
Blockade of L-Type Ca2+ Channel Activity Alleviates Oligodendrocyte Pathology following Brain Injury in Male Rats
 ,
,  and
and 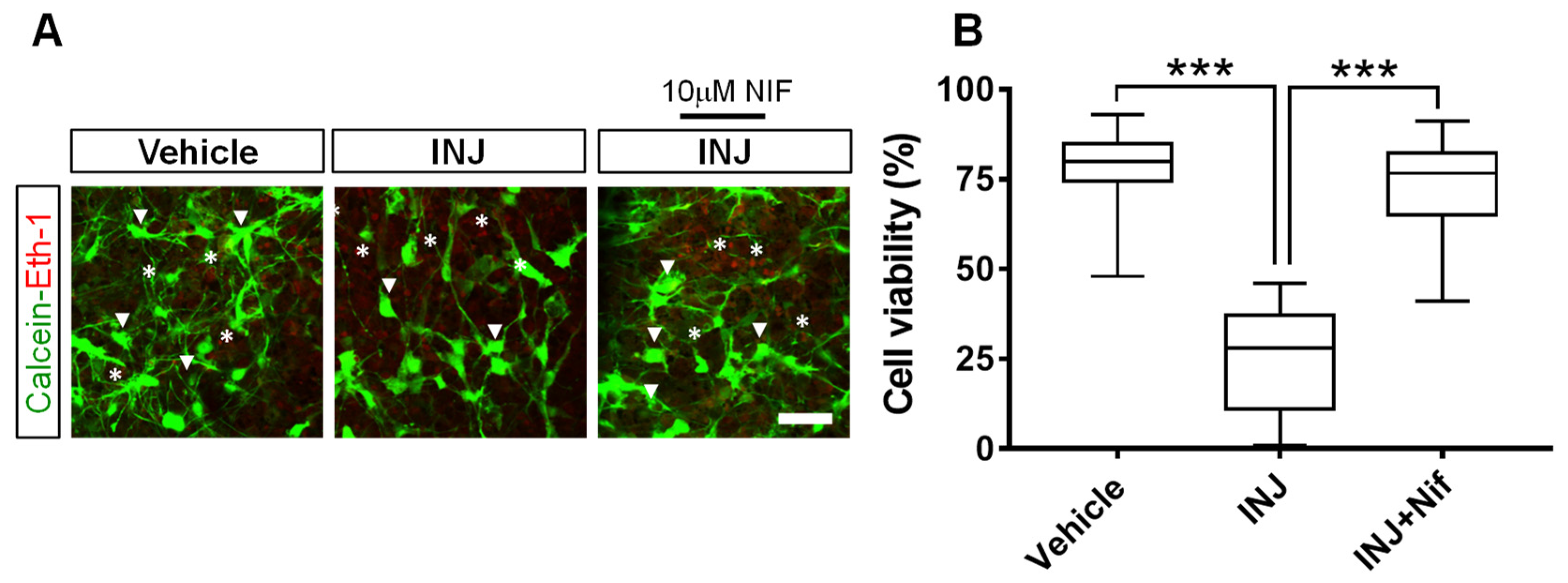{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Animals and Housing Conditions
2.3. Preparation of Cerebellar Slice Tissues
2.4. Induction of Cerebellar Slice Tissue Injury
2.5. Experimental Design
2.6. Cell Viability Assay
2.7. TUNEL Assay
2.8. Immunocytochemistry
2.9. Cell Proliferation Assay
2.10. Cell Quantification
2.11. Statistics
3. Results
3.1. L-Type Ca2+ Channel Inhibitor NIF Preserves Cell Viability following Brain Tissue Injury
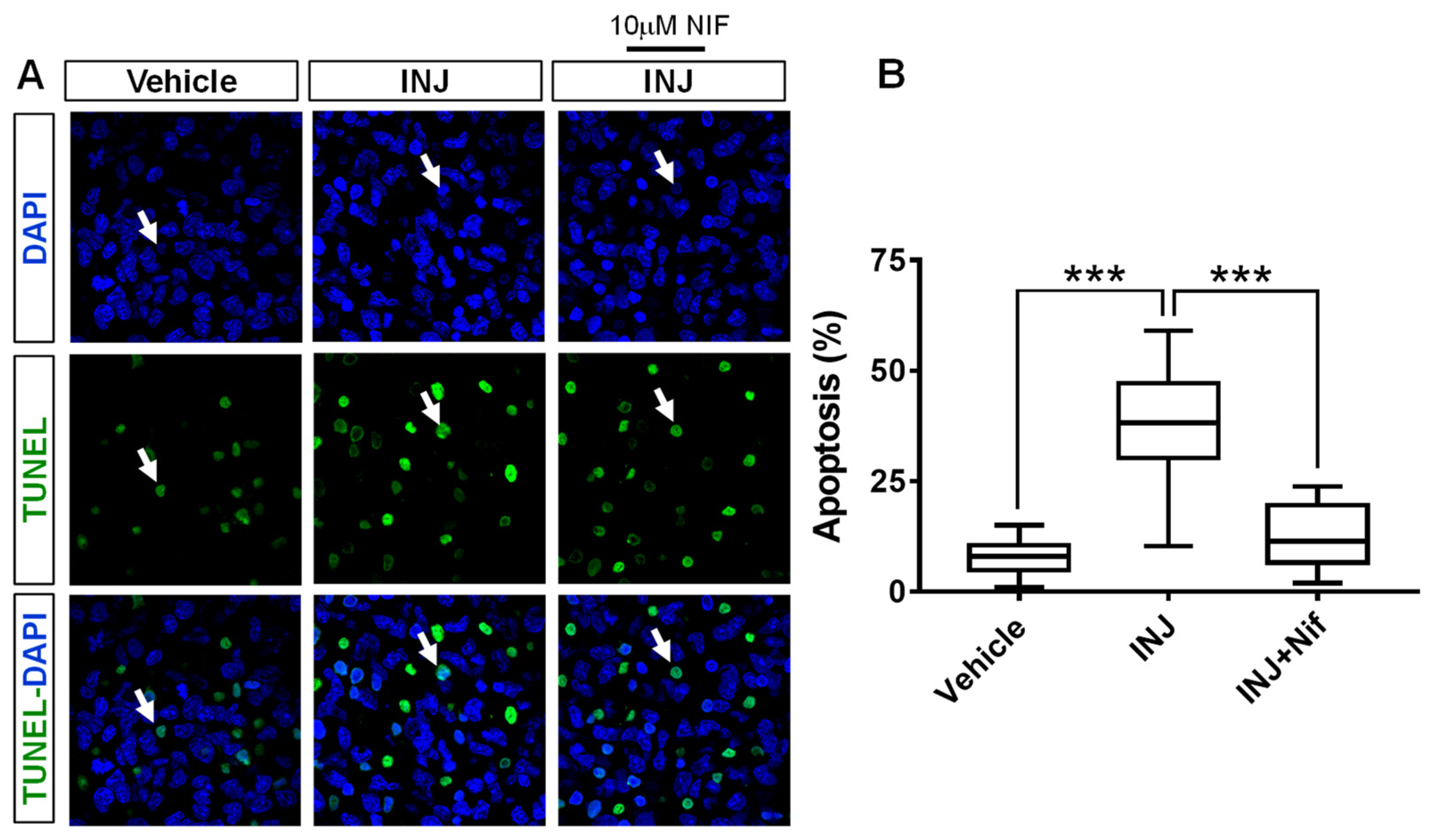
3.2. L-Type Ca2+ Channel Inhibitor NIF Minimizes Apoptosis following Brain Tissue Injury
3.3. L-Type Ca2+ Channel Inhibitor NIF Restores Mature OL Survival following Brain Tissue Injury
3.4. L-Type Ca2+ Channel Inhibitor NIF Enhances NG2+ OPC Survival following Injury
3.5. L-Type Ca2+ Channel Inhibitor NIF Preserves NG2+ OPC Mitotic Behavior following Injury
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Volpe, J.J. Brain injury in premature infants: A complex amalgam of destructive and developmental disturbances. Lancet Neurol. 2009, 8, 110–124. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr. Res. 2001, 50, 553–562. [Google Scholar] [CrossRef]
- Clarke, G.; Aatsinki, A.; O’Mahony, S.M. Brain development in premature infants: A bug in the programming system? Cell Host Microbe 2021, 29, 1477–1479. [Google Scholar] [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar] [CrossRef]
- Sherwin, C.; Fern, R. Acute lipopolysaccharide-mediated injury in neonatal white matter glia: Role of TNF-A, IL-1B, and calcium. J. Immunol. 2005, 175, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Han, B.H.; Luo, N.L.; Chricton, C.A.; Xanthoudakis, S.; Tam, J.; Arvin, K.L.; Holtzman, D.M. Selective vulnerability of late oligodendrocyte progenitors to hypoxia–ischemia. J. Neurosci. 2002, 22, 455–463. [Google Scholar] [CrossRef]
- Tekkök, S.B.; Ye, Z.; Ransom, B.R. Excitotoxic mechanisms of ischemic injury in myelinated white matter. J. Cereb. Blood Flow Metab. 2007, 27, 1540–1552. [Google Scholar] [CrossRef]
- Matute, C.; Alberdi, E.; Domercq, M.; Sánchez-Gómez, M.V.; Pérez-Samartín, A.; Rodríguez-Antigüedad, A.; Pérez-Cerdá, F. Excitotoxic damage to white matter. J. Anat. 2007, 210, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Matute, C. Glutamate and ATP signalling in white matter pathology. J. Anat. 2011, 219, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F.E. Role of glutamate receptors in periventricular leukomalacia. J. Child Neurol. 2005, 20, 950–959. [Google Scholar] [CrossRef]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000, 403, 316–321. [Google Scholar] [CrossRef] [PubMed]
- DeSilva, T.M.; Kabakov, A.Y.; Goldhoff, P.E.; Volpe, J.J.; Rosenberg, P.A. Regulation of glutamate transport in developing rat oligodendrocytes. J. Neurosci. 2009, 29, 7898–7908. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.G.; Fern, R. The mechanisms of acute ischemic injury in the cell processes of developing white matter astrocytes. J. Cereb. Blood Flow Metab. 2007, 28, 588–601. [Google Scholar] [CrossRef]
- Matute, C. Calcium dyshomeostasis in white matter pathology. Cell Calcium 2010, 47, 150–157. [Google Scholar] [CrossRef]
- Stys, P.K. White matter injury mechanisms. Curr. Mol. Med. 2004, 18, 113–130. [Google Scholar] [CrossRef]
- Paez, P.M.; Fulton, D.; Colwell, C.S.; Campagnoni, A.T. Voltage-operated Ca2+ and Na+ channels in the oligodendrocyte lineage. J. Neurosci. Res. 2009, 87, 3259–3266. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Tuo, Q.-Z.; Zhang, S.-T.; Lei, P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med. Res. Rev. 2022, 42, 259–305. [Google Scholar] [CrossRef]
- Arai, K.; Lo, E. Experimental models for analysis of oligodendrocyte pathophysiology in stroke. Exp. Transl. Stroke Med. 2009, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.M.; Reynoldsand, R.; Fawcett, J.W. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. 2001, 24, 39–47. [Google Scholar] [CrossRef]
- McTigue, D.M.; Wei, P.; Stokes, B.T. Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J. Neurosci. 2001, 21, 3392–3400. [Google Scholar] [CrossRef]
- Rivers, L.E.; Young, K.M.; Rizzi, M.; Jamen, F.; Psachoulia, K.; Wade, A.; Kessaris, N.; Richardson, W.D. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat. Neurosci. 2008, 11, 1392–1401. [Google Scholar] [CrossRef]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–926. [Google Scholar] [CrossRef]
- Suyama, K.; Watanabe, M.; Sakai, D.; Osada, T.; Imai, M.; Mochida, J. Nkx2.2 expression in differentiation of oligodendrocyte precursor cells and inhibitory factors for differentiation of oligodendrocytes after traumatic spinal cord injury. J. Neurotrauma 2007, 24, 1013–1025. [Google Scholar] [CrossRef]
- Al-Griw, M.A.; Alghazeer, R.O.; Awayn, A.; Shamlan, G.; Eskandrani, A.A.; Alnajeebi, A.M.; Babteen, N.A.; Alansari, W.S. Selective adenosine A2A receptor inhibitor SCH58261 reduces oligodendrocyte loss upon brain injury in young rats. Saudi J. Biol. Sci. 2021, 28, 310–316. [Google Scholar] [CrossRef]
- Al-Griw, M.A.; Wood, I.C.; Salter, M.G. Cerebellar Organotypic Slice Culture System: A Model of Developing Brain Ischaemia. Life Sci. J. 2017, 14, 89–98. [Google Scholar]
- Al-Griw, M.A.; Salter, M.G.; Wood, I.C. Inhibition of ionotropic GluR signaling preserves oligodendrocyte lineage and myelination in an ex vivo rat model of white matter ischemic injury. Acta Neurobiol. Exp. 2021, 81, 233–248. [Google Scholar] [CrossRef]
- Al-Griw, M.A.; Salter, M.G.; Wood, I.C. Blocking of NF-kB/p38MAPK pathways mitigates oligodendrocyte pathology in a model of neonatal white matter injury. Acta Neurobiol. Exp. 2022, 82, 52–64. [Google Scholar]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef]
- Meco, E.; Zheng, W.S.; Sharma, A.H.; Lampe, K.J. Guiding oligodendrocyte precursor cell maturation with Urokinase plasminogen activator-degradable elastin-like protein hydrogels. Biomacromolecules 2020, 21, 4724–4736. [Google Scholar] [CrossRef]
- Levine, J.M.; Card, J. Light and electron microscopic localization of a cell surface antigen (NG2) in the rat cerebellum: Association with smooth protoplasmic astrocytes. J. Neurosci. 1987, 7, 2711–2720. [Google Scholar] [CrossRef]
- Gupta, R.; Ambasta, R.K.; Kumar, P. Histone deacetylase in neuropathology. Adv. Clin. Chem. 2021, 104, 151–231. [Google Scholar]
- Shi, Z.; Wu, D.; Yao, J.P. Protection against Oxygen-Glucose Deprivation/Reperfusion Injury in Cortical Neurons by Combining Omega-3 Polyunsaturated Acid with Lyciumbarbarum Polysaccharide. Nutrients 2016, 8, 41. [Google Scholar] [CrossRef]
- Pietrobon, D. Calcium channels and channelopathies of the central nervous system. Mol. Neurobiol. 2002, 25, 31–50. [Google Scholar] [CrossRef]
- Boczek, T.; Zylinska, L. Receptor-Dependent and Independent Regulation of Voltage-Gated Ca2+ Channels and Ca2+-Permeable Channels by Endocannabinoids in the Brain. Int. J. Mol. Sci. 2021, 22, 8168. [Google Scholar] [CrossRef]
- Catterall, W.A. Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 2000, 16, 521–555. [Google Scholar] [CrossRef]
- Ureshino, R.P.; Rocha, K.K.; Lopes, G.S.; Bincoletto, C.; Smaili, S.S. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid. Redox Signal 2014, 21, 123–137. [Google Scholar] [CrossRef]
- Alberdi, E.; Sanchez-Gomez, M.V.; Matute, C. Calcium and glial cell death. Cell Calcium 2005, 38, 417–425. [Google Scholar] [CrossRef]
- Cho, S.G.; Choi, E.J. Apoptotic signaling pathways: Caspases and stress-activated protein kinases. J. Biochem. Mol. Biol. 2002, 35, 24–27. [Google Scholar] [CrossRef]
- Butt, A.M.; Bay, V. Axon-glial interactions in the central nervous system. J. Anat. 2011, 219, 1. [Google Scholar] [CrossRef]
- Cheli, V.T.; Santiago González, D.A.; Spreuer, V.; Paez, P.M. Voltage-gated Ca++ entry promotes oligodendrocyte progenitor cells maturation and myelination in vitro. Exp. Neurol. 2015, 265, 69–83. [Google Scholar] [CrossRef]
- Butt, A.M. Neurotransmitter-mediated calcium signalling in oligodendrocyte physiology and pathology. Glia 2006, 54, 666–675. [Google Scholar] [CrossRef]
- Fern, R. Intracellular calcium and cell death during ischemia in neonatal rat white matter astrocytes in situ. J. Neurosci. 1998, 18, 7232–7243. [Google Scholar] [CrossRef]
- Imaizumi, T.; Kocsis, J.D.; Waxman, S.G. The role of voltage-gated Ca21 channels in anoxic injury of spinal cord white matter. Brain Res. 1999, 817, 84–92. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Griw, M.A.; Alghazeer, R.; Ratemi, H.W.; Ben-Othman, M.E.; Tabagah, R.; Shamlan, G.; Habibullah, M.M.; Alnajeebi, A.M.; Babteen, N.A.; Eskandrani, A.A.; et al. Blockade of L-Type Ca2+ Channel Activity Alleviates Oligodendrocyte Pathology following Brain Injury in Male Rats. Curr. Issues Mol. Biol. 2023, 45, 3953-3964. https://doi.org/10.3390/cimb45050252
Al-Griw MA, Alghazeer R, Ratemi HW, Ben-Othman ME, Tabagah R, Shamlan G, Habibullah MM, Alnajeebi AM, Babteen NA, Eskandrani AA, et al. Blockade of L-Type Ca2+ Channel Activity Alleviates Oligodendrocyte Pathology following Brain Injury in Male Rats. Current Issues in Molecular Biology. 2023; 45(5):3953-3964. https://doi.org/10.3390/cimb45050252
Chicago/Turabian StyleAl-Griw, Mohamed A., Rabia Alghazeer, Haithm W. Ratemi, Mohamed E. Ben-Othman, Refaat Tabagah, Ghalia Shamlan, Mahmmoud M. Habibullah, Afnan M. Alnajeebi, Nouf A. Babteen, Areej A. Eskandrani, and et al. 2023. "Blockade of L-Type Ca2+ Channel Activity Alleviates Oligodendrocyte Pathology following Brain Injury in Male Rats" Current Issues in Molecular Biology 45, no. 5: 3953-3964. https://doi.org/10.3390/cimb45050252
APA StyleAl-Griw, M. A., Alghazeer, R., Ratemi, H. W., Ben-Othman, M. E., Tabagah, R., Shamlan, G., Habibullah, M. M., Alnajeebi, A. M., Babteen, N. A., Eskandrani, A. A., AL-Farga, A., & Alansari, W. S. (2023). Blockade of L-Type Ca2+ Channel Activity Alleviates Oligodendrocyte Pathology following Brain Injury in Male Rats. Current Issues in Molecular Biology, 45(5), 3953-3964. https://doi.org/10.3390/cimb45050252