
Lung Cancer Gene Regulatory Network of Transcription Factors Related to the Hallmarks of Cancer



Abstract
1. Introduction
2. Materials and Methods
3. Results
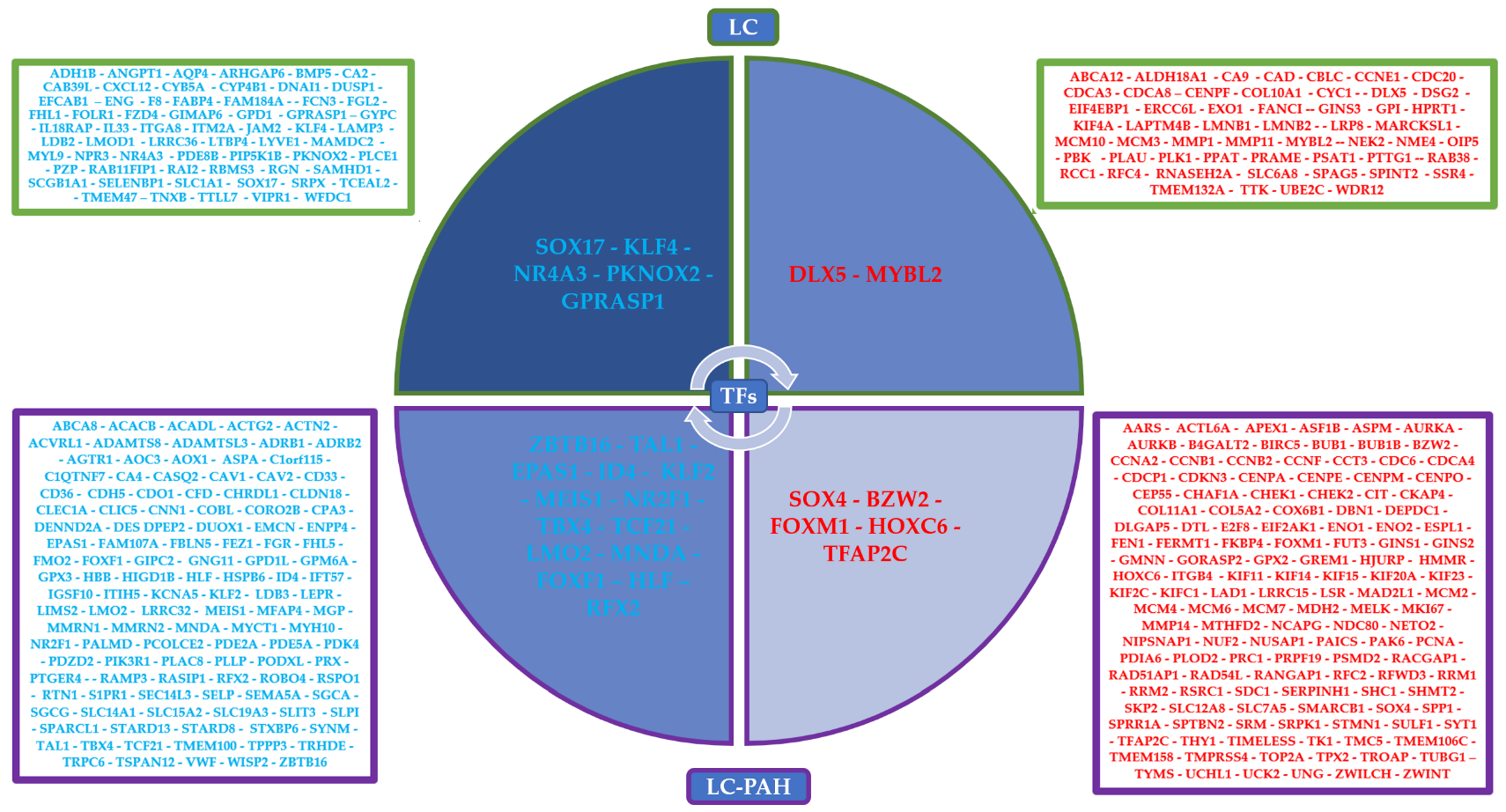
3.1. Co-Expression Networks Analysis
3.2. Gene Regulatory Networks Analysis
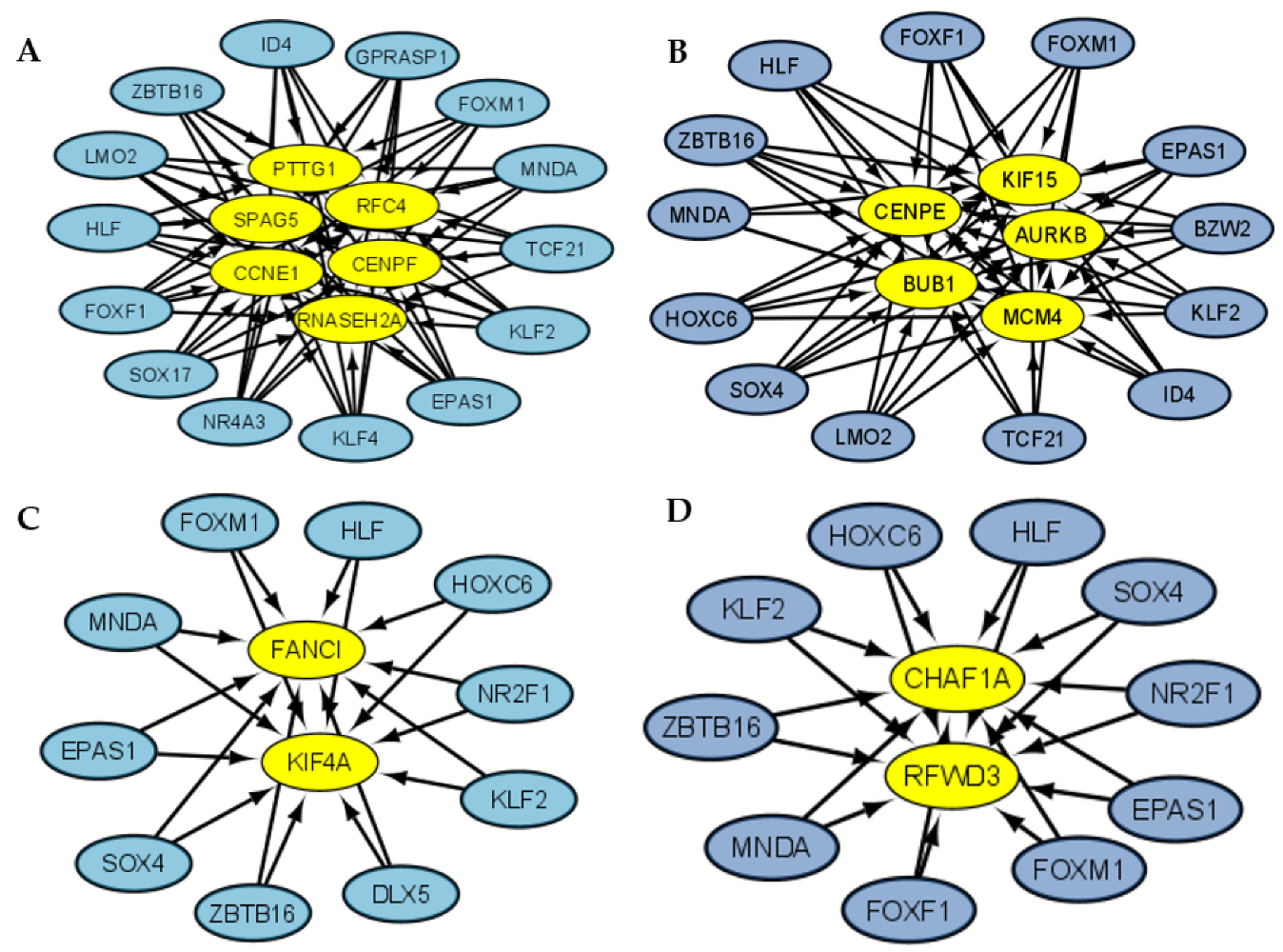
3.3. Coregulatory Networks and Fibration Symmetries Analysis
3.4. Transcriptional Regulatory Network Analysis of the Most Frequently Dysregulated Transcription Factors
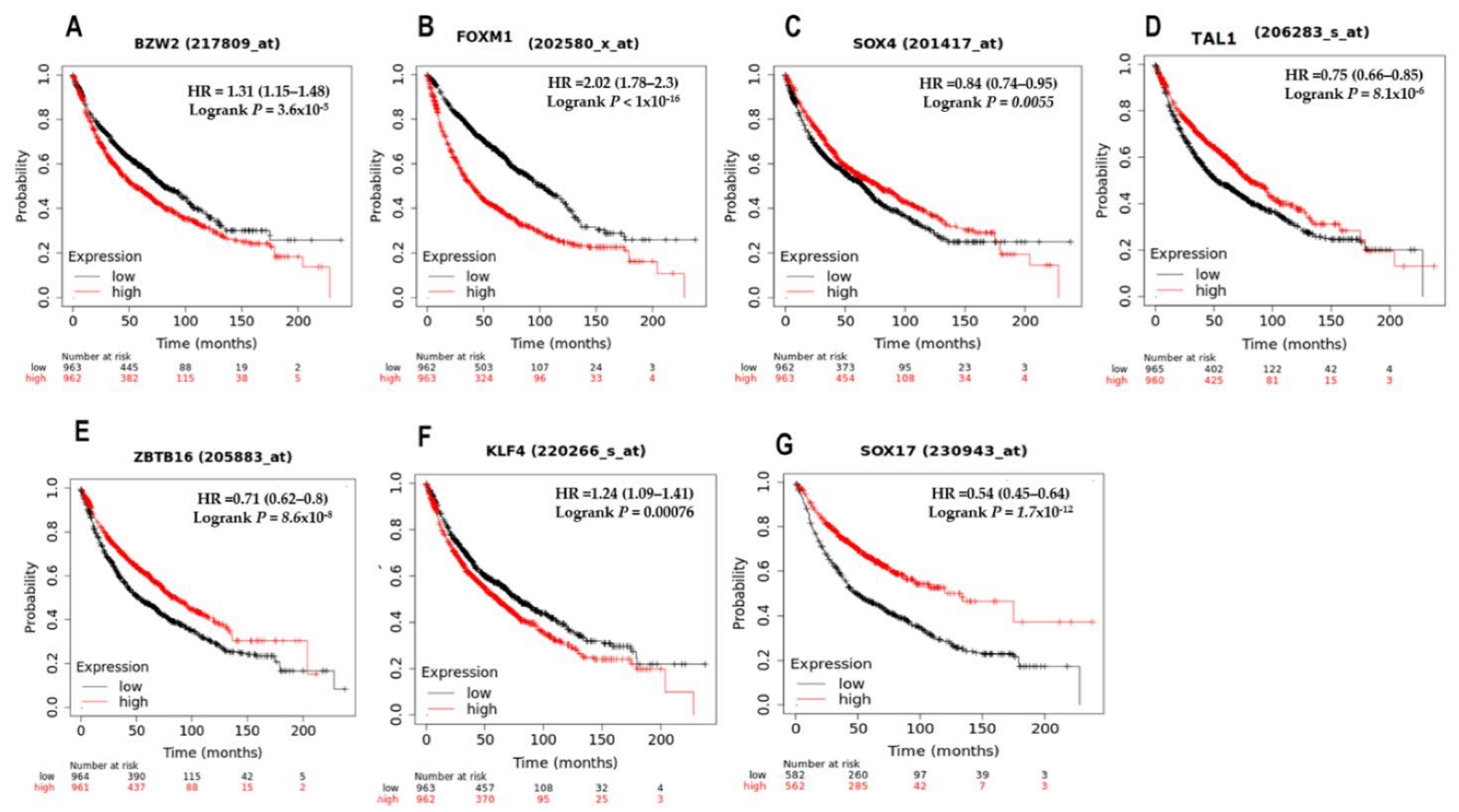
3.5. Survival Analysis of the Top Seven Deregulated Transcription Factors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siddiqui, F.; Vaqar, S.; Siddiqui, A.H. Lung Cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Aluru, J.S.; Barsouk, A. Epidemiology of Lung Cancer. Contemp. Oncol. 2021, 25, 45–52. [Google Scholar] [CrossRef]
- Benusiglio, P.R.; Fallet, V.; Sanchis-Borja, M.; Coulet, F.; Cadranel, J. Lung Cancer Is Also a Hereditary Disease. Eur. Respir. Rev. 2021, 30, 210045. [Google Scholar] [CrossRef]
- Otálora-Otálora, B.A.; Florez, M.; López-Kleine, L.; Canas Arboleda, A.; Grajales Urrego, D.M.; Rojas, A. Joint Transcriptomic Analysis of Lung Cancer and Other Lung Diseases. Front. Genet. 2019, 10, 1260. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.M.; Fountzilas, E.; Bleris, L.; Kurzrock, R. Transcriptomics and Solid Tumors: The next Frontier in Precision Cancer Medicine. Semin. Cancer Biol. 2022, 84, 50–59. [Google Scholar] [CrossRef]
- Morone, F.; Leifer, I.; Makse, H.A. Fibration Symmetries Uncover the Building Blocks of Biological Networks. Proc. Natl. Acad. Sci. USA 2020, 117, 8306–8314. [Google Scholar] [CrossRef]
- Leifer, I. Symmetry-Inspired Analysis of Biological Networks. Diss. Theses Capstone Proj. [Software Version 1.1]. 2022. Available online: https://github.com/makselab/fibrationSymmetries (accessed on 21 May 2022).
- Yang, X.; Wang, L.; Lin, L.; Liu, X. Elevated Pulmonary Artery Systolic Pressure Is Associated with Poor Survival of Patients with Non-Small Cell Lung Cancer. Cancer Manag. Res. 2020, 12, 6363–6371. [Google Scholar] [CrossRef]
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The Cancer Theory of Pulmonary Arterial Hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef]
- Otálora-Otálora, B.A.; Osuna-Garzón, D.A.; Carvajal-Parra, M.S.; Cañas, A.; Montecino, M.; López-Kleine, L.; Rojas, A. Identifying General Tumor and Specific Lung Cancer Biomarkers by Transcriptomic Analysis. Biology 2022, 11, 1082. [Google Scholar] [CrossRef]
- Tibshirani, A.R.; Seo, M.J.; Chu, G.; Narasimhan, B.; Li, J. Package “samr”. SAM: Significance Analysis of Microarrays. [Software Version 3.0]. 2018, pp. 1–31. Available online: https://cran.r-project.org/web/packages/samr/index.html (accessed on 10 June 2019).
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res. 2022, gkac194. [Google Scholar] [CrossRef]
- Wei, T.; Simko, V. R Package “Corrplot”: Visualization of a Correlation Matrix 2021. [Software Version 0.92]. Available online: https://cran.r-project.org/web/packages/corrplot/index.html (accessed on 21 May 2022).
- Henao, J.D. Coexnet: An R Package to Build CO-EXpression NETworks from Microarray Data 2018. [Software Version 1.15.0]. Available online: https://bioconductor.org/packages/coexnet/ (accessed on 21 May 2022).
- Ibragimov, R.; Malek, M.; Guo, J.; Baumbach, J. GEDEVO: An Evolutionary Graph Edit Distance Algorithm for Biological Network Alignment. In German Conference on Bioinformatics 2013; Schloss Dagstuhl-Leibniz-Zentrum fuer Informatik: Wadern, Germany, 2013. [Google Scholar]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal Complex Syst. 2005, 1695, 1–9. [Google Scholar]
- Janky, R.; Verfaillie, A.; Imrichová, H.; Van de Sande, B.; Standaert, L.; Christiaens, V.; Hulselmans, G.; Herten, K.; Naval Sanchez, M.; Potier, D.; et al. IRegulon: From a Gene List to a Gene Regulatory Network Using Large Motif and Track Collections. PLoS Comput. Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models. Genome Res. 2003, 13, 2498–2503. [Google Scholar] [CrossRef]
- Wu, G.; Dawson, E.; Duong, A.; Haw, R.; Stein, L. ReactomeFIViz: A Cytoscape App for Pathway and Network-Based Data Analysis. F1000Research 2014, 3, 146. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, R.; Radvanyi, F.; Elati, M. CoRegNet: Reconstruction and Integrated Analysis of Co-Regulatory Networks. Bioinformatics 2015, 31, 3066–3068. [Google Scholar] [CrossRef] [PubMed]
- Keenan, A.B.; Torre, D.; Lachmann, A.; Leong, A.K.; Wojciechowicz, M.L.; Utti, V.; Jagodnik, K.M.; Kropiwnicki, E.; Wang, Z.; Ma’ayan, A. ChEA3: Transcription Factor Enrichment Analysis by Orthogonal Omics Integration. Nucleic Acids Res. 2019, 47, W212–W224. [Google Scholar] [CrossRef]
- Groenevelt, C.; Gordon, R.; Wang, X.; Fletcher, M.; Markowetz, F.; Meyer, K.; Castro, M. Package ‘RTN.’ [Software Version 2.18.0]. 2021, pp. 1–70. Available online: http://bioconductor.org/packages/release/bioc/html/RTN.html (accessed on 21 May 2022).
- Győrffy, B.; Surowiak, P.; Budczies, J.; Lánczky, A. Online Survival Analysis Software to Assess the Prognostic Value of Biomarkers Using Transcriptomic Data in Non-Small-Cell Lung Cancer. PLoS ONE 2013, 8, e82241. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Durin, L.; Noël-Savina, E.; Héluain, V.; Mattei, P.; Mazières, J.; Prévot, G. Impact of Pulmonary Hypertension on Lung Cancer Management. Respir. Med. Res. 2022, 82, 100964. [Google Scholar] [CrossRef]
- Cool, C.D.; Kuebler, W.M.; Bogaard, H.J.; Spiekerkoetter, E.; Nicolls, M.R.; Voelkel, N.F. The Hallmarks of Severe Pulmonary Arterial Hypertension: The Cancer Hypothesis—Ten Years Later. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 318, L1115–L1130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qiao, W.; Shan, L. Expression and Functional Characterization of FOXM1 in Non-Small Cell Lung Cancer. OncoTargets Ther. 2018, 11, 3385–3393. [Google Scholar] [CrossRef] [PubMed]
- Koch, S. Regulation of Wnt Signaling by FOX Transcription Factors in Cancer. Cancers 2021, 13, 3446. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, Y.; Xue, J.; Gong, A.; Yu, G.; Zhou, A.; Lin, K.; Zhang, S.; Zhang, N.; Gottardi, C.J.; et al. Wnt-induced Deubiquitination FoxM1 Ensures Nucleus Β-catenin Transactivation. EMBO J. 2016, 35, 668–684. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.-C.; Wang, J.; Cheng, Y.; Zhang, X.-Y.; Ye, X.-Q. Overexpression of MYBL2 Promotes Proliferation and Migration of Non-Small-Cell Lung Cancer via Upregulating NCAPH. Mol. Cell. Biochem. 2020, 468, 185–193. [Google Scholar] [CrossRef]
- Morris, B.B.; Wages, N.A.; Grant, P.A.; Stukenberg, P.T.; Gentzler, R.D.; Hall, R.D.; Akerley, W.L.; Varghese, T.K.; Arnold, S.M.; Williams, T.M.; et al. MYBL2-Driven Transcriptional Programs Link Replication Stress and Error-Prone DNA Repair With Genomic Instability in Lung Adenocarcinoma. Front. Oncol. 2021, 10, 585551. [Google Scholar] [CrossRef]
- Ahmed, F. Integrated Network Analysis Reveals FOXM1 and MYBL2 as Key Regulators of Cell Proliferation in Non-Small Cell Lung Cancer. Front. Oncol. 2019, 9, 1011. [Google Scholar] [CrossRef]
- Mullen, D.J.; Yan, C.; Kang, D.S.; Zhou, B.; Borok, Z.; Marconett, C.N.; Farnham, P.J.; Offringa, I.A.; Rhie, S.K. TENET 2.0: Identification of Key Transcriptional Regulators and Enhancers in Lung Adenocarcinoma. PLoS Genet. 2020, 16, e1009023. [Google Scholar] [CrossRef]
- Sadasivam, S.; DeCaprio, J.A. The DREAM Complex: Master Coordinator of Cell Cycle Dependent Gene Expression. Nat. Rev. Cancer 2013, 13, 585–595. [Google Scholar] [CrossRef]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F Target Gene Analyses Identifies Cell Cycle Gene Regulatory Networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef]
- Zhao, X.; Zhou, L.L.; Li, X.; Ni, J.; Chen, P.; Ma, R.; Wu, J.; Feng, J. Overexpression of KIF20A Confers Malignant Phenotype of Lung Adenocarcinoma by Promoting Cell Proliferation and Inhibiting Apoptosis. Cancer Med. 2018, 7, 4678–4689. [Google Scholar] [CrossRef]
- Zhou, F.; Wang, M.; Aibaidula, M.; Zhang, Z.; Aihemaiti, A.; Aili, R.; Chen, H.; Dong, S.; Wei, W.; Maimaitiaili, A. TPX2 Promotes Metastasis and Serves as a Marker of Poor Prognosis in Non-Small Cell Lung Cancer. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2020, 26, e925147. [Google Scholar] [CrossRef] [PubMed]
- Kou, F.; Sun, H.; Wu, L.; Li, B.; Zhang, B.; Wang, X.; Yang, L. TOP2A Promotes Lung Adenocarcinoma Cells’ Malignant Progression and Predicts Poor Prognosis in Lung Adenocarcinoma. J. Cancer 2020, 11, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, Z.; Duan, R. Expression of ASPM in Lung Adenocarcinoma and Its Relationship with Development and Prognosis. Zhongguo Fei Ai Za Zhi Chin. J. Lung Cancer 2020, 23, 29–35. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, J.; Zheng, Y.; Liu, J.; Duan, T.; Tian, T. Overexpression of Abnormal Spindle-like Microcephaly-Associated (ASPM) Increases Tumor Aggressiveness and Predicts Poor Outcome in Patients with Lung Adenocarcinoma. Transl. Cancer Res. 2021, 10, 983–997. [Google Scholar] [CrossRef]
- Xia, C.; Xu, X.; Ding, Y.; Yu, C.; Qiao, J.; Liu, P. Abnormal Spindle-like Microcephaly-Associated Protein Enhances Cell Invasion through Wnt/β-Catenin-Dependent Regulation of Epithelial-Mesenchymal Transition in Non-Small Cell Lung Cancer Cells. J. Thorac. Dis. 2021, 13, 2460–2474. [Google Scholar] [CrossRef]
- Wen, X.; Wu, Y.; Awadasseid, A.; Tanaka, Y.; Zhang, W. New Advances in Canonical Wnt/β-Catenin Signaling in Cancer. Cancer Manag. Res. 2020, 12, 6987–6998. [Google Scholar] [CrossRef]
- Song, Y.-J.; Tan, J.; Gao, X.-H.; Wang, L.-X. Integrated Analysis Reveals Key Genes with Prognostic Value in Lung Adenocarcinoma. Cancer Manag. Res. 2018, 10, 6097–6108. [Google Scholar] [CrossRef]
- Tang, H.; Bai, Y.; Xiong, L.; Wei, Y.; Hu, W.; Xu, M.; Zhou, X.; Pan, G.; Zhang, L.; Zhu, M.; et al. Knockdown of CENPF Inhibits the Progression of Lung Adenocarcinoma. Aging 2021, 22, 2604. [Google Scholar]
- Sun, J.; Huang, J.; Lan, J.; Zhou, K.; Gao, Y.; Song, Z.; Deng, Y.; Liu, L.; Dong, Y.; Liu, X. Overexpression of CENPF Correlates with Poor Prognosis and Tumor Bone Metastasis in Breast Cancer. Cancer Cell Int. 2019, 19, 264. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, X.; Wang, L.; Wang, T.; Tang, X.; Su, X. Centromere Protein F (CENPF) Serves as a Potential Prognostic Biomarker and Target for Human Hepatocellular Carcinoma. J. Cancer 2021, 12, 2933–2951. [Google Scholar] [CrossRef]
- Dai, Y.; Liu, L.; Zeng, T.; Zhu, Y.-H.; Li, J.; Chen, L.; Li, Y.; Yuan, Y.-F.; Ma, S.; Guan, X.-Y. Characterization of the Oncogenic Function of Centromere Protein F in Hepatocellular Carcinoma. Biochem. Biophys. Res. Commun. 2013, 436, 711–718. [Google Scholar] [CrossRef]
- Gartel, A.L. FOXM1 in Cancer: Interactions and Vulnerabilities. Cancer Res. 2017, 77, 3135–3139. [Google Scholar] [CrossRef]
- Huang, R.; Gao, L. Identification of Potential Diagnostic and Prognostic Biomarkers in Non-Small Cell Lung Cancer Based on Microarray Data. Oncol. Lett. 2018, 15, 6436–6442. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Chan, C.-H.; Dubey, N.K.; Wei, H.-J.; Lu, J.-H.; Chang, C.-C.; Cheng, H.-C.; Ou, K.-L.; Deng, W.-P. Highly Expressed FOXF1 Inhibit Non-Small-Cell Lung Cancer Growth via Inducing Tumor Suppressor and G1-Phase Cell-Cycle Arrest. Int. J. Mol. Sci. 2020, 21, E3227. [Google Scholar] [CrossRef]
- Cao, P.; Walker, N.M.; Braeuer, R.R.; Mazzoni-Putman, S.; Aoki, Y.; Misumi, K.; Wheeler, D.S.; Vittal, R.; Lama, V.N. Loss of FOXF1 Expression Promotes Human Lung-Resident Mesenchymal Stromal Cell Migration via ATX/LPA/LPA1 Signaling Axis. Sci. Rep. 2020, 10, 21231. [Google Scholar] [CrossRef]
- Wei, H.-J.; Nickoloff, J.A.; Chen, W.-H.; Liu, H.-Y.; Lo, W.-C.; Chang, Y.-T.; Yang, P.-C.; Wu, C.-W.; Williams, D.F.; Gelovani, J.G.; et al. FOXF1 Mediates Mesenchymal Stem Cell Fusion-Induced Reprogramming of Lung Cancer Cells. Oncotarget 2014, 5, 9514–9529. [Google Scholar] [CrossRef]
- Wang, S.; Xiao, Z.; Hong, Z.; Jiao, H.; Zhu, S.; Zhao, Y.; Bi, J.; Qiu, J.; Zhang, D.; Yan, J.; et al. FOXF1 Promotes Angiogenesis and Accelerates Bevacizumab Resistance in Colorectal Cancer by Transcriptionally Activating VEGFA. Cancer Lett. 2018, 439, 78–90. [Google Scholar] [CrossRef]
- Zhang, W.; Duan, N.; Song, T.; Li, Z.; Zhang, C.; Chen, X. The Emerging Roles of Forkhead Box (FOX) Proteins in Osteosarcoma. J. Cancer 2017, 8, 1619–1628. [Google Scholar] [CrossRef]
- Saito, R.-A.; Micke, P.; Paulsson, J.; Augsten, M.; Peña, C.; Jönsson, P.; Botling, J.; Edlund, K.; Johansson, L.; Carlsson, P.; et al. Forkhead Box F1 Regulates Tumor-Promoting Properties of Cancer-Associated Fibroblasts in Lung Cancer. Cancer Res. 2010, 70, 2644–2654. [Google Scholar] [CrossRef]
- Gooskens, S.L.; Klasson, T.D.; Gremmels, H.; Logister, I.; Pieters, R.; Perlman, E.J.; Giles, R.H.; van den Heuvel-Eibrink, M.M. TCF21 Hypermethylation Regulates Renal Tumor Cell Clonogenic Proliferation and Migration. Mol. Oncol. 2018, 12, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zeng, C.; Ye, Y.; Wu, D.; Mu, Z.; Liu, J.; Xie, Y.; Wu, H. Promoter Methylation of TCF21 May Repress Autophagy in the Progression of Lung Cancer. J. Cell Commun. Signal. 2018, 12, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.T.; Lin, M.; Brena, R.M.; Lang, J.C.; Schuller, D.E.; Otterson, G.A.; Morrison, C.D.; Smiraglia, D.J.; Plass, C. Epigenetic Regulation of the Tumor Suppressor Gene TCF21 on 6q23-Q24 in Lung and Head and Neck Cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Karolak, J.A.; Gambin, T.; Szafranski, P.; Stankiewicz, P. Potential Interactions between the TBX4-FGF10 and SHH-FOXF1 Signaling during Human Lung Development Revealed Using ChIP-Seq. Respir. Res. 2021, 22, 26. [Google Scholar] [CrossRef] [PubMed]
- Nehme, E.; Rahal, Z.; Sinjab, A.; Khalil, A.; Chami, H.; Nemer, G.; Kadara, H. Epigenetic Suppression of the T-Box Subfamily 2 (TBX2) in Human Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1159. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Dekmak, B.; Boulos, F.; Kantrowitz, J.; Spira, A.; Fujimoto, J.; Kadara, H.; El-Hachem, N.; Nemer, G. Transcriptomic Alterations in Lung Adenocarcinoma Unveil New Mechanisms Targeted by the TBX2 Subfamily of Tumor Suppressor Genes. Front. Oncol. 2018, 8, 482. [Google Scholar] [CrossRef]
- Tang, T.; Shi, Y.; Opalenik, S.R.; Brantley-Sieders, D.M.; Chen, J.; Davidson, J.M.; Brandt, S.J. Expression of the TAL1/SCL Transcription Factor in Physiological and Pathological Vascular Processes. J. Pathol. 2006, 210, 121–129. [Google Scholar] [CrossRef]
- Meng, X.; Lu, P.; Bai, H.; Xiao, P.; Fan, Q. Transcriptional Regulatory Networks in Human Lung Adenocarcinoma. Mol. Med. Rep. 2012, 6, 961–966. [Google Scholar] [CrossRef]
- Terme, J.-M.; Lemaire, S.; Auboeuf, D.; Mocquet, V.; Jalinot, P. The Proto-Oncogenic Protein TAL1 Controls TGF-Β1 Signaling through Interaction with SMAD3. Biochim. Open 2016, 2, 69–78. [Google Scholar] [CrossRef]
- Ji, S.; Wen, S.-L.; Sun, Y.; Huang, P.; Wu, H.; He, M. The Biological Function and Clinical Significance of STIL in Osteosarcoma. Cancer Cell Int. 2021, 21, 218. [Google Scholar] [CrossRef]
- Yamada, Y.; Warren, A.J.; Dobson, C.; Forster, A.; Pannell, R.; Rabbitts, T.H. The T Cell Leukemia LIM Protein Lmo2 Is Necessary for Adult Mouse Hematopoiesis. Proc. Natl. Acad. Sci. USA 1998, 95, 3890–3895. [Google Scholar] [CrossRef]
- Yamada, Y.; Pannell, R.; Forster, A.; Rabbitts, T.H. The Oncogenic LIM-Only Transcription Factor Lmo2 Regulates Angiogenesis but Not Vasculogenesis in Mice. Proc. Natl. Acad. Sci. USA 2000, 97, 320–324. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Huang, D.; Wu, C.; Li, H.; Zhang, X.; Meng, B.; Li, Z.; Zhu, T.; Yang, S.; et al. LMO2 Promotes Tumor Cell Invasion and Metastasis in Basal-Type Breast Cancer by Altering Actin Cytoskeleton Remodeling. Oncotarget 2016, 8, 9513–9524. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, D.; Wang, Z.; Wu, C.; Zhang, Z.; Wang, D.; Li, Z.; Zhu, T.; Yang, S.; Sun, W. LMO2 Attenuates Tumor Growth by Targeting the Wnt Signaling Pathway in Breast and Colorectal Cancer. Sci. Rep. 2016, 6, 36050. [Google Scholar] [CrossRef]
- Liu, Y.; Yuan, M.; Wu, C.; Zhu, T.; Sun, W. A Comprehensive Function Analysis of LMO2 in Different Breast Cancer Subtypes. Oncotarget 2017, 9, 8911–8926. [Google Scholar] [CrossRef][Green Version]
- Cubedo, E.; Gentles, A.J.; Huang, C.; Natkunam, Y.; Bhatt, S.; Lu, X.; Jiang, X.; Romero-Camarero, I.; Freud, A.; Zhao, S.; et al. Identification of LMO2 Transcriptome and Interactome in Diffuse Large B-Cell Lymphoma. Blood 2012, 119, 5478–5491. [Google Scholar] [CrossRef]
- Jiang, W.; Xu, X.; Deng, S.; Luo, J.; Xu, H.; Wang, C.; Sun, T.; Lei, G.; Zhang, F.; Yang, C.; et al. Methylation of Kruppel-like Factor 2 (KLF2) Associates with Its Expression and Non-Small Cell Lung Cancer Progression. Am. J. Transl. Res. 2017, 9, 2024–2037, PMID: 28469808PMCID: PMC5411951. [Google Scholar]
- Wang, H.-G.; Cao, B.; Zhang, L.-X.; Song, N.; Li, H.; Zhao, W.-Z.; Li, Y.-S.; Ma, S.-M.; Yin, D.-J. KLF2 Inhibits Cell Growth via Regulating HIF-1α/Notch-1 Signal Pathway in Human Colorectal Cancer HCT116 Cells. Oncol. Rep. 2017, 38, 584–590. [Google Scholar] [CrossRef][Green Version]
- Pi, J.; Tao, T.; Zhuang, T.; Sun, H.; Chen, X.; Liu, J.; Cheng, Y.; Yu, Z.; Zhu, H.H.; Gao, W.-Q.; et al. A MicroRNA302-367-Erk1/2-Klf2-S1pr1 Pathway Prevents Tumor Growth via Restricting Angiogenesis and Improving Vascular Stability. Circ. Res. 2017, 120, 85–98. [Google Scholar] [CrossRef]
- Ma, Z.; Wei, K.; Yang, F.; Guo, Z.; Pan, C.; He, Y.; Wang, J.; Li, Z.; Chen, L.; Chen, Y.; et al. Tumor-Derived Exosomal MiR-3157-3p Promotes Angiogenesis, Vascular Permeability and Metastasis by Targeting TIMP/KLF2 in Non-Small Cell Lung Cancer. Cell Death Dis. 2021, 12, 840. [Google Scholar] [CrossRef]
- Shi, J.; Zhou, L.; Wang, X.; Du, J.; Jiang, M.; Song, Z.; Han, G.; Mai, Z. KLF2 Attenuates Bleomycin-Induced Pulmonary Fibrosis and Inflammation with Regulation of AP-1. Biochem. Biophys. Res. Commun. 2018, 495, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Fledderus, J.O.; Volger, O.L.; van Wanrooij, E.J.A.; Pardali, E.; Weesie, F.; Kuiper, J.; Pannekoek, H.; ten Dijke, P.; Horrevoets, A.J.G. KLF2 Suppresses TGF-β Signaling in Endothelium Through Induction of Smad7 and Inhibition of AP-1. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Jin-Xiang, Y.; Long, T.; Xiu-Rong, L.; Hong, G.; Jie-Cheng, Y.; Fei, Z. Kruppel-like Factor 2 Disturb Non-Small Cell Lung Cancer Energy Metabolism by Inhibited Glutamine Consumption. J. Pharm. Pharmacol. 2020, 72, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Y.; Jun Wei, J.W.; Liu, X. The Role of Sox Genes in Lung Morphogenesis and Cancer. Int. J. Mol. Sci. 2012, 13, 15767–15783. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, S.; Lee, S.; Kim, K.; Yang, Y.; Kim, J.H.; Adams, R.H.; Wells, J.M.; Morrison, S.J.; Koh, G.Y.; et al. Sox17 Promotes Tumor Angiogenesis and Destabilizes Tumor Vessels in Mice. J. Clin. Investig. 2013, 123, 418–431. [Google Scholar] [CrossRef]
- Lee, S.-H.; Lee, S.; Yang, H.; Song, S.; Kim, K.; Saunders, T.L.; Yoon, J.K.; Koh, G.Y.; Kim, I. Notch Pathway Targets Proangiogenic Regulator Sox17 to Restrict Angiogenesis. Circ. Res. 2014, 115, 215–226. [Google Scholar] [CrossRef]
- Anani, M.; Nobuhisa, I.; Taga, T. Sry-Related High Mobility Group Box 17 Functions as a Tumor Suppressor by Antagonizing the Wingless-Related Integration Site Pathway. J. Cancer Prev. 2020, 25, 204–212. [Google Scholar] [CrossRef]
- Park, K.-S.; Wells, J.M.; Zorn, A.M.; Wert, S.E.; Whitsett, J.A. Sox17 Influences the Differentiation of Respiratory Epithelial Cells. Dev. Biol. 2006, 294, 192–202. [Google Scholar] [CrossRef]
- Tan, D.S.; Holzner, M.; Weng, M.; Srivastava, Y.; Jauch, R. SOX17 in Cellular Reprogramming and Cancer. Semin. Cancer Biol. 2020, 67, 65–73. [Google Scholar] [CrossRef]
- Kuo, I.-Y.; Wu, C.-C.; Chang, J.-M.; Huang, Y.-L.; Lin, C.-H.; Yan, J.-J.; Sheu, B.-S.; Lu, P.-J.; Chang, W.-L.; Lai, W.-W.; et al. Low SOX17 Expression Is a Prognostic Factor and Drives Transcriptional Dysregulation and Esophageal Cancer Progression. Int. J. Cancer 2014, 135, 563–573. [Google Scholar] [CrossRef]
- Wang, S.; Bai, W.; Huang, J.; Lv, F.; Bai, H. Prognostic Significance of BZW2 Expression in Lung Adenocarcinoma Patients. Int. J. Clin. Exp. Pathol. 2019, 12, 4289–4296. [Google Scholar]
- Cheng, D.-D.; Li, S.-J.; Zhu, B.; Yuan, T.; Yang, Q.-C.; Fan, C.-Y. Downregulation of BZW2 Inhibits Osteosarcoma Cell Growth by Inactivating the Akt/MTOR Signaling Pathway. Oncol. Rep. 2017, 38, 2116–2122. [Google Scholar] [CrossRef]
- Xu, J.; Testa, J.R. DLX5 (Distal-Less Homeobox 5) Promotes Tumor Cell Proliferation by Transcriptionally Regulating MYC. J. Biol. Chem. 2009, 284, 20593–20601. [Google Scholar] [CrossRef]
- Zhang, T.-J.; Xu, Z.-J.; Gu, Y.; Wen, X.-M.; Ma, J.-C.; Zhang, W.; Deng, Z.-Q.; Leng, J.-Y.; Qian, J.; Lin, J.; et al. Identification and Validation of Prognosis-Related DLX5 Methylation as an Epigenetic Driver in Myeloid Neoplasms. Clin. Transl. Med. 2020, 10, e29. [Google Scholar] [CrossRef]
- Tan, Y.; Testa, J.R. DLX Genes: Roles in Development and Cancer. Cancers 2021, 13, 3005. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Xie, F.; Zhang, F.; Zhang, S.; Zhou, J.; Chen, D.; Liu, A. MiR-339-5p Negatively Regulates Loureirin A-induced Hair Follicle Stem Cell Differentiation by Targeting DLX5. Mol. Med. Rep. 2018, 18, 1279–1286. [Google Scholar] [CrossRef]
- Dy, P.; Penzo-Méndez, A.; Wang, H.; Pedraza, C.E.; Macklin, W.B.; Lefebvre, V. The Three SoxC Proteins—Sox4, Sox11 and Sox12—Exhibit Overlapping Expression Patterns and Molecular Properties. Nucleic Acids Res. 2008, 36, 3101–3117. [Google Scholar] [CrossRef]
- Peng, X.; Liu, G.; Peng, H.; Chen, A.; Zha, L.; Wang, Z. SOX4 Contributes to TGF-β-Induced Epithelial–Mesenchymal Transition and Stem Cell Characteristics of Gastric Cancer Cells. Genes Dis. 2017, 5, 49–61. [Google Scholar] [CrossRef]
- Medina, P.P.; Castillo, S.D.; Blanco, S.; Sanz-Garcia, M.; Largo, C.; Alvarez, S.; Yokota, J.; Gonzalez-Neira, A.; Benitez, J.; Clevers, H.C.; et al. The SRY-HMG Box Gene, SOX4, Is a Target of Gene Amplification at Chromosome 6p in Lung Cancer†. Hum. Mol. Genet. 2009, 18, 1343–1352. [Google Scholar] [CrossRef]
- Quan, X.; Li, X.; Yin, Z.; Ren, Y.; Zhou, B. P53/MiR-30a-5p/ SOX4 Feedback Loop Mediates Cellular Proliferation, Apoptosis, and Migration of Non-Small-Cell Lung Cancer. J. Cell. Physiol. 2019, 234, 22884–22895. [Google Scholar] [CrossRef]
- Hur, W.; Rhim, H.; Jung, C.K.; Kim, J.D.; Bae, S.H.; Jang, J.W.; Yang, J.M.; Oh, S.-T.; Kim, D.G.; Wang, H.J.; et al. SOX4 Overexpression Regulates the P53-Mediated Apoptosis in Hepatocellular Carcinoma: Clinical Implication and Functional Analysis in Vitro. Carcinogenesis 2010, 31, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Xu, X.; Li, S.; Ma, J.; Shi, Q.; Guo, S.; Liu, L.; Guo, W.; Xu, P.; He, Y.; et al. SOX4 Promotes Proliferative Signals by Regulating Glycolysis through AKT Activation in Melanoma Cells. J. Investig. Dermatol. 2017, 137, 2407–2416. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yang, P.; Jiang, Y. CD147 Promotes Melanoma Cell Growth via SOX4-Mediated Glycolytic Metabolism. Trop. J. Pharm. Res. 2020, 19, 2521–2527. [Google Scholar] [CrossRef]
- Patel, D.; Morton, D.J.; Carey, J.; Havrda, M.C.; Chaudhary, J. Inhibitor of Differentiation 4 (ID4): From Development to Cancer. Biochim. Biophys. Acta 2015, 1855, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.-M.; Jin, X.; Lee, J.-S.; Oh, S.-Y.; Sohn, Y.-W.; Park, H.-J.; Joo, K.M.; Park, W.-Y.; Nam, D.-H.; DePinho, R.A.; et al. Inhibitor of Differentiation 4 Drives Brain Tumor-Initiating Cell Genesis through Cyclin E and Notch Signaling. Genes Dev. 2008, 22, 2028–2033. [Google Scholar] [CrossRef]
- Jeon, H.-M.; Kim, S.-H.; Jin, X.; Park, J.B.; Kim, S.H.; Joshi, K.; Nakano, I.; Kim, H. Crosstalk between Glioma-Initiating Cells and Endothelial Cells Drives Tumor Progression. Cancer Res. 2014, 74, 4482–4492. [Google Scholar] [CrossRef]
- Donzelli, S.; Milano, E.; Pruszko, M.; Sacconi, A.; Masciarelli, S.; Iosue, I.; Melucci, E.; Gallo, E.; Terrenato, I.; Mottolese, M.; et al. Expression of ID4 Protein in Breast Cancer Cells Induces Reprogramming of Tumour-Associated Macrophages. Breast Cancer Res. 2018, 20, 59. [Google Scholar] [CrossRef]
- Wang, C.-C.; Hsu, Y.-L.; Chang, C.-J.; Wang, C.-J.; Hsiao, T.-H.; Pan, S.-H. Inhibitor of DNA-Binding Protein 4 Suppresses Cancer Metastasis through the Regulation of Epithelial Mesenchymal Transition in Lung Adenocarcinoma. Cancers 2019, 11, E2021. [Google Scholar] [CrossRef]
- Chen, H.-J.; Yu, Y.; Sun, Y.-X.; Huang, C.-Z.; Li, J.-Y.; Liu, F.; Guo, G.-X.; Ye, Y.-B. Id4 Suppresses the Growth and Invasion of Colorectal Cancer HCT116 Cells through CK18-Related Inhibition of AKT and EMT Signaling. J. Oncol. 2021, 2021, e6660486. [Google Scholar] [CrossRef]
- Liang, D.; Hu, H.; Li, S.; Dong, J.; Wang, X.; Wang, Y.; He, L.; He, Z.; Gao, Y.; Gao, S.J.; et al. Oncogenic Herpesvirus KSHV Hijacks BMP-Smad1-Id Signaling to Promote Tumorigenesis. PLoS Pathog. 2014, 10, e1004253. [Google Scholar] [CrossRef]
- Xu, S.; Wang, Y.; Li, Y.; Zhang, L.; Wang, C.; Wu, X. Comprehensive Analysis of Inhibitor of Differentiation/DNA-Binding Gene Family in Lung Cancer Using Bioinformatics Methods. Biosci. Rep. 2020, 40, BSR20193075. [Google Scholar] [CrossRef]
- Wu, R.; Roy, A.M.; Tokumaru, Y.; Gandhi, S.; Asaoka, M.; Oshi, M.; Yan, L.; Ishikawa, T.; Takabe, K. NR2F1, a Tumor Dormancy Marker, Is Expressed Predominantly in Cancer-Associated Fibroblasts and Is Associated with Suppressed Breast Cancer Cell Proliferation. Cancers 2022, 14, 2962. [Google Scholar] [CrossRef]
- Rodriguez-Tirado, C.; Kale, N.; Carlini, M.J.; Shrivastava, N.; Rodrigues, A.A.; Khalil, B.D.; Bravo-Cordero, J.J.; Hong, Y.; Alexander, M.; Ji, J.; et al. NR2F1 Is a Barrier to Dissemination of Early-Stage Breast Cancer Cells. Cancer Res. 2022, 82, 2313–2326. [Google Scholar] [CrossRef]
- Wu, R.; Oshi, M.; Asaoka, M.; Tokumaru, Y.; Ishikawa, T.; Takabe, K. Association of NR2F1, a Tumor Dormancy Marker, with Cancer Cell Proliferation and Lymph Node Metastasis, and Expression in Cancer-Associated Fibroblasts in Breast Cancer. J. Clin. Oncol. 2022, 40, e12550. [Google Scholar] [CrossRef]
- Pickens, B.S.; Teets, B.W.; Soprano, K.J.; Soprano, D.R. Role of COUP-TFI during Retinoic Acid-Induced Differentiation of P19 Cells to Endodermal Cells. J. Cell. Physiol. 2013, 228, 791–800. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Cai, K.; Zheng, D.; Zhu, C.; Li, L.; Wang, F.; He, Z.; Yu, C.; Sun, C. Hypoxia-Induced Long Noncoding RNA NR2F1-AS1 Maintains Pancreatic Cancer Proliferation, Migration, and Invasion by Activating the NR2F1/AKT/MTOR Axis. Cell Death Dis. 2022, 13, 232. [Google Scholar] [CrossRef]
- Jin, L.; Chen, C.; Huang, L.; Sun, Q.; Bu, L. Long Noncoding RNA NR2F1-AS1 Stimulates the Tumorigenic Behavior of Non-Small Cell Lung Cancer Cells by Sponging MiR-363-3p to Increase SOX4. Open Med. 2022, 17, 87–95. [Google Scholar] [CrossRef]
- Satija, S.; Kaur, H.; Tambuwala, M.M.; Sharma, P.; Vyas, M.; Khurana, N.; Sharma, N.; Bakshi, H.A.; Charbe, N.B.; Zacconi, F.C.; et al. Hypoxia-Inducible Factor (HIF): Fuel for Cancer Progression. Curr. Mol. Pharmacol. 2021, 14, 321–332. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Bai, S.; Zhang, J.; Liu, Z.; Yang, J. NR2F1-AS1/MiR-140/HK2 Axis Regulates Hypoxia-Induced Glycolysis and Migration in Hepatocellular Carcinoma. Cancer Manag. Res. 2021, 13, 427–437. [Google Scholar] [CrossRef]
- Peng, J.; Zhang, L.; Drysdale, L.; Fong, G.-H. The Transcription Factor EPAS-1/Hypoxia-Inducible Factor 2α Plays an Important Role in Vascular Remodeling. Proc. Natl. Acad. Sci. USA 2000, 97, 8386–8391. [Google Scholar] [CrossRef]
- Kim, W.Y.; Perera, S.; Zhou, B.; Carretero, J.; Yeh, J.J.; Heathcote, S.A.; Jackson, A.L.; Nikolinakos, P.; Ospina, B.; Naumov, G.; et al. HIF2alpha Cooperates with RAS to Promote Lung Tumorigenesis in Mice. J. Clin. Investig. 2009, 119, 2160–2170. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Simon, M.C. Hypoxia-Inducible Factors: Central Regulators of the Tumor Phenotype. Curr. Opin. Genet. Dev. 2007, 17, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-H.; Bao, Y.; Wang, X.; Yan, F.; Guo, S.; Ma, Y.; Xu, D.; Jin, L.; Xu, J.; Wang, J. Hypoxic-Stabilized EPAS1 Proteins Transactivate DNMT1 and Cause Promoter Hypermethylation and Transcription Inhibition of EPAS1 in Non-Small Cell Lung Cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 6694–6705. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, S.; Tanimoto, K.; Nishio, Y.; Putra, A.C.; Fuchita, H.; Ohe, M.; Sutani, A.; Kuraki, T.; Hiyama, K.; Murakami, I.; et al. Association of EPAS1 Gene Rs4953354 Polymorphism with Susceptibility to Lung Adenocarcinoma in Female Japanese Non-Smokers. J. Thorac. Oncol. 2014, 9, 1709–1713. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, Y.; Zhang, R.; Su, L.; Gogarten, S.M.; Liu, G.; Brennan, P.; Field, J.K.; McKay, J.D.; Lissowska, J.; et al. Multi-Omics Analysis Reveals a HIF Network and Hub Gene EPAS1 Associated with Lung Adenocarcinoma. eBioMedicine 2018, 32, 93–101. [Google Scholar] [CrossRef]
- Triner, D.; Shah, Y.M. Hypoxia-Inducible Factors: A Central Link between Inflammation and Cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef]
- Shen, H.; Zhan, M.; Zhang, Y.; Huang, S.; Xu, S.; Huang, X.; He, M.; Yao, Y.; Man, M.; Wang, J. PLZF Inhibits Proliferation and Metastasis of Gallbladder Cancer by Regulating IFIT2. Cell Death Dis. 2018, 9, 71. [Google Scholar] [CrossRef]
- He, J.; Wu, M.; Xiong, L.; Gong, Y.; Yu, R.; Peng, W.; Li, L.; Li, L.; Tian, S.; Wang, Y.; et al. BTB/POZ Zinc Finger Protein ZBTB16 Inhibits Breast Cancer Proliferation and Metastasis through Upregulating ZBTB28 and Antagonizing BCL6/ZBTB27. Clin. Epigenet. 2020, 12, 82. [Google Scholar] [CrossRef]
- Xiao, G.-Q.; Li, F.; Findeis-Hosey, J.; Hyrien, O.; Unger, P.D.; Xiao, L.; Dunne, R.; Kim, E.S.; Yang, Q.; McMahon, L.; et al. Down-Regulation of Cytoplasmic PLZF Correlates with High Tumor Grade and Tumor Aggression in Non–Small Cell Lung Carcinoma. Hum. Pathol. 2015, 46, 1607–1615. [Google Scholar] [CrossRef]
- Liu, T.M.; Lee, E.H.; Lim, B.; Shyh-Chang, N. Concise Review: Balancing Stem Cell Self-Renewal and Differentiation with PLZF. Stem Cells Dayt. Ohio 2016, 34, 277–287. [Google Scholar] [CrossRef]
- Shiraishi, K.; Yamasaki, K.; Nanba, D.; Inoue, H.; Hanakawa, Y.; Shirakata, Y.; Hashimoto, K.; Higashiyama, S. Pre-B-Cell Leukemia Transcription Factor 1 Is a Major Target of Promyelocytic Leukemia Zinc-Finger-Mediated Melanoma Cell Growth Suppression. Oncogene 2007, 26, 339–348. [Google Scholar] [CrossRef]
- Shaknovich, R.; Yeyati, P.L.; Ivins, S.; Melnick, A.; Lempert, C.; Waxman, S.; Zelent, A.; Licht, J.D. The Promyelocytic Leukemia Zinc Finger Protein Affects Myeloid Cell Growth, Differentiation, and Apoptosis. Mol. Cell. Biol. 1998, 18, 5533–5545. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Guo, S.; Bao, Y.; Ma, Y.; Yan, F.; Xu, K.; Xu, Z.; Jin, L.; Lu, D.; et al. Hypermethylation Reduces Expression of Tumor-Suppressor PLZF and Regulates Proliferation and Apoptosis in Non-Small-Cell Lung Cancers. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 4194–4203. [Google Scholar] [CrossRef]
- La, H.M.; Hobbs, R.M. Mechanisms Regulating Mammalian Spermatogenesis and Fertility Recovery Following Germ Cell Depletion. Cell. Mol. Life Sci. 2019, 76, 4071–4102. [Google Scholar] [CrossRef]
- Nitkin, C.R.; Xia, S.; Menden, H.; Yu, W.; Xiong, M.; Heruth, D.P.; Ye, S.Q.; Sampath, V. FOSL1 Is a Novel Mediator of Endotoxin/Lipopolysaccharide-Induced Pulmonary Angiogenic Signaling. Sci. Rep. 2020, 10, 13143. [Google Scholar] [CrossRef]
- Šeda, O.; Šedová, L.; Včelák, J.; Vaňková, M.; Liška, F.; Bendlová, B. ZBTB16 and Metabolic Syndrome: A Network Perspective. Physiol. Res. 2017, 66, S357–S365. [Google Scholar] [CrossRef]
- Zhuang, S.-H.; Meng, C.-C.; Fu, J.-J.; Huang, J. Long Non-Coding RNA ELFN1-AS1-Mediated ZBTB16 Inhibition Augments the Progression of Gastric Cancer by Activating the PI3K/AKT Axis. Kaohsiung J. Med. Sci. 2022, 38, 621–632. [Google Scholar] [CrossRef]
- Hsieh, C.-L.; Botta, G.; Gao, S.; Li, T.; Van Allen, E.M.; Treacy, D.J.; Cai, C.; He, H.H.; Sweeney, C.J.; Brown, M.; et al. PLZF, a Tumor Suppressor Genetically Lost in Metastatic Castration Resistant Prostate Cancer, Is a Mediator of Resistance to Androgen Deprivation Therapy. Cancer Res. 2015, 75, 1944–1948. [Google Scholar] [CrossRef]
- Sadler, A.J.; Rossello, F.J.; Yu, L.; Deane, J.A.; Yuan, X.; Wang, D.; Irving, A.T.; Kaparakis-Liaskos, M.; Gantier, M.P.; Ying, H.; et al. BTB-ZF Transcriptional Regulator PLZF Modifies Chromatin to Restrain Inflammatory Signaling Programs. Proc. Natl. Acad. Sci. USA 2015, 112, 1535–1540. [Google Scholar] [CrossRef]
- Gu, L.; Casserly, D.; Brady, G.; Carpenter, S.; Bracken, A.P.; Fitzgerald, K.A.; Unterholzner, L.; Bowie, A.G. Myeloid Cell Nuclear Differentiation Antigen Controls the Pathogen-Stimulated Type I Interferon Cascade in Human Monocytes by Transcriptional Regulation of IRF7. Nat. Commun. 2022, 13, 14. [Google Scholar] [CrossRef]
- Zhongxiang Tang Abnormal Gene Expression Regulation Mechanism of Myeloid Cell Nuclear Differentiation Antigen in Lung Adenocarcinoma. Biology 2022, 11, 1047. [CrossRef] [PubMed]
- Sun, C.; Liu, C.; Dong, J.; Li, D.; Li, W. Effects of the Myeloid Cell Nuclear Differentiation Antigen on the Proliferation, Apoptosis and Migration of Osteosarcoma Cells. Oncol. Lett. 2014, 7, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Briggs, J.A.; Briggs, R.C. Human Hematopoietic Cell Specific Nuclear Protein MNDA Interacts with the Multifunctional Transcription Factor YY1 and Stimulates YY1 DNA Binding. J. Cell. Biochem. 1998, 70, 489–506. [Google Scholar] [CrossRef]
- Huang, T.; Wang, G.; Yang, L.; Peng, B.; Wen, Y.; Ding, G.; Wang, Z. Transcription Factor YY1 Modulates Lung Cancer Progression by Activating LncRNA-PVT1. DNA Cell Biol. 2017, 36, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Kaufhold, S.; Garbán, H.; Bonavida, B. Yin Yang 1 Is Associated with Cancer Stem Cell Transcription Factors (SOX2, OCT4, BMI1) and Clinical Implication. J. Exp. Clin. Cancer Res. CR 2016, 35, 84. [Google Scholar] [CrossRef]
- Yang, Y.; Tang, X.; Song, X.; Tang, L.; Cao, Y.; Liu, X.; Wang, X.; Li, Y.; Yu, M.; Wan, H.; et al. Evidence for an Oncogenic Role of HOXC6 in Human Non-Small Cell Lung Cancer. PeerJ 2019, 7, e6629. [Google Scholar] [CrossRef]
- Huang, H.; Huo, Z.; Jiao, J.; Ji, W.; Huang, J.; Bian, Z.; Xu, B.; Shao, J.; Sun, J. HOXC6 Impacts Epithelial-Mesenchymal Transition and the Immune Microenvironment through Gene Transcription in Gliomas. Cancer Cell Int. 2022, 22, 170. [Google Scholar] [CrossRef]
- Tang, L.; Cao, Y.; Song, X.; Wang, X.; Li, Y.; Yu, M.; Li, M.; Liu, X.; Huang, F.; Chen, F.; et al. HOXC6 Promotes Migration, Invasion and Proliferation of Esophageal Squamous Cell Carcinoma Cells via Modulating Expression of Genes Involved in Malignant Phenotypes. PeerJ 2019, 7, e6607. [Google Scholar] [CrossRef]
- Fujiki, K.; Duerr, E.; Kikuchi, H.; Ng, A.; Xavier, R.J.; Mizukami, Y.; Imamura, T.; Kulke, M.H.; Chung, D.C. Hoxc6 Is Overexpressed in Gastrointestinal Carcinoids and Interacts With JunD to Regulate Tumor Growth. Gastroenterology 2008, 135, 907–916.e2. [Google Scholar] [CrossRef]
- Hamid, A.R.A.H.; Hoogland, A.M.; Smit, F.; Jannink, S.; van Rijt-van de Westerlo, C.; Jansen, C.F.J.; van Leenders, G.J.L.H.; Verhaegh, G.W.; Schalken, J.A. The Role of HOXC6 in Prostate Cancer Development. Prostate 2015, 75, 1868–1876. [Google Scholar] [CrossRef]
- Zhang, F.; Ren, C.-C.; Liu, L.; Chen, Y.-N.; Yang, L.; Zhang, X.-A. HOXC6 Gene Silencing Inhibits Epithelial-Mesenchymal Transition and Cell Viability through the TGF-β/Smad Signaling Pathway in Cervical Carcinoma Cells. Cancer Cell Int. 2018, 18, 204. [Google Scholar] [CrossRef]
- Chen, J.; Liu, A.; Lin, Z.; Wang, B.; Chai, X.; Chen, S.; Lu, W.; Zheng, M.; Cao, T.; Zhong, M.; et al. Downregulation of the Circadian Rhythm Regulator HLF Promotes Multiple-Organ Distant Metastases in Non-Small Cell Lung Cancer through PPAR/NF-ΚB Signaling. Cancer Lett. 2020, 482, 56–71. [Google Scholar] [CrossRef]
- Waters, K.M.; Sontag, R.L.; Weber, T.J. Hepatic Leukemia Factor Promotes Resistance to Cell Death: Implications for Therapeutics and Chronotherapy. Toxicol. Appl. Pharmacol. 2013, 268, 141–148. [Google Scholar] [CrossRef]
- Taniwaki, M.; Daigo, Y.; Ishikawa, N.; Takano, A.; Tsunoda, T.; Yasui, W.; Inai, K.; Kohno, N.; Nakamura, Y. Gene Expression Profiles of Small-Cell Lung Cancers: Molecular Signatures of Lung Cancer. Int. J. Oncol. 2006, 29, 567–575. [Google Scholar] [CrossRef]
- Fedorova, O.; Petukhov, A.; Daks, A.; Shuvalov, O.; Leonova, T.; Vasileva, E.; Aksenov, N.; Melino, G.; Barlev, N.A. Orphan Receptor NR4A3 Is a Novel Target of P53 That Contributes to Apoptosis. Oncogene 2019, 38, 2108–2122. [Google Scholar] [CrossRef]
- Son, B.; Jeon, J.; Lee, S.; Kim, H.; Kang, H.; Youn, H.; Jo, S.; Youn, B. Radiotherapy in Combination with Hyperthermia Suppresses Lung Cancer Progression via Increased NR4A3 and KLF11 Expression. Int. J. Radiat. Biol. 2019, 95, 1696–1707. [Google Scholar] [CrossRef]
- Haller, F.; Bieg, M.; Will, R.; Körner, C.; Weichenhan, D.; Bott, A.; Ishaque, N.; Lutsik, P.; Moskalev, E.A.; Mueller, S.K.; et al. Enhancer Hijacking Activates Oncogenic Transcription Factor NR4A3 in Acinic Cell Carcinomas of the Salivary Glands. Nat. Commun. 2019, 10, 368. [Google Scholar] [CrossRef]
- Crean, D.; Murphy, E.P. Targeting NR4A Nuclear Receptors to Control Stromal Cell Inflammation, Metabolism, Angiogenesis, and Tumorigenesis. Front. Cell Dev. Biol. 2021, 9, 589770. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Sousa, L.; Pankonien, I.; Clarke, L.A.; Silva, I.; Kunzelmann, K.; Amaral, M.D. KLF4 Acts as a Wt-CFTR Suppressor through an AKT-Mediated Pathway. Cells 2020, 9, 1607. [Google Scholar] [CrossRef]
- Yu, T.; Chen, X.; Zhang, W.; Liu, J.; Avdiushko, R.; Napier, D.L.; Liu, A.X.; Neltner, J.M.; Wang, C.; Cohen, D.; et al. KLF4 Regulates Adult Lung Tumor-Initiating Cells and Represses K-Ras-Mediated Lung Cancer. Cell Death Differ. 2016, 23, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Hofstetter, W.L.; Li, H.; Zhou, Y.; He, Y.; Pataer, A.; Wang, L.; Xie, K.; Swisher, S.G.; Fang, B. Putative Tumor-Suppressive Function of Krüppel-Like Factor 4 in Primary Lung Carcinoma. Clin. Cancer Res. 2009, 15, 5688–5695. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lin, L.; Wang, X.; Li, Y.; Liu, Z.; Ye, W.; Huang, W.; Lin, G.; Liu, H.; Zhang, J.; et al. Overexpression of Krüppel-Like Factor 4 Suppresses Migration and Invasion of Non-Small Cell Lung Cancer Through c-Jun-NH2-Terminal Kinase/Epithelial-Mesenchymal Transition Signaling Pathway. Front. Pharmacol. 2020, 10, 1512. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huang, L.; Gu, J.; Wu, J.; Ou, W.; Feng, J.; Liu, B.; Cui, X.; Zhou, Y. Restoration of KLF4 Inhibits Invasion and Metastases of Lung Adenocarcinoma through Suppressing MMP2. J. Cancer 2017, 8, 3480–3489. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Mostow, K.; Jackett, K.; Kelty, E.; Dakpa, T.; Ryan, C.; Hagos, E. KLF4 Regulates Metabolic Homeostasis in Response to Stress. Cells 2021, 10, 830. [Google Scholar] [CrossRef]
- Xiong, M.; Wang, F.; Zhau, H.E.; Huang, X.; Chung, L.; Zhang, J.; Lu, Y. Abstract 3295: GPRASP1:A Novel Potential Biomarker for Neuroendocrine Carcinoma. Cancer Res. 2019, 79, 3295. [Google Scholar] [CrossRef]
- Li, W.; Huang, K.; Guo, H.; Cui, G. Meis1 Regulates Proliferation of Non-Small-Cell Lung Cancer Cells. J. Thorac. Dis. 2014, 6, 850–855. [Google Scholar] [CrossRef]
- Zhu, J.; Cui, L.; Xu, A.; Yin, X.; Li, F.; Gao, J. MEIS1 Inhibits Clear Cell Renal Cell Carcinoma Cells Proliferation and in Vitro Invasion or Migration. BMC Cancer 2017, 17, 176. [Google Scholar] [CrossRef]
- Li, Y.; Gan, Y.; Liu, J.; Li, J.; Zhou, Z.; Tian, R.; Sun, R.; Liu, J.; Xiao, Q.; Li, Y.; et al. Downregulation of MEIS1 Mediated by ELFN1-AS1/EZH2/DNMT3a Axis Promotes Tumorigenesis and Oxaliplatin Resistance in Colorectal Cancer. Signal Transduct. Target. Ther. 2022, 7, 87. [Google Scholar] [CrossRef]
- Rauch, T.A.; Wang, Z.; Wu, X.; Kernstine, K.H.; Riggs, A.D.; Pfeifer, G.P. DNA Methylation Biomarkers for Lung Cancer. Tumor Biol. 2012, 33, 287–296. [Google Scholar] [CrossRef]
- Hisa, T.; Spence, S.E.; Rachel, R.A.; Fujita, M.; Nakamura, T.; Ward, J.M.; Devor-Henneman, D.E.; Saiki, Y.; Kutsuna, H.; Tessarollo, L.; et al. Hematopoietic, Angiogenic and Eye Defects in Meis1 Mutant Animals. EMBO J. 2004, 23, 450–459. [Google Scholar] [CrossRef]
- Girgin, B.; Karadağ-Alpaslan, M.; Kocabaş, F. Oncogenic and Tumor Suppressor Function of MEIS and Associated Factors. Turk. J. Biol. 2020, 44, 328–355. [Google Scholar] [CrossRef]
- Zargari, S.; Negahban Khameneh, S.; Rad, A.; Forghanifard, M.M. MEIS1 Promotes Expression of Stem Cell Markers in Esophageal Squamous Cell Carcinoma. BMC Cancer 2020, 20, 789. [Google Scholar] [CrossRef]
- Jiang, M.; Xu, S.; Bai, M.; Zhang, A. The Emerging Role of MEIS1 in Cell Proliferation and Differentiation. Am. J. Physiol.-Cell Physiol. 2021, 320, C264–C269. [Google Scholar] [CrossRef]
- Kocabas, F.; Zheng, J.; Thet, S.; Copeland, N.G.; Jenkins, N.A.; DeBerardinis, R.J.; Zhang, C.; Sadek, H.A. Meis1 Regulates the Metabolic Phenotype and Oxidant Defense of Hematopoietic Stem Cells. Blood 2012, 120, 4963–4972. [Google Scholar] [CrossRef]
- Meng, L.; Tian, Z.; Wang, J.; Liu, X.; Zhang, W.; Hu, M.; Wang, M.; Zhang, Y. Effect of Myeloid Ecotropic Viral Integration Site (MEIS) Family Genes on Tumor Microenvironment Remodeling and Its Potential Therapeutic Effect. Transl. Androl. Urol. 2021, 10, 594–608. [Google Scholar] [CrossRef]
- Li, X.; Xie, M.; Yin, S.; Xiong, Z.; Mao, C.; Zhang, F.; Chen, H.; Jin, L.; Lan, P.; Lian, L. Identification and Validation of a Six Immune-Related Genes Signature for Predicting Prognosis in Patients With Stage II Colorectal Cancer. Front. Genet. 2021, 12, 717. [Google Scholar] [CrossRef]
- VanOpstall, C.; Perike, S.; Brechka, H.; Gillard, M.; Lamperis, S.; Zhu, B.; Brown, R.; Bhanvadia, R.; Vander Griend, D.J. MEIS-Mediated Suppression of Human Prostate Cancer Growth and Metastasis through HOXB13-Dependent Regulation of Proteoglycans. eLife 2020, 9, e53600. [Google Scholar] [CrossRef]
- Do, H.; Kim, D.; Kang, J.; Son, B.; Seo, D.; Youn, H.; Youn, B.; Kim, W. TFAP2C Increases Cell Proliferation by Downregulating GADD45B and PMAIP1 in Non-Small Cell Lung Cancer Cells. Biol. Res. 2019, 52, 35. [Google Scholar] [CrossRef]
- Kang, J.; Kim, W.; Lee, S.; Kwon, D.; Chun, J.; Son, B.; Kim, E.; Lee, J.-M.; Youn, H.; Youn, B. TFAP2C Promotes Lung Tumorigenesis and Aggressiveness through MiR-183- and MiR-33a-Mediated Cell Cycle Regulation. Oncogene 2017, 36, 1585–1596. [Google Scholar] [CrossRef]
- Kim, W.; Kim, E.; Lee, S.; Kim, D.; Chun, J.; Park, K.H.; Youn, H.; Youn, B. TFAP2C-Mediated Upregulation of TGFBR1 Promotes Lung Tumorigenesis and Epithelial-Mesenchymal Transition. Exp. Mol. Med. 2016, 48, e273. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.E.; Toniolo, P.A.; Frank, D.A. Targeting Transcription Factors: Promising New Strategies for Cancer Therapy. Curr. Opin. Oncol. 2013, 25, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shen, J.-X.; Wen, X.-F.; Guo, Y.-X.; Zhang, G.-J. Targeting Notch Degradation System Provides Promise for Breast Cancer Therapeutics. Crit. Rev. Oncol. Hematol. 2016, 104, 21–29. [Google Scholar] [CrossRef]
- Liao, G.-B.; Li, X.-Z.; Zeng, S.; Liu, C.; Yang, S.-M.; Yang, L.; Hu, C.-J.; Bai, J.-Y. Regulation of the Master Regulator FOXM1 in Cancer. Cell Commun. Signal. CCS 2018, 16, 57. [Google Scholar] [CrossRef]
- Petrovic, V.; Costa, R.H.; Lau, L.F.; Raychaudhuri, P.; Tyner, A.L. Negative Regulation of the Oncogenic Transcription Factor FoxM1 by Thiazolidinediones and Mithramycin. Cancer Biol. Ther. 2010, 9, 1008–1016. [Google Scholar] [CrossRef]
- Dong, G.-Z.; Jeong, J.H.; Lee, Y.-I.; Lee, S.Y.; Zhao, H.-Y.; Jeon, R.; Lee, H.J.; Ryu, J.-H. Diarylheptanoids Suppress Proliferation of Pancreatic Cancer PANC-1 Cells through Modulating Shh-Gli-FoxM1 Pathway. Arch. Pharm. Res. 2017, 40, 509–517. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Code | Samples |
|---|---|
| GSE19804 | Normal (60) vs. Cancer NSCLC (60) |
| GSE10072 | Normal (49) vs. Cancer-Lung adenocarcinoma (58) |
| GSE3268 | Normal (5) vs. Cancer-Squamous lung cancer cells (5) |
| GSE108055 | Normal (9) vs. Cancer-Small-cell lung cancer (54) |
| E-MTAB-5231 | Normal (18) vs. Cancer-NSCLC (22) |
| E-MTAB-3950 | Normal (30) vs. Cancer-Early Squamous Carcinoma (30) |
| GSE52248 | Normal (6) vs. Lung adenocarcinoma (12) |
| GSE70089 | Normal (3) vs. Lung carcinoma (3) |
| GSE81089 | Normal (19) vs. Cancer NSCLC (199) |
| GSE84776 | Normal (9) vs. Squamous lung cancer (9) |
| GSE113439 | Normal (11) vs. Pulmonary arterial hypertension (PAH) (15) |
| Transcription Factor | Lung Cancer | Fold Change | Pulmonary Arterial Hypertension | Total | |
|---|---|---|---|---|---|
| 1 | SOX4 | 10 | 2.04040127 | PAH | 11 |
| 2 | SOX17 | 10 | 0.50804133 | - | 10 |
| 3 | BZW2 | 9 | 1.86844644 | PAH | 10 |
| 4 | FOXM1 | 9 | 2.92595918 | PAH | 10 |
| 5 | ZBTB16 | 9 | 0.36129524 | PAH | 10 |
| 6 | TAL1 | 9 | 0.71569014 | PAH | 10 |
| 7 | KLF4 | 9 | 0.34963135 | - | 9 |
| 8 | EPAS1 | 8 | 0.29798652 | PAH | 9 |
| 9 | HOXC6 | 8 | 1.64583097 | PAH | 9 |
| 10 | ID4 | 8 | 0.58410489 | PAH | 9 |
| 11 | KLF2 | 8 | 0.35253951 | PAH | 9 |
| 12 | MEIS1 | 8 | 0.53393586 | PAH | 9 |
| 13 | NR2F1 | 8 | 0.65817465 | PAH | 9 |
| 14 | TBX4 | 8 | 0.66704351 | PAH | 9 |
| 15 | TCF21 | 8 | 0.25969303 | PAH | 9 |
| 16 | TFAP2C | 8 | 1.86076131 | PAH | 9 |
| 17 | LMO2 | 8 | 0.43204536 | PAH | 9 |
| 18 | MNDA | 8 | 0.43069329 | PAH | 9 |
| 19 | FOXF1 | 8 | 0.25699375 | PAH | 9 |
| 20 | HLF | 8 | 0.51723087 | PAH | 9 |
| 21 | RFX2 | 8 | 0.80986159 | PAH | 9 |
| 22 | DLX5 | 8 | 1.76223624 | − | 8 |
| 23 | MYBL2 | 8 | 1.79966863 | − | 8 |
| 24 | NR4A3 | 8 | 0.39647141 | − | 8 |
| 25 | PKNOX2 | 8 | 0.64308105 | − | 8 |
| 26 | GPRASP1 | 8 | 0.77475237 | − | 8 |
| CCPs | CCPs | FOXM1 | MYBL2 | |||||
|---|---|---|---|---|---|---|---|---|
| Nodes | Edges | Targets | BM | NES | Targets | BM | NES | |
| ALL LC—PAH | 9 | 11 | 9 | 6 | 10.190 | 8 | 1 | 5.598 |
| MA LC—PAH | 39 | 91 | 30 | 6 | 10.318 | 19 | 1 | 6.552 |
| RNAS LC—PAH | 32 | 36 | 24 | 6 | 11.294 | 13 | 1 | 4.127 |
| ALL LC | 29 | 39 | 21 | 6 | 10.427 | 19 | 1 | 6.352 |
| MA LC | 94 | 555 | 47 | 6 | 9.595 | 40 | 1 | 5.509 |
| RNAS LC | 118 | 370 | 53 | 6 | 11.199 | 26 | 1 | 4.395 |
| Alignment | Number of Alignments | Percentage | Median |
|---|---|---|---|
| ASPM—ASPM | 6 | 60% | 0.512949636 |
| CENPF—CENPF | 6 | 60% | 0.525433165 |
| PRC1—PRC1 | 6 | 60% | 0.446116868 |
| TPX2—TPX2 | 6 | 60% | 0.570271269 |
| TOP2A—TOP2A | 5 | 50% | 0.551322899 |
| KIF20A—KIF20A | 4 | 40% | 0.444933142 |
| KIF2C—KIF2C | 4 | 40% | 0.469979467 |
| NUSAP1—NUSAP1 | 4 | 40% | 0.484566649 |
| NSCLC | SCLC | |||||||
|---|---|---|---|---|---|---|---|---|
| LC-PAH | LC | LC-PAH | LC | |||||
| TF | Targets | TF | Targets | TF | Targets | TF | Targets | |
| 1 | TCF21 | 176 | TCF21 | 76 | MNDA | 127 | DLX5 | 67 |
| 2 | ZBTB16 | 175 | ZBTB16 | 74 | ZBTB16 | 123 | MNDA | 66 |
| 3 | FOXF1 | 173 | FOXF1 | 73 | KLF2 | 120 | KLF2 | 64 |
| 4 | FOXM1 | 172 | NR4A3 | 73 | EPAS1 | 113 | EPAS1 | 62 |
| 5 | EPAS1 | 164 | KLF2 | 73 | SOX4 | 90 | ZBTB16 | 55 |
| 6 | KLF2 | 164 | KLF4 | 73 | NR2F1 | 90 | NR2F1 | 50 |
| 7 | ID4 | 151 | EPAS1 | 71 | FOXF1 | 87 | FOXF1 | 39 |
| 8 | MNDA | 143 | ID4 | 67 | HLF | 63 | SOX4 | 36 |
| 9 | HLF | 82 | SOX17 | 67 | HOXC6 | 57 | TCF21 | 35 |
| 10 | LMO2 | 52 | GPRASP1 | 51 | FOXM1 | 47 | HLF | 26 |
| 11 | HOXC6 | 47 | MNDA | 50 | TCF21 | 45 | HOXC6 | 20 |
| 12 | SOX4 | 24 | HLF | 31 | LMO2 | 37 | FOXM1 | 18 |
| 13 | TFAP2C | 12 | FOXM1 | 23 | ID4 | 34 | LMO2 | 10 |
| 14 | BZW2 | 10 | LMO2 | 21 | RFX2 | 8 | RFX2 | 9 |
| 15 | MEIS1 | 10 | HOXC6 | 12 | ID4 | 8 | ||
| 16 | TAL1 | 3 | MEIS1 | 10 | ||||
| 17 | SOX4 | 6 | ||||||
| 18 | TFAP2C | 3 | ||||||
| 19 | TAL1 | 1 | ||||||
| 20 | PKNOX2 | 1 | ||||||
| 21 | RFX2 | 1 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otálora-Otálora, B.A.; López-Kleine, L.; Rojas, A. Lung Cancer Gene Regulatory Network of Transcription Factors Related to the Hallmarks of Cancer. Curr. Issues Mol. Biol. 2023, 45, 434-464. https://doi.org/10.3390/cimb45010029
Otálora-Otálora BA, López-Kleine L, Rojas A. Lung Cancer Gene Regulatory Network of Transcription Factors Related to the Hallmarks of Cancer. Current Issues in Molecular Biology. 2023; 45(1):434-464. https://doi.org/10.3390/cimb45010029
Chicago/Turabian StyleOtálora-Otálora, Beatriz Andrea, Liliana López-Kleine, and Adriana Rojas. 2023. "Lung Cancer Gene Regulatory Network of Transcription Factors Related to the Hallmarks of Cancer" Current Issues in Molecular Biology 45, no. 1: 434-464. https://doi.org/10.3390/cimb45010029
APA StyleOtálora-Otálora, B. A., López-Kleine, L., & Rojas, A. (2023). Lung Cancer Gene Regulatory Network of Transcription Factors Related to the Hallmarks of Cancer. Current Issues in Molecular Biology, 45(1), 434-464. https://doi.org/10.3390/cimb45010029