

A Clinical Qualification Protocol Highlights Overlapping Genomic Influences and Neuro-Autonomic Mechanisms in Ehlers–Danlos and Long COVID-19 Syndromes


Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. DNA Testing
2.3. Patient and DNA Databases
2.4. Classification of Gene Products, Impacts on Tissue Elements and Processes
2.5. Statistics
3. Results
3.1. EDS Patients and Their Parallel Tissue Laxity, Neurologic, and Autonomic Findings

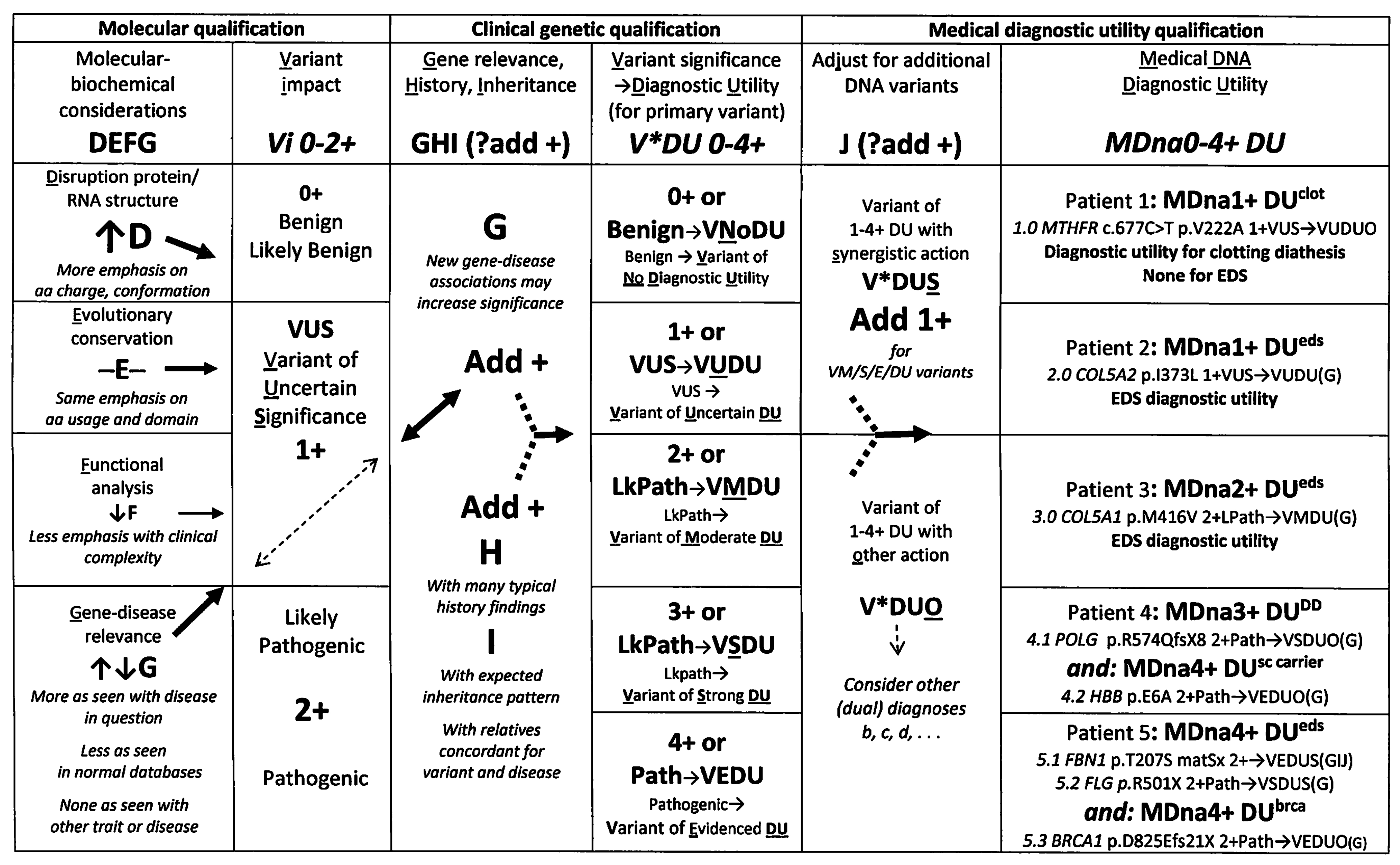
3.2. A Novel Clinical Protocol for DNA Variant Qualification
3.3. Genes Relevant to EDS
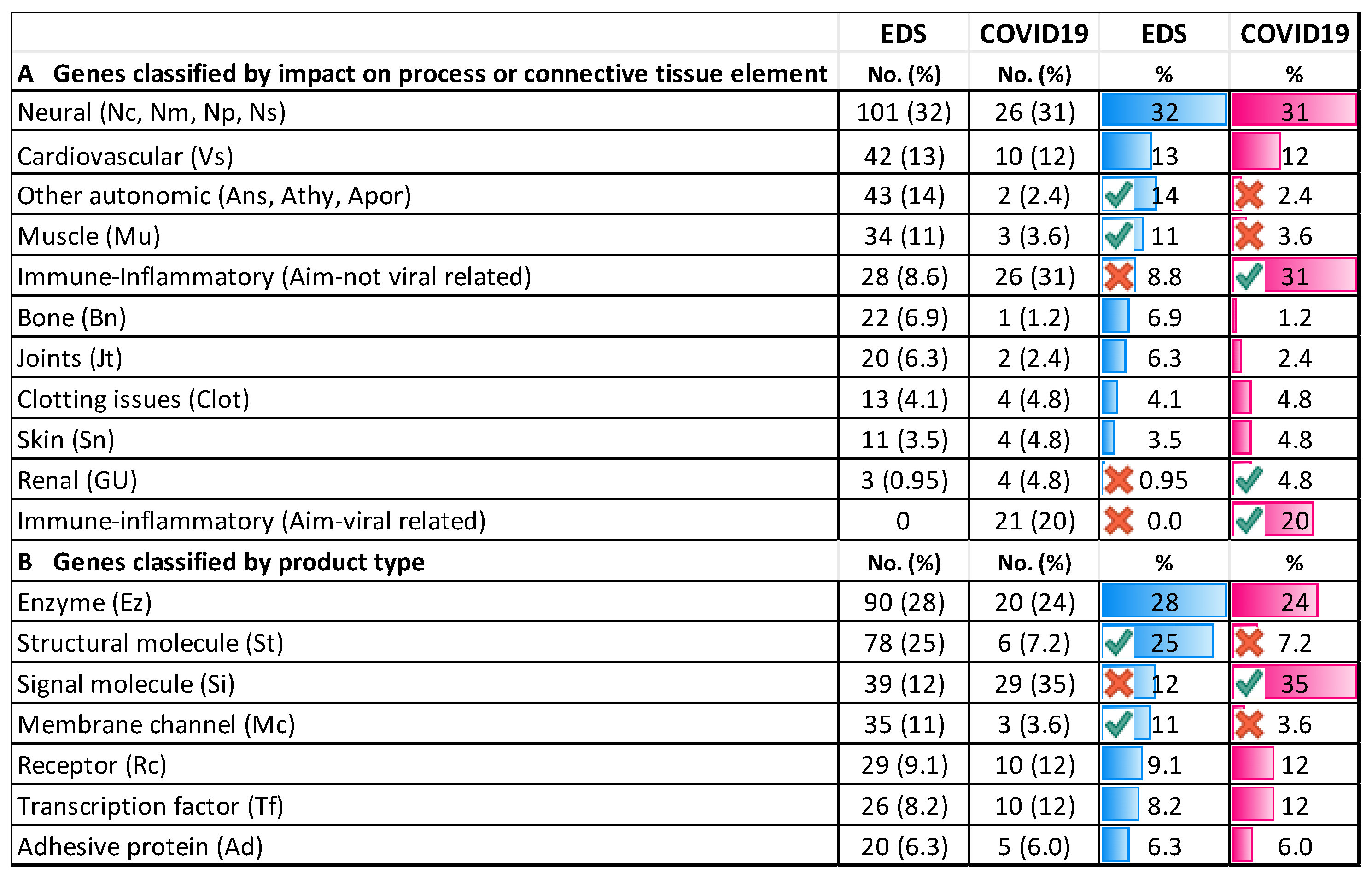
3.4. Genes Conferring Susceptibility to Severe COVID-19 Infection
3.4.1. Comparing COVID-19/EDS Gene Type and Distribution
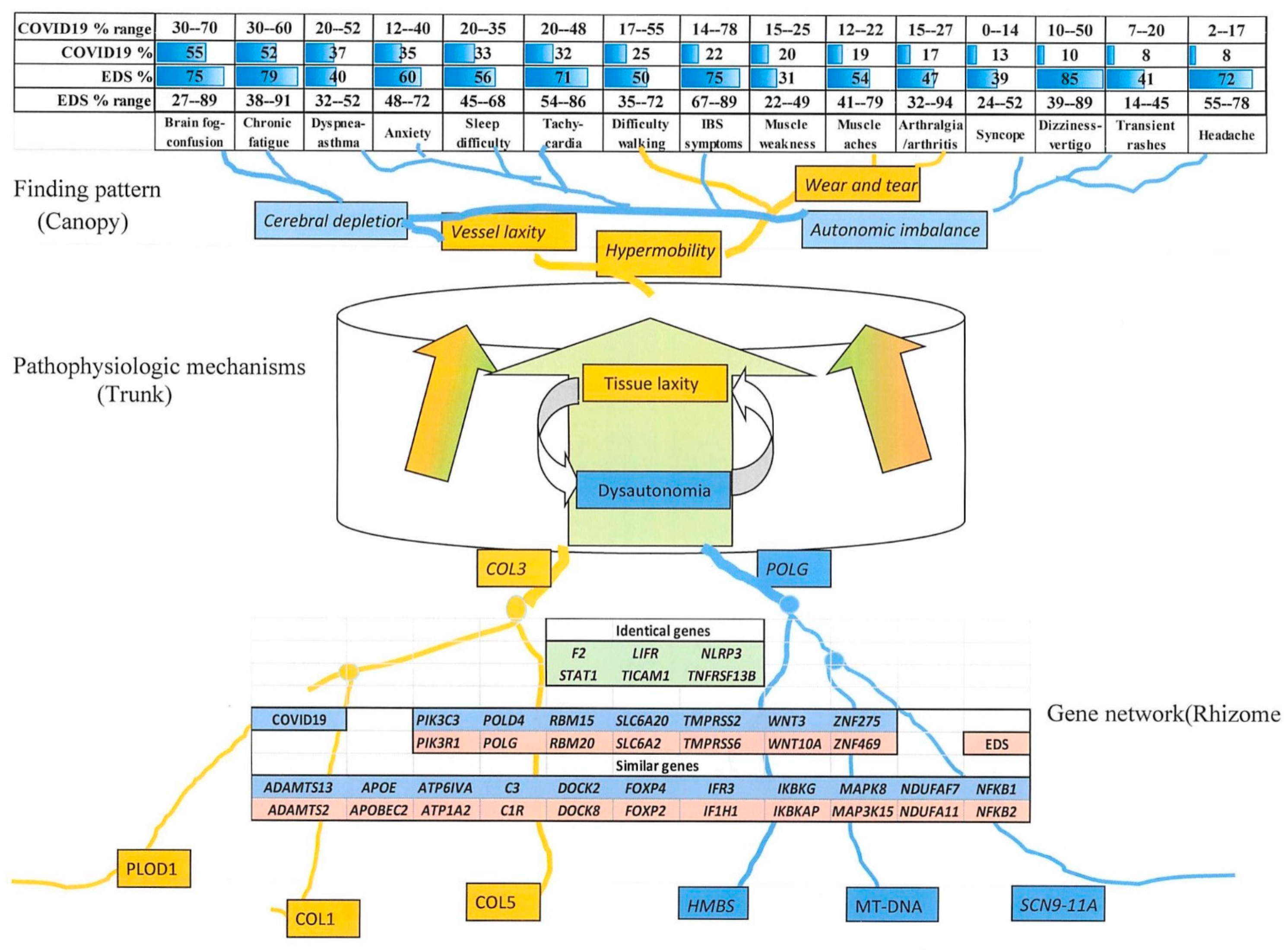
3.4.2. Comparing Individual Symptoms and Genes Relevant to COVID-19 with Those of Eds
3.4.3. Comparison of EDS and Long COVID-19 Symptoms
3.4.4. Similar Genes Relevant to EDS and COVID-19 Severity
4. Discussion
4.1. Clinical–DNA Correlation in EDS
4.1.1. Examples of Clinical Qualification
4.1.2. Advantages of Molecular and Clinical Qualification
4.2. Distribution and Nature of EDS and COVID-19-Related Genes
4.3. Connections of Signs and Symptoms to Genes as “Entomes”
4.4. Implications for Future Research
4.4.1. Expanded Studies on EDS and Long COVID-19 Symptoms and Outcomes
4.4.2. Future Therapies for EDS, COVID-19, and the Related Symptoms of Aging
5. Conclusions
- The prevalence of dysautonomia findings is equal to those of tissue laxity in 1261 EDS patients with systematic evaluation, with their underlying articulo-autonomic dysplasia mechanisms being essential for comparisons to long COVID-19.
- Comparison of EDS to COVID-19 included 15 overlapping dysautonomia–musculoskeletal symptoms in those with persisting symptoms and 24 identical or similar genes among the 84 moderating the severity of acute infection.
- These many gene changes are hypothesized to act through a network or entome to produce overlapping Ehlers–Danlos or long COVID-19 syndrome profiles, their disease symptoms (canopy), genes (rhizome), and connecting pathogenetic mechanisms (trunk-phloem) visualized as entomes by analogy to Tolkien’s Ents.
- If a better characterization of the natural history and genetic predispositions to long COVID-19 validates the common musculoskeletal, neuro-autonomic, and immune/inflammatory mechanisms hypothesized in this article, then EDS-proven exercise, dietary, and medication therapies may be tried in the 6.2% of COVID-19 patients who endure persisting symptoms.
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weerakkody, R.A.; Vandrovcova, J.; Kanonidou, C.; Mueller, M.; Gampawar, P.; Ibrahim, Y.; Norsworthy, P.; Biggs, J.; Abdullah, A.; Ross, D.; et al. Targeted next-generation sequencing makes new molecular diagnoses and expands genotype–phenotype relationship in Ehlers–Danlos syndrome. Genet Med. 2016, 18, 1119–1127. [Google Scholar] [CrossRef]
- Wilson, G.N. Genomic Analysis of 727 Patients with Ehlers-Danlos Syndrome I: Clinical Perspective Relates 23 Genes to a Maternally Influenced Arthritis-Adrenaline Disorder. J. Biosci. Med. 2019, 7, 181–204. [Google Scholar] [CrossRef]
- Junkiert-Czarnecka, A.; Pilarska-Deltow, M.; Bąk, A.; Heise, M.; Latos-Bieleńska, A.; Zaremba, J.; Bartoszewska-Kubiak, A.; Haus, O. Next-Generation Sequencing of Connective Tissue Genes in Patients with Classical Ehlers-Danlos Syndrome. Curr. Issues Mol. Biol. 2022, 44, 1472–1478. [Google Scholar] [CrossRef]
- Tinkle, B.T.; Levy, H.P. Symptomatic joint hypermobility: The hypermobile type of Ehlers-Danlos syndrome and the hypermobility spec-trum disorders. Med. Clin. North Am. 2019, 103, 1021–1033. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef]
- Bowen, J.M.; Sobey, G.J.; Burrows, N.P.; Colombi, M.; Lavallee, M.E.; Malfait, F.; Francomano, C.A. Ehlers-Danlos syndrome, classical type. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 27–39. [Google Scholar] [CrossRef]
- Byers, P.H.; Belmont, J.; Black, J.; De Backer, J.; Frank, M.; Jeunemaitre, X.; Johnson, D.; Pepin, M.; Robert, L.; Sanders, L.; et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 40–47. [Google Scholar] [CrossRef]
- McKusick, V.A. Heritable disorders of connective tissue: I. The clinical behavior of hereditary syndromes. J. Chronic Dis. 1955, 2, 491–499. [Google Scholar] [CrossRef]
- Gazit, Y.; Nahir, A.M.; Grahame, R.; Jacob, G. Dysautonomia in the joint hypermobility syndrome. Am. J. Med. 2003, 115, 33–40. [Google Scholar] [CrossRef] [PubMed]
- De Wandele, I.; Rombaut, L.; Leybaert, L.; Van de Borne, P.; De Backer, T.; Malfait, F.; De Paepe, A.; Calders, P. Dysautonomia and its underlying mechanisms in the hypermobility type of Ehlers–Danlos syndrome. Semin. Arthr. Rheum. 2014, 44, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.N. Clinical Analysis Supports Articulo-Autonomic Dysplasia as a Unifying Pathogenic Mechanism in Ehlers-Danlos Syndrome and Related Conditions. J. Biosci. Med. 2019, 7, 149–168. [Google Scholar] [CrossRef]
- Cazzato, D.; Castori, M.; Lombardi, R.; Caravello, F.; Bella, E.D.; Petrucci, A.; Grammatico, P.; Dordoni, C.; Colombi, M.; Lauria, G. Small fiber neuropathy is a common feature of Ehlers-Danlos syndromes. Neurology 2016, 87, 155–159. [Google Scholar] [CrossRef]
- Henderson, F.C.; Austin, C.; Benzel, E.; Bolognese, P.; Ellenbogen, R.; Francomano, C.A.; Ireton, C.; Klinge, P.; Koby, M.; Long, D.; et al. Neurological and spinal manifestations of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Wegener, H.; Leineweber, S.; Seeger, K. The vWFA2 domain of type VII collagen is responsible for collagen binding. Biochem. Biophys. Res. Commun. 2013, 430, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T.; Raynal, N.; Groeneveld, D.; Maddox, B.; Peachey, A.R.; Huizinga, E.G.; de Groot, P.G.; Farndale, R.W. A single high-affinity binding site for von Willebrand factor in collagen III, identified using synthetic triple-helical peptides. Blood 2006, 108, 3753–3756. [Google Scholar] [CrossRef] [PubMed]
- Lorton, D.; Bellinger, D.L. Molecular Mechanisms Underlying β-Adrenergic Receptor-Mediated Cross-Talk between Sympathetic Neurons and Immune Cells. Int. J. Mol. Sci. 2015, 16, 5635–5665. [Google Scholar] [CrossRef]
- Wilson, G.N.; Tonk, V.S. Mitochondrial Dysfunction Contributes to Ehlers-Danlos Syndrome—A Patient Presentation. J. Biol. Life Sci. 2020, 11, 190–202. [Google Scholar] [CrossRef]
- Gaudó, P.; Emperador, S.; Garrido-Pérez, N.; Ruiz-Pesini, E.; Yubero, D.; García-Cazorla, A.; Artuch, R.; Montoya, J.; Bayona-Bafaluy, M.P. Infectious stress triggers a POLG-related mitochondrial disease. Neurogenetics 2020, 21, 19–27. [Google Scholar] [CrossRef]
- Vernino, S.; Bourne, K.M.; Stiles, L.E.; Grubb, B.P.; Fedorowski, A.; Stewart, J.M.; Arnold, A.C.; Pace, L.A.; Axelsson, J.; Boris, J.R.; et al. Postural orthostatic tachycardia syndrome (POTS): State of the science and clinical care from a 2019 National Institutes of Health Expert Consensus Meeting—1. Auton. Neurosci. 2021, 235, 102828. [Google Scholar] [CrossRef]
- Benarroch, E.E. Postural Tachycardia Syndrome: A Heterogeneous and Multifactorial Disorder. Mayo Clin. Proc. 2012, 87, 1214–1225. [Google Scholar] [CrossRef]
- Wang, E.; Ganti, T.; Vaou, E.; Hohler, A. The relationship between mast cell activation syndrome, postural tachycardia syndrome, and Ehlers-Danlos syndrome. Allergy Asthma Proc. 2021, 42, 243–246. [Google Scholar] [CrossRef]
- Monaco, A.; Choi, D.; Uzun, S.; Maitland, A.; Riley, B. Association of mast-cell-related conditions with hypermobile syn-dromes: A review of the literature. Immunol. Res. 2022, 70, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Z.; Wang, Q.Q.; Yang, Q.Q.; Gu, H.Y.; Yin, Y.Q.; Li, Y.D.; Hou, J.-C.; Chen, R.; Sun, Q.-Q.; Sun, Y.-F.; et al. NG2 glia regulate brain innate immunity via TGF-β2/TGFBR2 axis. BMC Med. 2019, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Thwaites, P.A.; Gibson, P.R.; Burgell, R.E. Hypermobile Ehlers–Danlos syndrome and disorders of the gastrointestinal tract: What the gastroenterologist needs to know. J. Gastroenterol. Hepatol. 2022, 37, 1693–1709. [Google Scholar] [CrossRef] [PubMed]
- Deer, R.R.; Rock, M.A.; Vasilevsky, N.; Carmody, L.; Rando, H.; Anzalone, A.J.; Basson, M.D.; Bennett, T.D.; Bergquist, T.; Boudreau, E.A.; et al. Characterizing Long COVID: Deep Phenotype of a Complex Condition. Ebiomedicine 2021, 74, 103722. [Google Scholar] [CrossRef]
- Dani, M.; Dirksen, A.; Taraborrelli, P.; Torocastro, M.; Panagopoulos, D.; Sutton, R.; Lim, P.B. Autonomic dysfunction in ‘long COVID’: Rationale, physiology and management strategies. Clin. Med. 2020, 21, e63–e67. [Google Scholar] [CrossRef]
- Raj, S.R.; Arnold, A.C.; Barboi, A.; Claydon, V.E.; Limberg, J.K.; Lucci, V.M.; Numan, M.; Peltier, A.; Snapper, H.; Vernino, S.; et al. Long-COVID postural tachycardia syndrome: An American Autonomic Society statement. Clin. Auton. Res. 2021, 31, 365–368. [Google Scholar] [CrossRef]
- Ceban, F.; Ling, S.; Lui, L.M.W.; Lee, Y.; Gill, H.; Teopiz, K.M.; Rodrigues, N.B.; Subramaniapillai, M.; Di Vincenzo, J.D.; Cao, B.; et al. Fatigue and cognitive impairment in Post-COVID-19 Syndrome: A systematic review and meta-analysis. Brain Behav. Immun. 2022, 101, 93–135. [Google Scholar] [CrossRef]
- Dotan, A.; David, P.; Arnheim, D.; Shoenfeld, Y. The autonomic aspects of the post-COVID19 syndrome. Autoimmun. Rev. 2022, 21, 103071. [Google Scholar] [CrossRef]
- Batiha, G.E.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Welson, N.N. Pathophysiology of Post-COVID syndromes: A new perspective. Virol. J. 2022, 19, 158. [Google Scholar] [CrossRef]
- Thaweethai, T.; Jolley, S.E.; Karlson, E.W.; Levitan, E.B.; Levy, B.; McComsey, G.A.; McCorkell, L.; Nadkarni, G.N.; Parthasarathy, S.; Singh, U.; et al. Recover Consortium. Development of a Definition of Postacute Sequelae of SARS-CoV-2 Infection. JAMA 2023, 329, 1934–1946. [Google Scholar] [CrossRef] [PubMed]
- Global Burden of Disease Long COVID Collaborators; Wulf Hanson, S.; Abbafati, C.; Aerts, J.G.; Al-Aly, Z.; Ashbaugh, C.; Ballouz, T.; Blyuss, O.; Bobkova, P.; Bonsel, G.; et al. Estimated Global Proportions of Individuals with Persistent Fatigue, Cognitive, and Respiratory Symptom Clusters following Symptomatic COVID-19 in 2020 and 2021. JAMA 2022, 328, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Ye, L.; Pan, K.; Chen, Y.; Xing, D.; Yan, K.; Chen, Z.; Ding, N.; Li, W.; Huang, H.; et al. New Insights of Emerging SARS-CoV-2: Epidemiology, Etiology, Clinical Features, Clinical Treatment, and Prevention. Front. Cell Dev. Biol. 2020, 8, 410. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://covid19.who.int (accessed on 22 January 2023).
- Dale, L. Neurological Complications of COVID-19: A Review of the Literature. Cureus 2022, 14, e27633. [Google Scholar] [CrossRef]
- Tossetta, G.; Fantone, S.; Delli Muti, N.; Balercia, G.; Ciavattini, A.; Giannubilo, S.R.; Marzioni, D. Preeclampsia and severe acute respiratory syndrome coronavirus 2 infection: A systematic review. J. Hypertens. 2022, 40, 1629–1638. [Google Scholar] [CrossRef]
- Delli Muti, N.; Finocchi, F.; Tossetta, G.; Salvio, G.; Cutini, M.; Marzioni, D.; Balercia, G. Could SARS-CoV-2 infection affect male fertility and sexuality? Apmis 2022, 130, 243–252. [Google Scholar] [CrossRef]
- Williams, F.M.K.; Freidin, M.B.; Mangino, M.; Couvreur, S.; Visconti, A.; Bowyer, R.C.E.; Le Roy, C.I.; Falchi, M.; Mompeó, O.; Sudre, C.; et al. Self-Reported Symptoms of COVID-19, Including Symptoms Most Predictive of SARS-CoV-2 Infection, Are Heritable. Twin Res. Hum. Genet. 2020, 23, 316–321. [Google Scholar] [CrossRef]
- Asano, T.; Boisson, B.; Onodi, F.; Matuozzo, D.; Moncada-Velez, M.; Maglorius Renkilaraj, M.R.L.; Zhang, P.; Meertens, L.; Bolze, A.; Materna, M.; et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021, 6, eabl4348. [Google Scholar] [CrossRef]
- Ishaq, U.; Malik, A.; Malik, J.; Mehmood, A.; Qureshi, A.; Laique, T.; Zaidi, S.M.J.; Javaid, M.; Rana, A.S. Association of ABO blood group with COVID-19 severity, acute phase reactants and mortality. PLoS ONE 2021, 16, e0261432. [Google Scholar] [CrossRef]
- Dieter, C.; Brondani, L.A.; Leitão, C.B.; Gerchman, F.; Lemos, N.E.; Crispim, D. Genetic polymorphisms associated with susceptibility to COVID-19 disease and severity: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0270627. [Google Scholar] [CrossRef]
- Sharma, P.; Pandey, A.K.; Bhattacharyya, D.K. Determining crucial genes associated with COVID-19 based on COPD Findings. Comput. Biol. Med. 2021, 128, 104126. [Google Scholar] [CrossRef]
- Philippe, A.; Gendron, N.; Bory, O.; Beauvais, A.; Mirault, T.; Planquette, B.; Sanchez, O.; Diehl, J.-L.; Chocron, R.; Smadja, D.M. Von Willebrand factor collagen-binding capacity predicts in-hospital mortality in COVID-19 patients: Insight from VWF/ADAMTS13 ratio imbalance. Angiogenesis 2021, 24, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. Increased VWF and Decreased ADAMTS-13 in COVID-19: Creating a Milieu for (Micro)Thrombosis. Semin. Thromb. Hemost. 2021, 47, 400–418. [Google Scholar] [CrossRef]
- Lynch, S.M.; Guo, G.; Gibson, D.S.; Bjourson, A.J.; Rai, T.S. Role of Senescence and Aging in SARS-CoV-2 Infection and COVID-19 Disease. Cells 2021, 10, 3367. [Google Scholar] [CrossRef] [PubMed]
- Amital, M.; Ben-Shabat, N.; Amital, H.; Buskila, D.; Cohen, A.D.; Amital, D. COVID-19 associated hospitalization in 571 patients with fibromyalgia—A population-based study. PLoS ONE 2021, 16, e0261772. [Google Scholar] [CrossRef] [PubMed]
- Barros, A.; Queiruga-Piñeiro, J.; Lozano-Sanroma, J.; Alcalde, I.; Gallar, J.; Fernández-Vega Cueto, L.; Alfonso, J.F.; Quirós, L.M.; Merayo-Lloves, J. Small fiber neuropathy in the cornea of Covid-19 patients associated with the generation of ocular surface disease. Ocul. Surf. 2022, 23, 40–48. [Google Scholar] [CrossRef]
- McFarland, A.J.; Yousuf, M.S.; Shiers, S.; Price, T.J. Neurobiology of SARS-CoV-2 interactions with the peripheral nervous system: Implications for COVID-19 and pain. PAIN Rep. 2021, 6, e885. [Google Scholar] [CrossRef]
- Díaz-Alberola, I.; Espuch-Oliver, A.; García-Aznar, J.M.; Ganoza-Gallardo, C.; Aguilera-Franco, M.; Sampedro, A.; Jiménez, P.; López-Nevot, M. Common Variable Immunodeficiency Associated with a De Novo IKZF1 Variant and a Low Humoral Immune Response to the SARS-CoV-2 Vaccine. J. Clin. Med. 2022, 11, 2303. [Google Scholar] [CrossRef]
- Tutal, E.; Ozaras, R.; Leblebicioglu, H. Systematic review of COVID-19 and autoimmune thyroiditis. Travel Med. Infect. Dis. 2022, 47, 102314. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef]
- Bamshad, M.J.; Ng, S.B.; Bigham, A.W.; Tabor, H.K.; Emond, M.J.; Nickerson, D.A.; Shendure, J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011, 12, 745–755. [Google Scholar] [CrossRef]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Microarray analysis performed by a variety of commercial laboratories for patients with developmental disability and autism, including the author-associated laboratory at Texas Tech University that used standard methods and interpretation as outlined. In Chromosome Structure and Variation: Heteromorphism, Polymorphism, and Pathogenesis; Wyandt, H.E., Wilson, G.N., Tonk, V.S., Eds.; Springer Nature: New York, NY, USA, 2017; Chapters 9–10. [Google Scholar]
- Retterer, K.; Scuffins, J.; Schmidt, D.; Lewis, R.; Pineda-Alvarez, D.; Stafford, A.; Schmidt, L.; Warren, S.; Gibellini, F.; Kondakova, A.; et al. Assessing copy number from exome sequencing and exome array CGH based on CNV spectrum in a large clinical cohort. Genet Med. 2015, 17, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.N. A network of genes (entome) is responsible for congruent tissue laxity and dysautonomia findings in hypermobility spectrum/Ehlers-Danlos syndrome disorders. Manuscript in preparation. July 2023. [Google Scholar]
- MedCalc Software Ltd. Available online: https://www.medcalc.org/calc (accessed on 22 January 2023).
- Ehlers-Danlos Society. Criteria for EDS types. 2017. Available online: https://www.ehlers-danlos.com/2017-eds-international-classification/ (accessed on 22 January 2023).
- The Ehlers-Danlos Society. Beighton Maneuvers Illustrated. Available online: https://www.ehlers-danlos.com/assessing-joint-hypermobility/ (accessed on 22 January 2023).
- Yonko, E.A.; LoTurco, H.M.; Carter, E.M.; Raggio, C.L. Orthopedic considerations and surgical outcomes in Ehlers–Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2021, 187, 458–465. [Google Scholar] [CrossRef]
- Ng, P.C.; Levy, S.; Huang, J.; Stockwell, T.B.; Walenz, B.P.; Li, K.; Axelrod, N.; Busam, D.A.; Strausberg, R.L.; Venter, J.C. Genetic Variation in an Individual Human Exome. PLOS Genet. 2008, 4, e1000160. [Google Scholar] [CrossRef]
- Wilson, G.N.; Tonk, V.S. A protocol for qualifying DNA variants associated with complex diseases like Ehlers-Danlos syndrome. GEN Protocols 2021—An open access component of the genengnews.com website that has now been discontinued.
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for investigating causality of sequence variants in human disease. Nature 2014, 24, 469–476. [Google Scholar] [CrossRef]
- Tam, B.; Sinha, S.; Wang, S.M. Combining Ramachandran plot and molecular dynamics simulation for structural-based variant classification: Using TP53 variants as model. Comput. Struct. Biotechnol. J. 2020, 18, 4033–4039. [Google Scholar] [CrossRef]
- Genome Browser, University of California Santa Clara. Available online: https://genome.ucsc.edu/ (accessed on 22 January 2023).
- Hu, Z.; Yu, C.; Furutsuki, M.; Andreoletti, G.; Ly, M.; Hoskins, R.; Adhikari, A.N.; Brenner, S.E. VIPdb, a genetic Variant Impact Predictor Database. Hum. Mutat. 2019, 40, 1202–1214. [Google Scholar] [CrossRef]
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 22 January 2023).
- MITOMAP: A Human Mitochondrial Genome Database. 2019. Available online: http://www.mitomap.org (accessed on 22 January 2023).
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Gnomad. Available online: https://gnomad.broadinstitute.org/ (accessed on 22 January 2023).
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Noda, K.; Kitagawa, K.; Miki, T.; Horiguchi, M.; Akama, T.O.; Taniguchi, T.; Taniguchi, H.; Takahashi, K.; Ogra, Y.; Mecham, R.P.; et al. A matricellular protein fibulin-4 is essential for the activation of lysyl oxidase. Sci. Adv. 2020, 6, eabc1404. [Google Scholar] [CrossRef] [PubMed]
- Torre-Fuentes, L.; Matías-Guiu, J.; Hernández-Lorenzo, L.; Montero-Escribano, P.; Pytel, V.; Porta-Etessam, J.; Gómez-Pinedo, U.; Matías-Guiu, J.A. ACE2, TMPRSS2, and Furin variants and SARS-CoV-2 infection in Madrid, Spain. J. Med. Virol. 2021, 93, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Weis, M.A.; Hudson, D.M.; Kim, L.; Scott, M.; Wu, J.J.; Eyre, D.R. Location of 3-Hydroxyproline Residues in Collagen Types I, II, III, and V/XI Implies a Role in Fibril Supramolecular Assembly. J. Biol. Chem. 2010, 285, 2580–2590. [Google Scholar] [CrossRef]
- Human Phenotype Ontology. Available online: https://hpo.jax.org/app/ (accessed on 22 January 2023).
- Wilson, G.N.; Tonk, V.S. Demon Genes May Deform Common Syndromes: Collagen VI Gene Change in Down Syndrome Unifies the Medical and Molecular Approach to Hypermobility Disorders. J. Biosci. Med. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Serjeant, G.R.; Vichinsky, E. Variability of homozygous sickle cell disease: The role of alpha and beta globin chain variation and other factors. Blood Cells, Mol. Dis. 2018, 70, 66–77. [Google Scholar] [CrossRef]
- Woźniak, E.; Owczarczyk-Saczonek, A.; Lange, M.; Czarny, J.; Wygonowska, E.; Placek, W.; Nedoszytko, B. The Role of Mast Cells in the Induction and Maintenance of Inflammation in Selected Skin Diseases. Int. J. Mol. Sci. 2023, 24, 7021. [Google Scholar] [CrossRef]
- Abdalla, E.M.; Rohrbach, M.; Bürer, C.; Kraenzlin, M.; El-Tayeby, H.; Elbelbesy, M.F.; Nabil, A.; Giunta, C. Kyphoscoliotic type of Ehlers-Danlos Syndrome (EDS VIA) in six Egyptian patients presenting with a homogeneous clinical phenotype. Eur. J. Pediatr. 2015, 174, 105–112. [Google Scholar] [CrossRef]
- Hunter, D.J.; Drazen, J.M. Has the Genome Granted Our Wish Yet? N. Engl. J. Med. 2019, 380, 2391–2393. [Google Scholar] [CrossRef]
- Jang, J.Y.; Blum, A.; Liu, J.; Finkel, T. The role of mitochondria in aging. J. Clin. Investig. 2018, 128, 3662–3670. [Google Scholar] [CrossRef]
- Shenoy, S. Coronavirus (Covid-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020, 69, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.H.; Li, Y.Y.; Jin, J. The essential functions of mitochondrial dynamics in immune cells. Cell. Mol. Immunol. 2020, 17, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Field, C.S.; Baixauli, F.; Kyle, R.L.; Puleston, D.J.; Cameron, A.M.; Sanin, D.E.; Hippen, K.L.; Loschi, M.; Thangavelu, G.; Corrado, M.; et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab. 2020, 31, 422–437. [Google Scholar] [CrossRef]
- Weinfurt, K.P.; Reeve, B.B. Patient-Reported Outcome Measures in Clinical Research. JAMA 2022, 328, 472–473. [Google Scholar] [CrossRef]
- Talarico, R.; Aguilera, S.; Alexander, T.; Amoura, Z.; Antunes, A.M.; Arnaud, L.; Avcin, T.; Beretta, L.; Bombardieri, S.; Burmester, G.R.; et al. The impact of COVID-19 on rare and complex connective tissue diseases: The experience of ERN ReCONNET. Nat. Rev. Rheumatol. 2021, 17, 177–184. [Google Scholar] [CrossRef]
- Wilson, G.N.; Tonk, V.S. Do severe complications like aneurysms from Kawasaki and Kawasaki-like infections arise in children with underlying articulo-autonomic dysplasia/Ehlers-Danlos syndrome? Ann. Ped. Res. 2020, 4, 1–4. Available online: www.remedypublications.com/open-access/do-severe-complications-like-aneurysms-from-kawasaki-and-kawasaki-like-infections-6248.pdf (accessed on 15 July 2023).
- D’agnelli, S.; Arendt-Nielsen, L.; Gerra, M.C.; Zatorri, K.; Boggiani, L.; Baciarello, M.; Bignami, E. Fibromyalgia: Genetics and epigenetics insights may provide the basis for the development of diagnostic biomarkers. Mol. Pain 2018, 15. [Google Scholar] [CrossRef]
- Yue, F.; Era, T.; Yamaguchi, T.; Kosho, T. Pathophysiological Investigation of Skeletal Deformities of Musculocontractural Ehlers–Danlos Syndrome Using Induced Pluripotent Stem Cells. Genes 2023, 14, 730. [Google Scholar] [CrossRef]
- Lewis, M.T.; Levitsky, Y.; Bazil, J.N.; Wiseman, R.W. Measuring Mitochondrial Function: From Organelle to Organism. Methods Mol Biol. 2022, 2497, 141–172. [Google Scholar] [CrossRef]
- Grünewald, A.; Voges, L.; Rakovic, A.; Kasten, M.; Vandebona, H.; Hemmelmann, C.; Lohmann, K.; Orolicki, S.; Ramirez, A.; Schapira, A.H.V.; et al. Mutant Parkin Impairs Mitochondrial Function and Morphology in Human Fibroblasts. PLoS ONE 2010, 5, e12962. [Google Scholar] [CrossRef] [PubMed]
- Russek, L.N.; Block, N.P.; Byrne, E.; Chalela, S.; Chan, C.; Comerford, M.; Frost, N.; Hennessey, S.; McCarthy, A.; Nicholson, L.L.; et al. Presentation and physical therapy management of upper cervical instability in patients with symptomatic generalized joint hypermobility: International expert consensus recommendations. Front. Med. 2023, 9, 1072764. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.; Barlow, C.E. Review of the Role of Exercise in Improving Quality of Life in Healthy Individuals and in Those with Chronic Diseases. Curr. Sports Med. Rep. 2011, 10, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Nancarrow-Lei, R.; Mafi, P.; Mafi, R.; Khan, W. A Systemic Review of Adult Mesenchymal Stem Cell Sources and their Multilineage Differentiation Potential Relevant to Musculoskeletal Tissue Repair and Regeneration. Curr. Stem Cell Res. Ther. 2017, 12, 601–610. [Google Scholar] [CrossRef]
- van Der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of genetic variants among young men with severe COVID-19. JAMA 2020, 324, 663–673. [Google Scholar] [CrossRef]
- Delorey, T.M.; Ziegler, C.G.; Heimberg, G.; Normand, R.; Yang, Y.; Segerstolpe, Å.; Abbondanza, D.; Fleming, S.J.; Subramanian, A.; Montoro, D.T.; et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 2021, 595, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Namkoong, H.; Edahiro, R.; Takano, T.; Nishihara, H.; Shirai, Y.; Sonehara, K.; Tanaka, H.; Azekawa, S.; Mikami, Y.; Lee, H.; et al. DOCK2 is involved in the host genetics and biology of severe COVID-19. Nature 2022, 609, 754–760. [Google Scholar] [CrossRef]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef]
- Chu, J.; Xing, C.; Du, Y.; Duan, T.; Liu, S.; Zhang, P.; Cheng, C.; Henley, J.; Liu, X.; Qian, C.; et al. Pharmacological inhibition of fatty acid synthesis blocks SARS-CoV-2 replication. Nat. Metab. 2021, 3, 1466–1475. [Google Scholar] [CrossRef]
- Feng, S.; Song, F.; Guo, W.; Tan, J.; Zhang, X.; Qiao, F.; Guo, J.; Zhang, L.; Jia, X. Potential genes associated with COVID-19 and comorbidity. Int. J. Med. Sci. 2022, 19, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Baldassarri, M.; Picchiotti, N.; Fava, F.; Fallerini, C.; Benetti, E.; Daga, S.; Valentino, F.; Doddato, G.; Furini, S.; Giliberti, A.; et al. Shorter androgen receptor polyQ alleles protect against life-threatening COVID19 disease in European males. EBioMedicine 2021, 65, 103246. [Google Scholar] [CrossRef] [PubMed]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 2021, 184, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Boussier, J.; Yatim, N.; Marchal, A.; Hadjadj, J.; Charbit, B.; El Sissy, C.; Carlier, N.; Pène, F.; Mouthon, L.; Tharaux, P.L.; et al. Severe COVID-19 is associated with hyperactivation of the alternative complement pathway. J. Allergy Clin. Immunol. 2022, 149, 550–556. [Google Scholar] [CrossRef]
- Nakanishi, T.; Pigazzini, S.; Degenhardt, F.; Cordioli, M.; Butler-Laporte, G.; Maya-Miles, D.; Bujanda, L.; Bouysran, Y.; Niemi, M.E.; Palom, A.; et al. Age-dependent impact of the major common genetic risk factor for COVID-19 on severity and mortality. J. Clin. Investig. 2021, 131, e152386. [Google Scholar] [CrossRef]
- Niemi, M.E.K.; Daly, M.J.; Ganna, A. The human genetic epidemiology of COVID-19. Nat. Rev. Genet. 2022, 23, 533–546. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Saponi-Cortes, J.M.; Rivas, M.D.; Calle-Alonso, F.; Sanchez, J.F.; Costo, A.; Martin, C.; Zamorano, J. IFNL4 genetic variant can predispose to COVID-19. Sci. Rep. 2021, 11, 21185. [Google Scholar] [CrossRef]
- Wu, L.; Zhu, J.; Liu, D.; Sun, Y.; Wu, C. An integrative multiomics analysis identifies putative causal genes for COVID-19 severity. Genet. Med. 2021, 23, 2076–2086. [Google Scholar] [CrossRef]
- Wu, P.; Ding, L.; Li, X.; Liu, S.; Cheng, F.; He, Q.; Xiao, M.; Wu, P.; Hou, H.; Jiang, M.; et al. Trans-ethnic genome-wide association study of severe COVID-19. Commun. Biol. 2021, 4, 1034. [Google Scholar] [CrossRef] [PubMed]
- Patrick, M.T.; Zhang, H.; Wasikowski, R.; Prens, E.P.; Weidinger, S.; Gudjonsson, J.E.; Elder, J.T.; He, K.; Tsoi, L.C. Associations between COVID-19 and skin conditions identified through epidemiology and genomic studies. J. Allergy Clin. Immunol. 2021, 147, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Zhang, Q.; Wang, K.; Zhang, X.; Yang, R.; Bi, K.; Chen, W.; Diao, H. RBM15-mediated N6-methyladenosine modification affects COVID-19 severity by regulating the expression of multitarget genes. Cell Death Dis. 2021, 12, 732. [Google Scholar] [CrossRef] [PubMed]
- Grimaudo, S.; Amodio, E.; Pipitone, R.M.; Maida, C.M.; Pizzo, S.; Prestileo, T.; Tramuto, F.; Sardina, D.; Vitale, F.; Casuccio, A.; et al. PNPLA3 and TLL-1 polymorphisms as potential predictors of disease severity in patients with COVID-19. Front. Cell. Dev. Biol 2021, 9, 627914. [Google Scholar] [CrossRef] [PubMed]
- Latini, A.; Agolini, E.; Novelli, A.; Borgiani, P.; Giannini, R.; Gravina, P.; Smarrazzo, A.; Dauri, M.; Andreoni, M.; Rogliani, P.; et al. COVID-19 and genetic variants of protein involved in the SARS-CoV-2 entry into the host cells. Genes 2020, 11, 1010. [Google Scholar] [CrossRef]
- Wang, F.; Huang, S.; Gao, R.; Zhou, Y.; Lai, C.; Li, Z.; Xian, W.; Qian, X.; Li, Z.; Huang, Y.; et al. Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discov. 2020, 6, 83. [Google Scholar] [CrossRef]
- Biering, S.B.; Sarnik, S.A.; Wang, E.; Zengel, J.R.; Leist, S.R.; Schäfer, A.; Sathyan, V.; Hawkins, P.; Okuda, K.; Tau, C.; et al. Genome-wide bidirectional CRISPR screens identify mucins as host factors modulating SARS-CoV-2 infection. Nat. Genet. 2022, 54, 1078–1089. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Patient Group | All EDS | Female | Male | No EDS |
|---|---|---|---|---|
| Total number (No.) T | 1899 | 1553 | 346 | 80 |
| Number with systematic evaluations (%T) | 1261 (66) | 1064 (69) | 197 (57) | 64 (80) |
| “ with DNA testing (%T) | 967 (51) | 816 (53) | 151 (44) | 23 (29) |
| “ with potentially significant DNA variant (V) | 568 | 480 | 88 | 4 |
| “ with variant relevant to EDS/DD by Co. (%V) | 20 (3.5) | 16 (3.3) | 4 (4.5) | 0 |
| “ with variant relevant to other Dx by Co. (%V) | 181 (32) | 154 (32) | 27 (31) | 0 |
| “ with variant relevant to EDS/DD by Au. (%V) | 566 (99) | 478 (99) | 88 (100) | 4 (100) |
| “ with primary MT DNA variant (%V) | 93 (16) | 79 (16) | 14 (16) | 0 |
| “ with primary nuclear to MT DNA variant (%V) | 19 (3.4) | 18 (3.8) | 1 (1.1) | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, G.N. A Clinical Qualification Protocol Highlights Overlapping Genomic Influences and Neuro-Autonomic Mechanisms in Ehlers–Danlos and Long COVID-19 Syndromes. Curr. Issues Mol. Biol. 2023, 45, 6003-6023. https://doi.org/10.3390/cimb45070379
Wilson GN. A Clinical Qualification Protocol Highlights Overlapping Genomic Influences and Neuro-Autonomic Mechanisms in Ehlers–Danlos and Long COVID-19 Syndromes. Current Issues in Molecular Biology. 2023; 45(7):6003-6023. https://doi.org/10.3390/cimb45070379
Chicago/Turabian StyleWilson, Golder N. 2023. "A Clinical Qualification Protocol Highlights Overlapping Genomic Influences and Neuro-Autonomic Mechanisms in Ehlers–Danlos and Long COVID-19 Syndromes" Current Issues in Molecular Biology 45, no. 7: 6003-6023. https://doi.org/10.3390/cimb45070379
APA StyleWilson, G. N. (2023). A Clinical Qualification Protocol Highlights Overlapping Genomic Influences and Neuro-Autonomic Mechanisms in Ehlers–Danlos and Long COVID-19 Syndromes. Current Issues in Molecular Biology, 45(7), 6003-6023. https://doi.org/10.3390/cimb45070379