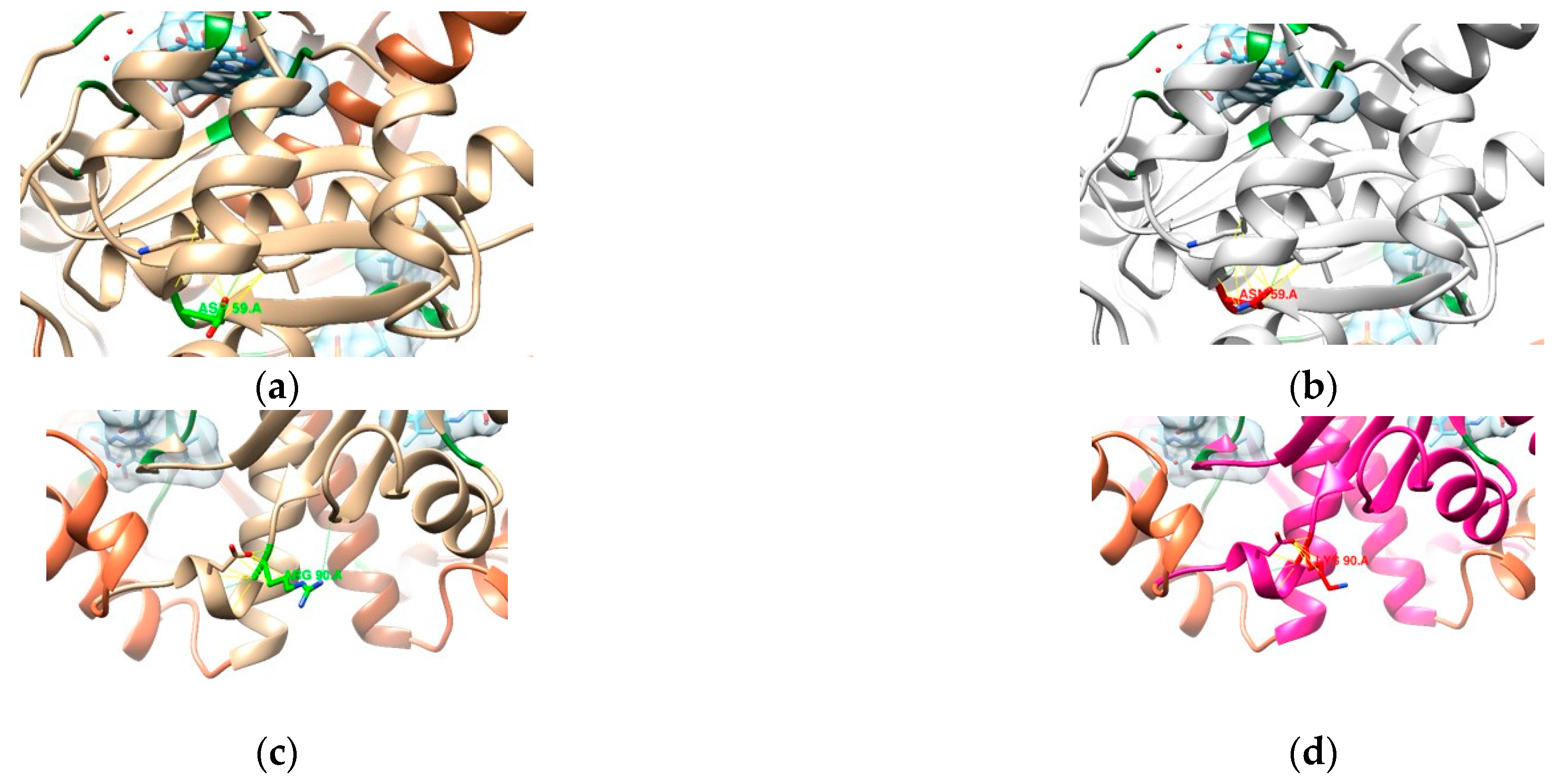
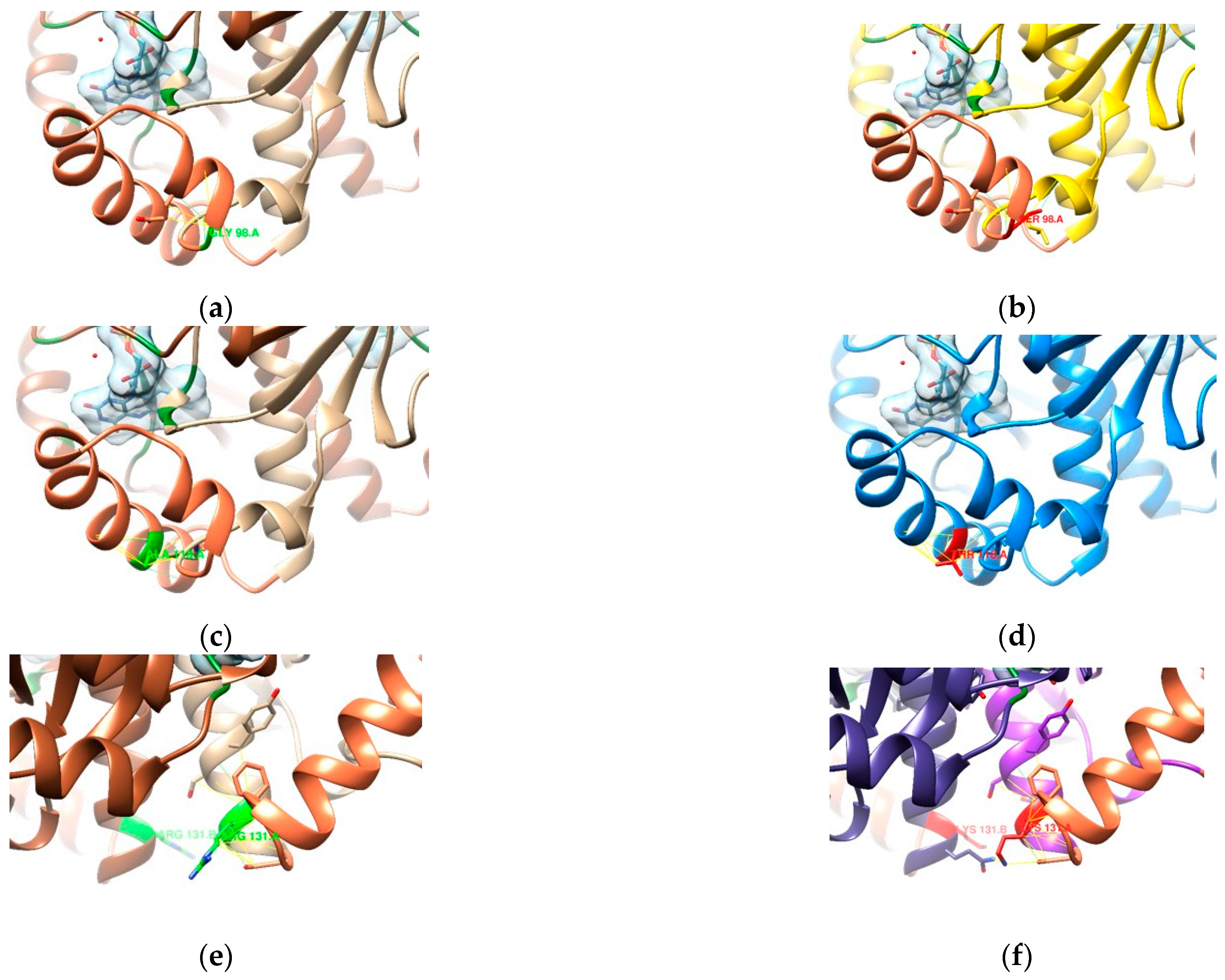
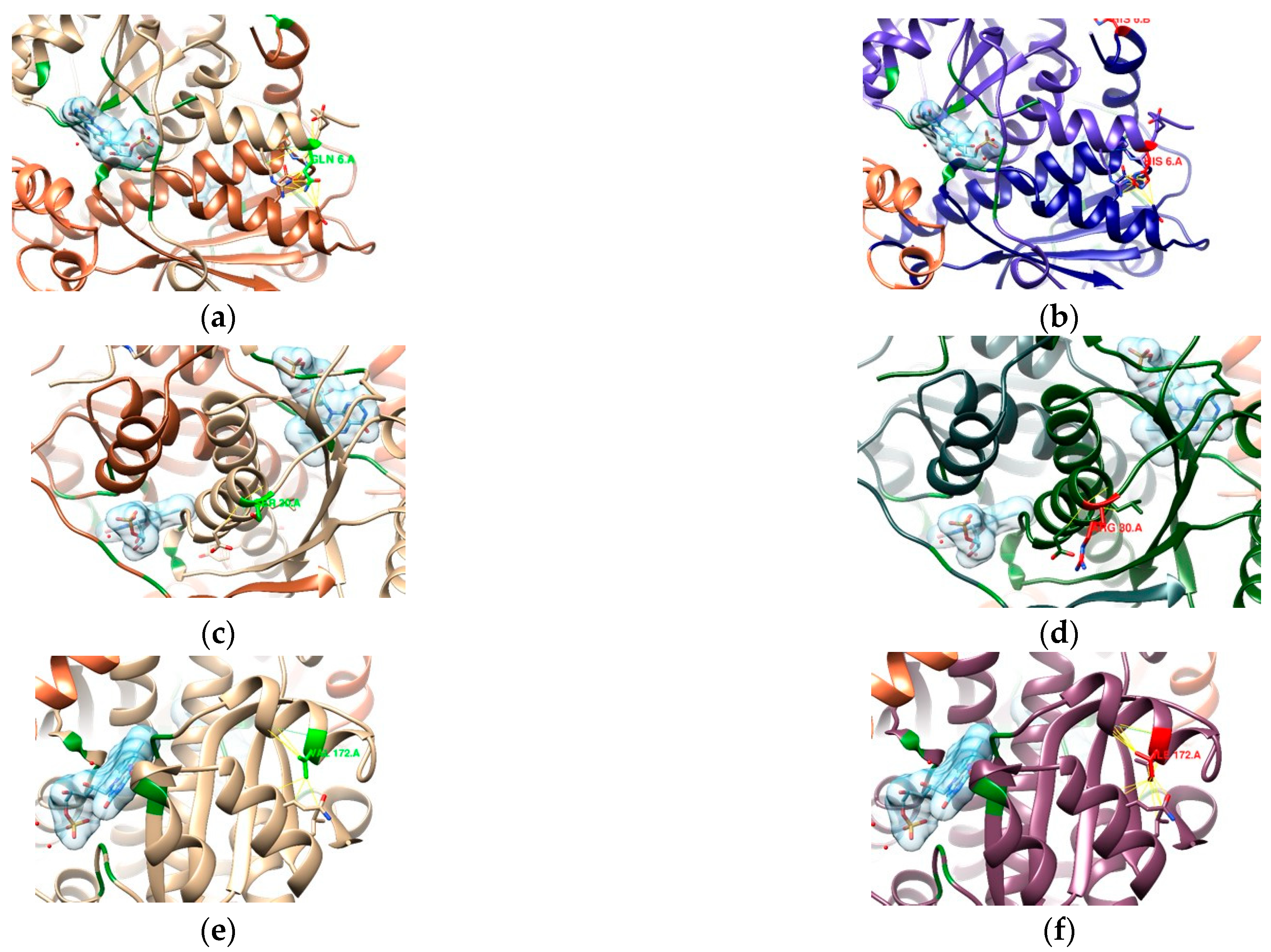
3.2. In Silico Mutational Analysis
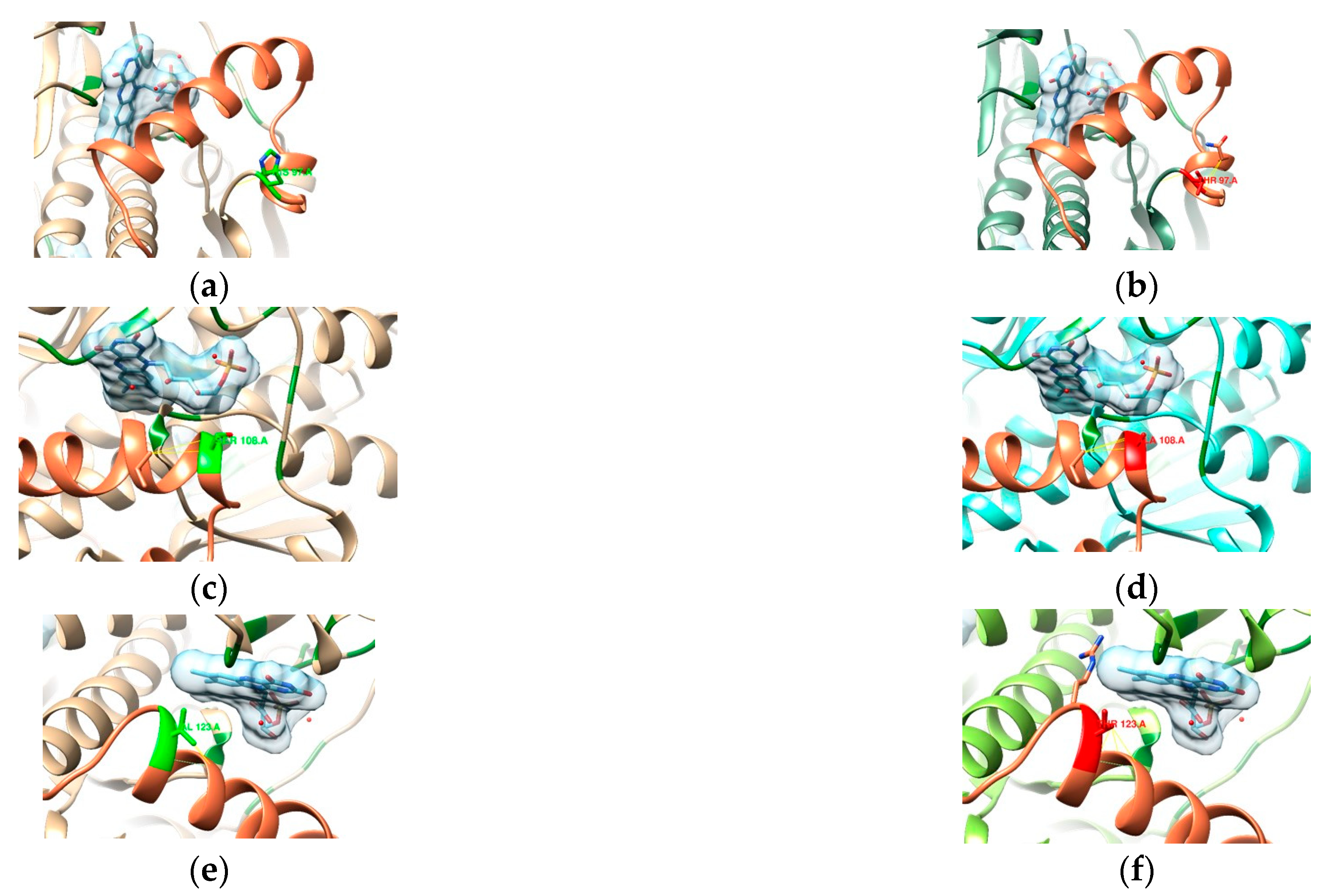
3.2.1. 23S rRNA
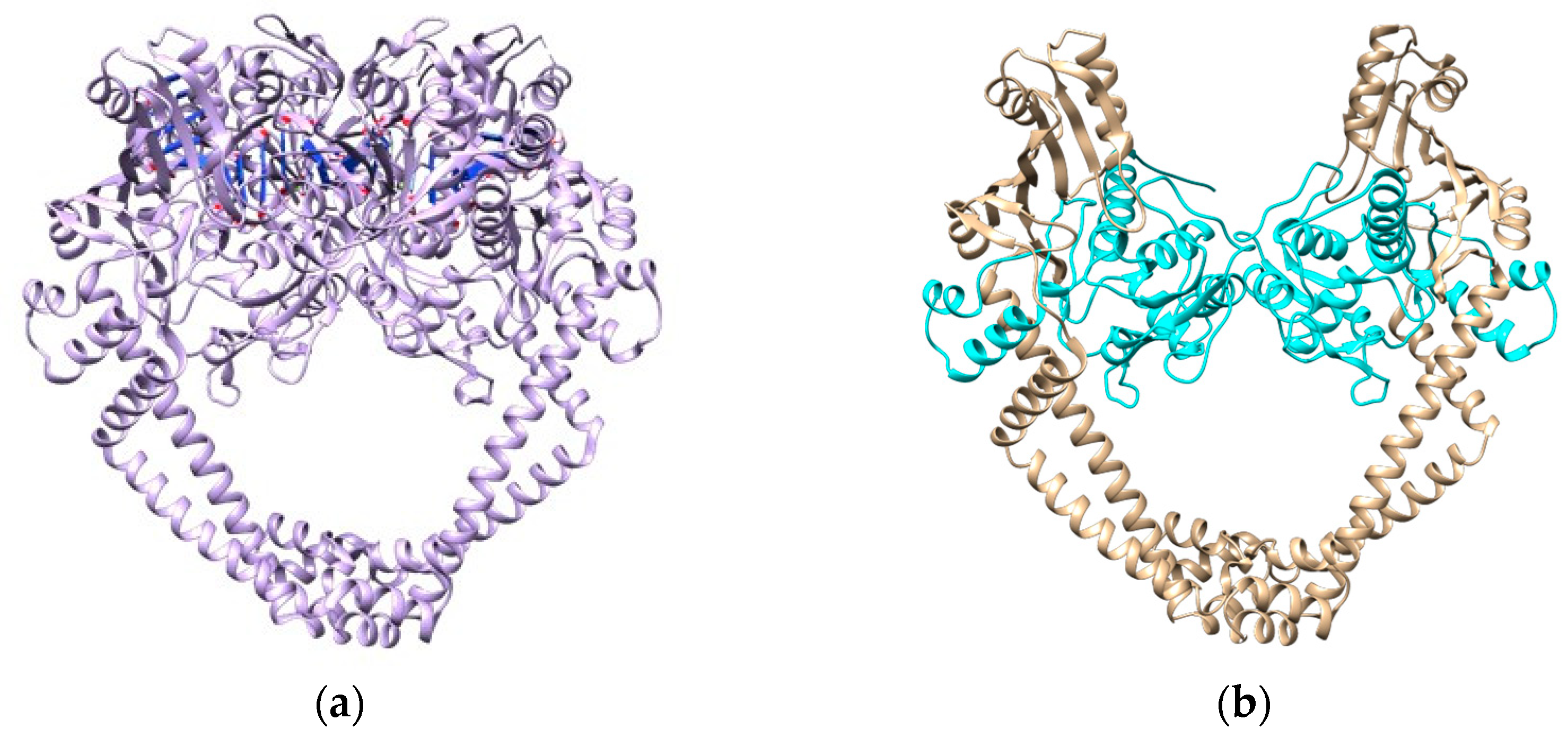
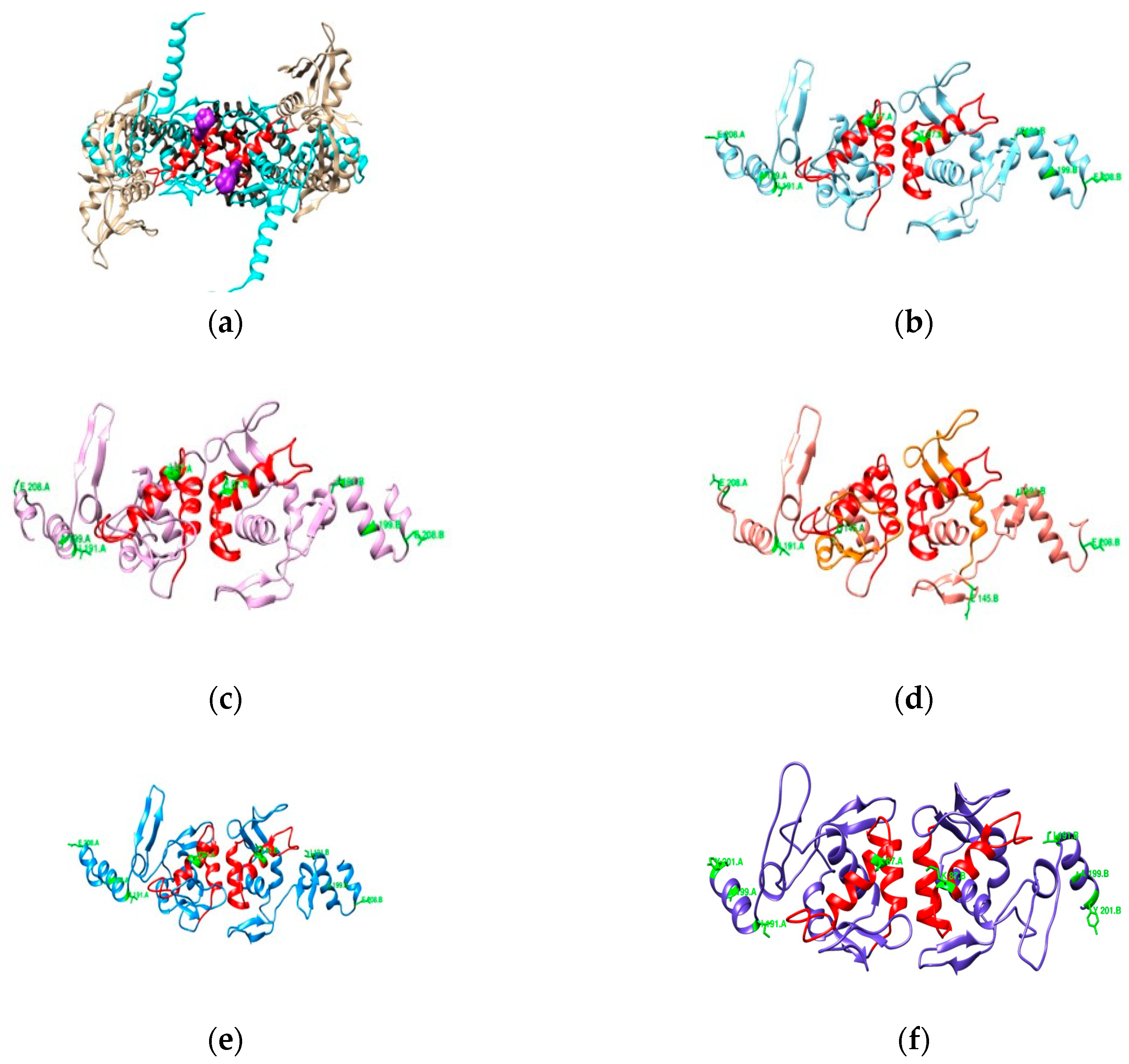
3.2.2. DNA Gyrase Subunit A Homology Modeling
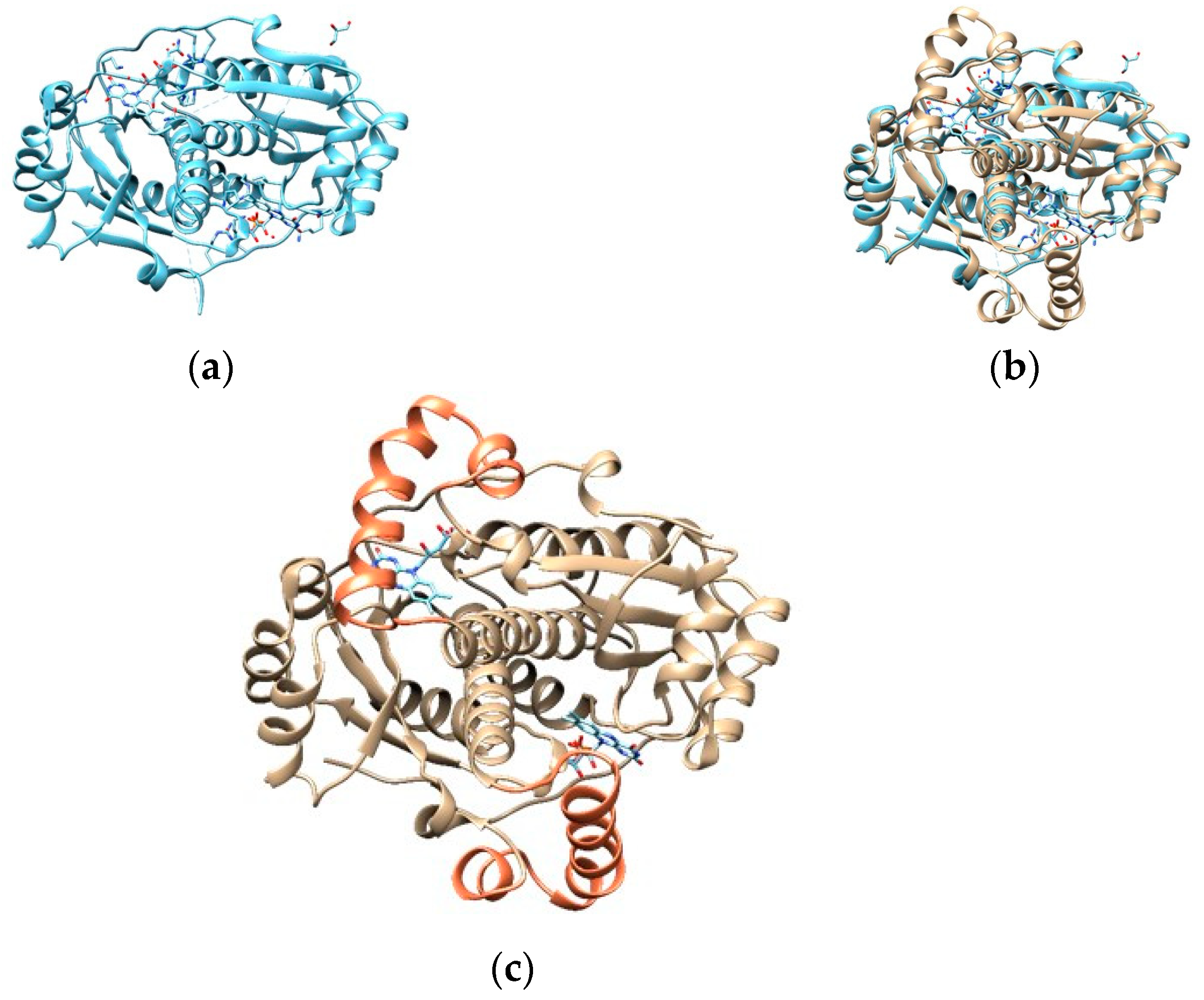
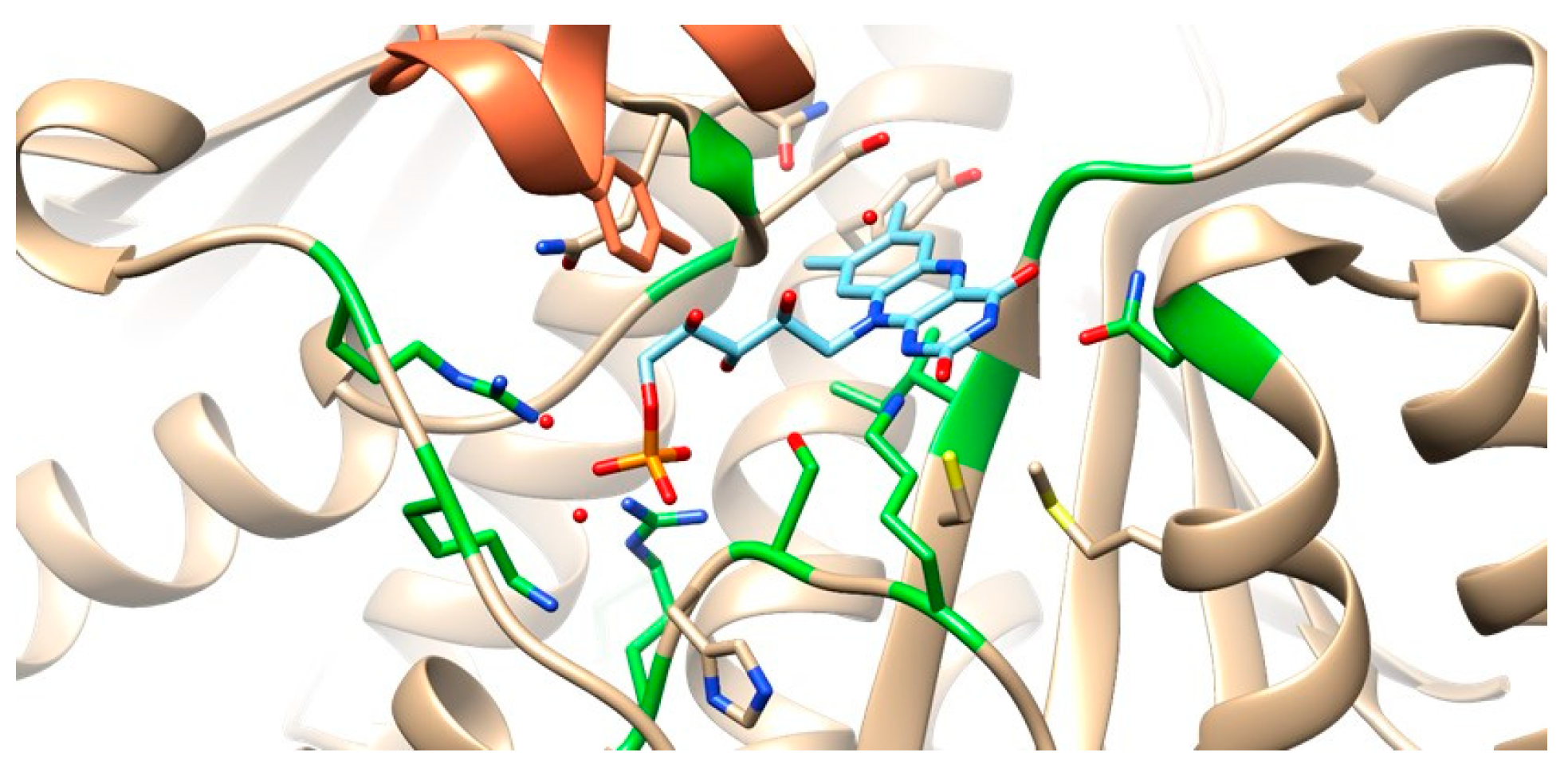
3.2.3. RdxA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yulizal, O.; Tarigan, S.; Isnainul, O.; Muttaqin, Z. Gastric histopathological features after the administration of omeprazole; amoxicillin; and clarithromycin in gastritis Helicobacter pylori rat model. J. Adv. Vet. Anim. Res. 2021, 8, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.E.; Omorogbe, J.; Hussey, M.; Tighe, D.; Holleran, G.; O’Morain, C.; Smith, S.M.; McNamara, D. Molecular detection of helicobacter pylori antibiotic resistance in stool vs biopsy samples. World J. Gastroenterol. 2016, 22, 9214–9221. [Google Scholar] [CrossRef]
- Singh, H.; Chung, H.; Awan, I.; Mone, A.; Cooper, R.; Patel, S.; Grigoryan, Z.; Ishida, Y.; Papachristou, C.; Judge, T.; et al. Designing strategies for eradication of Helicobacter pylori based on prevalence patterns of infection and antibiotic resistance in a low-income; medically underserved community in the United States. Helicobacter 2021, 26, e12769. [Google Scholar] [CrossRef]
- Savoldi, A.; Carrara, E.; Graham, D.Y.; Conti, M.; Tacconelli, E. Prevalence of antibiotic resistance in Helicobacter pylori, a systematic review and meta-analysis in World Health Organization regions. Gastroenterology 2018, 155, 1372–1382.e17. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.C.; Iyer, P.G.; Moss, S.F. AGA clinical practice update on the management of refractory Helicobacter pylori infection, expert review. Gastroenterology 2021, 160, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Sjomina, O.; Pavlova, J.; Niv, Y.; Leja, M. Epidemiology of Helicobacter pylori infection. Helicobacter 2018, 23, e12514. [Google Scholar] [CrossRef]
- Santos, M.L.C.; de Brito, B.B.; da Silva, F.A.F.; Sampaio, M.M.; Marques, H.S.; Oliveira, E.; Silva, N.; de Magalhães Queiroz, D.M.; de Melo, F.F. Helicobacter pylori infection, Beyond gastric manifestations. World J. Gastroenterol. 2020, 26, 4076–4093. [Google Scholar] [CrossRef]
- Lopo, I.; Libânio, D.; Pita, I.; Dinis-Ribeiro, M.; Pimentel-Nunes, P. Helicobacter pylori antibiotic resistance in Portugal, Systematic review and meta-analysis. Helicobacter 2018, 23, e12493. [Google Scholar] [CrossRef]
- Nogueira, C.; Mota, M.; Gradiz, R.; Cipriano, M.A.; Caramelo, F.; Cruz, H.; Alarcão, A.; E Sousa, F.C.; Oliveira, F.; Martinho, F. Prevalence and characteristics of Epstein–Barr virus-associated gastric carcinomas in Portugal. Infect. Agent. Cancer 2017, 12, 41. [Google Scholar] [CrossRef]
- de Arbulo, M.G.R.; Tamayo, E.; Bujanda, L.; Mendibil, L.; Mendiola, J.; Cilla, G.; Montes, M. Surveillance of Helicobacter pylori resistance over 22 years (2000–2021) in northern Spain. J. Glob. Antimicrob. Resist. 2023, 34, 127–133. [Google Scholar] [CrossRef]
- Crowe, S.E. Helicobacter pylori infection. N. Engl. J. Med. 2019, 380, 1158–1165. [Google Scholar] [CrossRef]
- Tshibangu-Kabamba, E.; Yamaoka, Y. Helicobacter pylori infection and antibiotic resistance—From biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 613–629. [Google Scholar] [CrossRef]
- Megraud, F.; Coenen, S.; Versporten, A.; Kist, M.; Lopez-Brea, M.; Hirschl, A.M.; Andersen, L.P.; Goossens, H.; Glupczynski, Y.; Study Group participants. Helicobacter pylori resistance to antibiotics in Europe and its relationship to antibiotic consumption. Gut 2013, 62, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Boyanova, L.; Markovska, R.; Medeiros, J.; Gergova, G.; Mitov, I. Delafloxacin against Helicobacter pylori; a potential option for improving eradication success? Diagn. Microbiol. Infect. Dis. 2020, 96, 114980. [Google Scholar] [CrossRef] [PubMed]
- Mégraud, F.; Lehours, P. Helicobacter pylori detection and antimicrobial susceptibility testing. Clin. Microbiol. Rev. 2007, 20, 280–322. [Google Scholar] [CrossRef]
- Albasha, A.M.; Elnosh, M.M.; Osman, E.H.; Zeinalabdin, D.M.; Fadl, A.A.M.; Ali, M.A.; Altayb, H.N. Helicobacter pylori 23S rRNA gene A2142G, A2143G, T2182C, and C2195T mutations associated with clarithromycin resistance detected in Sudanese patients. BMC Microbiol. 2021, 21, 38. [Google Scholar] [CrossRef] [PubMed]
- Domanovich-Asor, T.; Motro, Y.; Khalfin, B.; Craddock, H.A.; Peretz, A.; Moran-Gilad, J. Genomic analysis of antimicrobial resistance genotype-to-phenotype agreement in helicobacter pylori. Microorganisms 2021, 9, 2. [Google Scholar] [CrossRef]
- Losurdo, G.; Giorgio, F.; Pricci, M.; Girardi, B.; Russo, F.; Riezzo, G.; Martulli, M.; Piazzolla, M.; Cocomazzi, F.; Abbruzzi, F.; et al. Helicobacter pylori primary and secondary genotypic resistance to clarithromycin and levofloxacin detection in stools, A 4-year scenario in Southern Italy. Antibiotics 2020, 9, 723. [Google Scholar] [CrossRef]
- Aguilera-Correa, J.J.; Urruzuno, P.; Barrio, J.; Martinez, M.J.; Agudo, S.; Somodevilla, A.; Llorca, L.; Alarcón, T. Detection of Helicobacter pylori and the genotypes of resistance to clarithromycin and the heterogeneous genotype to this antibiotic in biopsies obtained from symptomatic children. Diagn. Microbiol. Infect. Dis. 2017, 87, 150–153. [Google Scholar] [CrossRef]
- Wang, L.H.; Cheng, H.; Hu, F.L.; Li, J. Distribution of gyrA mutations in fluoroquinolone-resistant Helicobacter pylori strains. World J. Gastroenterol. 2010, 16, 2272–2277. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, M.; Lu, B.; Dai, J. Helicobacter pylori and Antibiotic Resistance; A Continuing and Intractable Problem. Helicobacter 2016, 21, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Liu, S.; Ho, B.; Yan, Z.; Yan, X. Alterations in rdxA and frxA genes and their upstream regions in metronidazole-resistant Helicobacter pylori isolates. Res. Microbiol. 2007, 158, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Joo, Y.M.; Lee, H.S.; Chung, I.S.; Yoo, Y.J.; Merrell, D.S.; Cha, J.H. Genetic analysis of Helicobacter pylori clinical isolates suggests resistance to metronidazole can occur without the loss of functional rdxA. J. Antibiot. 2009, 62, 43–50. [Google Scholar] [CrossRef]
- Podzorski, R.P.; Podzorski, D.S.; Wuerth, A.; Tolia, V. Analysis of the vacA, cagA, cagE, iceA, and babA2 genes in Helicobacter pylori from sixty-one pediatric patients from the Midwestern United States. Diagn. Microbiol. Infect. Dis. 2003, 46, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Miftahussurur, M.; Shrestha, P.K.; Subsomwong, P.; Sharma, R.P.; Yamaoka, Y. Emerging Helicobacter pylori levofloxacin resistance and novel genetic mutation in Nepal. BMC Microbiol. 2016, 16, 256. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Morgulis, A.; Coulouris, G.; Raytselis, Y.; Madden, T.L.; Agarwala, R.; Schäffer, A.A. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Penel, S.; Arigon, A.M.; Dufayard, J.F.; Sertier, A.S.; Daubin, V.; Duret, L.; Gouy, M.; Perrière, G. Databases of homologous gene families for comparative genomics. BMC Bioinform. 2009, 10, S3. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Guerrero, C.E.; Ciria, R.; Abreu-Goodger, C.; Moreno-Hagelsieb, G.; Merino, E. GeConT 2: Gene context analysis for orthologous proteins, conserved domains and metabolic pathways. Nucleic Acids Res. 2008, 36, W176–W180. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast; scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Šali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Martínez-Júlvez, M.; Rojas, A.L.; Olekhnovich, I.; Espinosa Angarica, V.; Hoffman, P.S.; Sancho, J. Structure of RdxA—An oxygen-insensitive nitroreductase essential for metronidazole activation in Helicobacter pylori. FEBS J. 2012, 279, 4306–4317. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Versalovic, J.; Shortridge, D.; Kibler, K.; Griffy, M.V.; Beyer, J.; Flamm, R.K.; Tanaka, S.K.; Graham, D.Y.; Go, M.F. Mutations in 23S rRNA are associated with clarithromycin resistance in Helicobacter pylori. Antimicrob. Agents Chemother. 1996, 40, 477–480. [Google Scholar] [CrossRef]
- Wang, G.; Taylor, D.E. Site-specific mutations in the 23S rRNA gene of Helicobacter pylori confer two types of resistance to macrolide-lincosamide-streptogramin B antibiotics. Antimicrob. Agents Chemother. 1998, 42, 1952–1958. [Google Scholar] [CrossRef]
- Higgins, P.G.; Fluit, A.C.; Schmitz, F. Fluoroquinolones: Structure and target sites. Curr. Drug Targets 2003, 4, 181–190. [Google Scholar] [CrossRef]
- Correia, S.; Poeta, P.; Hébraud, M.; Capelo, J.L.; Igrejas, G. Mechanisms of quinolone action and resistance, where do we stand? J. Med. Microbiol. 2017, 66, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Butlop, T.R.; Mungkote, N.T.; Chaichanawongsaroj, N.T. Analysis of allelic variants of rdxA associated with metronidazole resistance in Helicobacter pylori: Detection of common genotypes in rdxA by multiplex allele-specific polymerase chain reaction. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef]
- Mirzaei, N.; Poursina, F.; Moghim, S.; Rahimi, E.; Safaei, H.G. The mutation of the rdxA gene in metronidazole-resistant Helicobacter pylori clinical isolates. Adv. Biomed. Res. 2014, 3, 90. [Google Scholar] [CrossRef]
- Alba, C.; Blanco, A.; Alarcón, T. Antibiotic resistance in Helicobacter pylori. Curr. Opin. Infect. Dis. 2017, 30, 489–497. [Google Scholar] [CrossRef]
- Arslan, N.; Yılmaz, Ö.; Demiray-Gürbüz, E. Importance of antimicrobial susceptibility testing for the management of eradication in Helicobacter pylori infection. World J. Gastroenterol. 2017, 23, 2854. [Google Scholar] [CrossRef]
- Murakami, K.; Okimoto, T.; Kodama, M.; Tanahashi, J.; Fujioka, T.; Ikeda, F.; Muraoka, H.; Takigawa, M.; Saika, T.; Hasegawa, M.; et al. Sitafloxacin activity against Helicobacter pylori isolates, including those with gyrA mutations. Antimicrob. Agents Chemother. 2009, 53, 3097–3099. [Google Scholar] [CrossRef] [PubMed]
- Hofreuter, D.; Behrendt, J.; Franz, A.; Meyer, J.; Jansen, A.; Bluemel, B.; Glocker, E.O. Antimicrobial resistance of Helicobacter pylori in an eastern German region. Helicobacter 2021, 26, e12765. [Google Scholar] [CrossRef]
- Spiteri, J.A.; Zahra, G.; Schembri, J.; Pisani, A.; Borg, E.; Spiteri, N.; Bianco, E.Z.; Caruana, P.; Gauci, J.; Muscat, M.; et al. Identification of antibiotic resistance patterns in Helicobacter pylori strains isolated from gastric biopsies using real-time PCR and genotypic analysis. Ann. Gastroenterol. 2021, 34, 501–509. [Google Scholar] [CrossRef]
- The, X.; Khosravi, Y.; Lee, W.C.; Leow, A.H.; Loke, M.F.; Vadivelu, J.; Goh, K.L. Functional and molecular surveillance of Helicobacter pylori antibiotic resistance in Kuala Lumpur. PLoS ONE 2014, 9, e101481. [Google Scholar] [CrossRef]
- Marques, B.; Donato, M.M.; Cardoso, O.; Luxo, C.; Martinho, A.; Almeida, N. Study of rdxA and frxA genes mutations in metronidazole-resistant and-susceptible Helicobacter pylori clinical isolates from the central region of Portugal. J. Glob. Antimicrob. Resist. 2019, 17, 300–304. [Google Scholar] [CrossRef]
- Wang, Y.H.; Wang, F.F.; Gong, X.L.; Yan, L.L.; Zhao, Q.Y.; Song, Y.P.; Zhao, R.L.; He, Y.J.; Zhou, L.; Liu, D.S.; et al. Genotype profiles of Helicobacter pylori from gastric biopsies and strains with antimicrobial-induced resistance. Therap. Adv. Gastroenterol. 2020, 13, 1756284820952596. [Google Scholar] [CrossRef]
- Tanih, N.F.; Ndip, L.M.; Ndip, R.N. Characterisation of the genes encoding resistance to metronidazole (rdxA and frxA) and clarithromycin (the 23S-rRNA genes) in South African isolates of Helicobacter pylori. Ann. Trop. Med. Parasitol. 2011, 105, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Mendz, G.L.; Mégraud, F. Is the molecular basis of metronidazole resistance in microaerophilic organisms understood? Trends Microbiol. 2002, 10, 370–375. [Google Scholar] [CrossRef]
- Zurita, J.; Sevillano, G.; Paz y Miño, A.; Zurita-Salinas, C.; Peñaherrera, V.; Echeverrıá, M.; Helicobacter pylori Research Group; Navarrete, H. Mutations associated with Helicobacter pylori antimicrobial resistance in the Ecuadorian population. J. Appl. Microbiol. 2022, 132, 2694–2704. [Google Scholar] [CrossRef] [PubMed]
- Solcà, N.M.; Bernasconi, M.V.; Piffaretti, J.C. Mechanism of metronidazole resistance in Helicobacter pylori, comparison of the rdxA gene sequences in 30 strains. Antimicrob. Agents Chemother. 2000, 44, 2207–2210. [Google Scholar] [CrossRef]
- Yang, Y.J.; Wu, J.J.; Sheu, B.S.; Kao, A.W.; Huang, A.H. The rdxA gene plays a more major role than frxA gene mutation in high-level metronidazole resistance of Helicobacter pylori in Taiwan. Helicobacter 2004, 9, 400–407. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valada, P.; Mata, A.; Brito, R.M.M.; Gonçalves, T.; Medeiros, J.A.; Nogueira, C. Detection of Helicobacter pylori and the Genotypes of Resistance to Clarithromycin, Fluoroquinolones, and Metronidazole in Gastric Biopsies: An In Silico Analysis to Help Understand Antibiotic Resistance. Curr. Issues Mol. Biol. 2025, 47, 187. https://doi.org/10.3390/cimb47030187
Valada P, Mata A, Brito RMM, Gonçalves T, Medeiros JA, Nogueira C. Detection of Helicobacter pylori and the Genotypes of Resistance to Clarithromycin, Fluoroquinolones, and Metronidazole in Gastric Biopsies: An In Silico Analysis to Help Understand Antibiotic Resistance. Current Issues in Molecular Biology. 2025; 47(3):187. https://doi.org/10.3390/cimb47030187
Chicago/Turabian StyleValada, Pedro, Ana Mata, Rui M. M. Brito, Teresa Gonçalves, José A. Medeiros, and Célia Nogueira. 2025. "Detection of Helicobacter pylori and the Genotypes of Resistance to Clarithromycin, Fluoroquinolones, and Metronidazole in Gastric Biopsies: An In Silico Analysis to Help Understand Antibiotic Resistance" Current Issues in Molecular Biology 47, no. 3: 187. https://doi.org/10.3390/cimb47030187
APA StyleValada, P., Mata, A., Brito, R. M. M., Gonçalves, T., Medeiros, J. A., & Nogueira, C. (2025). Detection of Helicobacter pylori and the Genotypes of Resistance to Clarithromycin, Fluoroquinolones, and Metronidazole in Gastric Biopsies: An In Silico Analysis to Help Understand Antibiotic Resistance. Current Issues in Molecular Biology, 47(3), 187. https://doi.org/10.3390/cimb47030187