
A Link between Mitochondrial Dysregulation and Idiopathic Autism Spectrum Disorder (ASD): Alterations in Mitochondrial Respiratory Capacity and Membrane Potential



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. High–Resolution Respirometry
2.4. Cytochrome c Oxidase (CIV) Activity Assay
2.5. Respiration
2.6. Mitochondrial Membrane Potential
2.7. Data Analysis
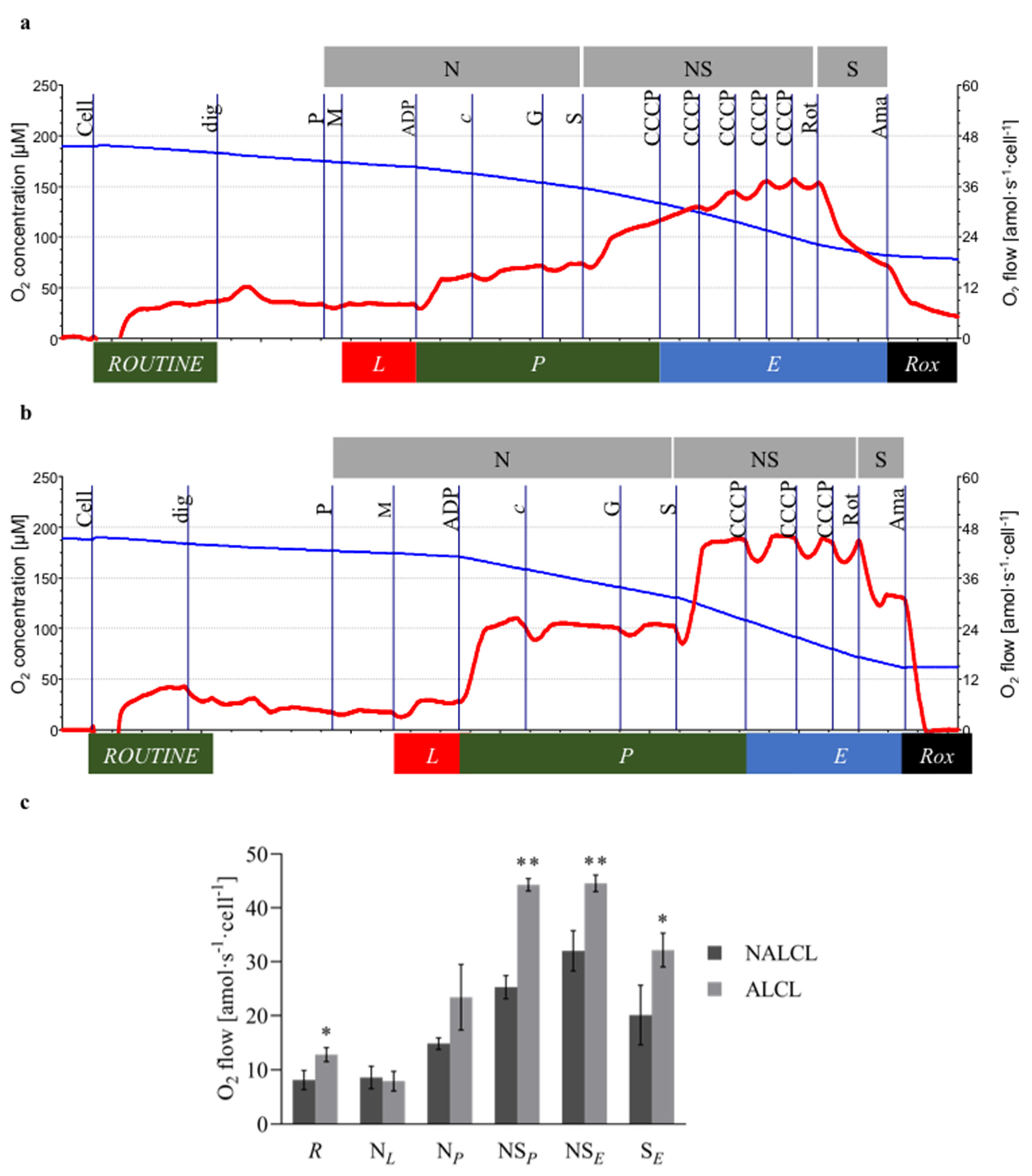
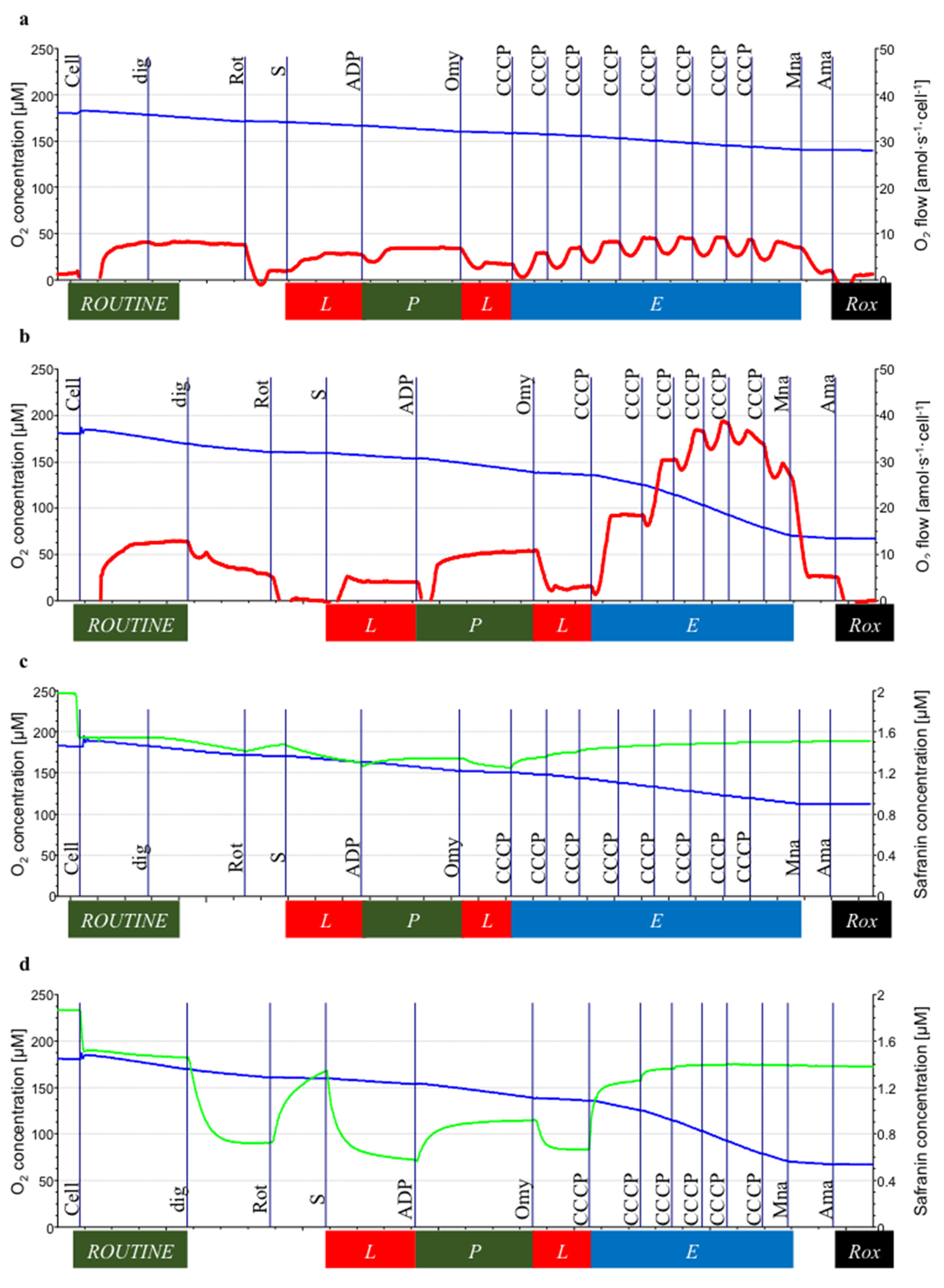
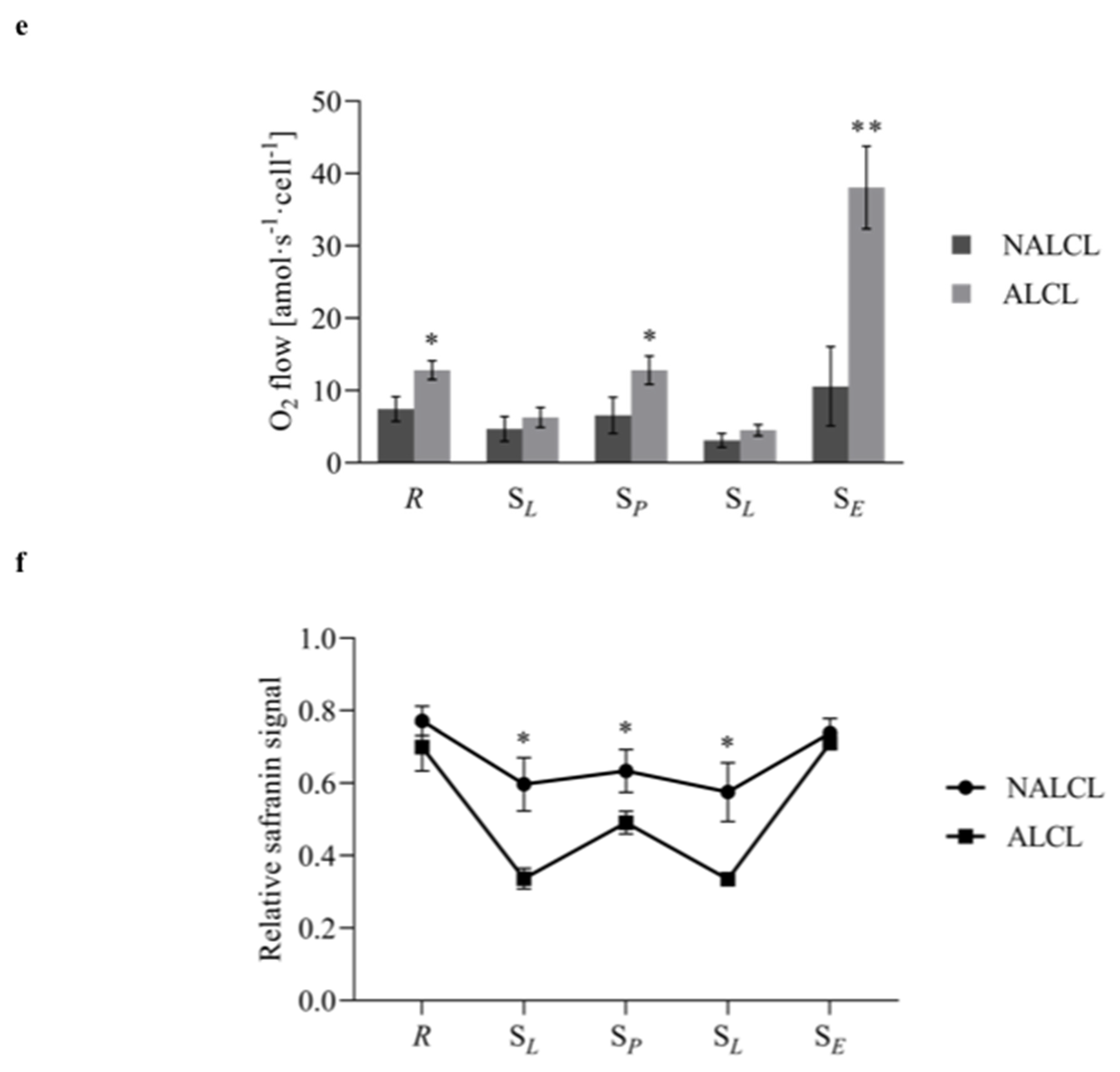
3. Results
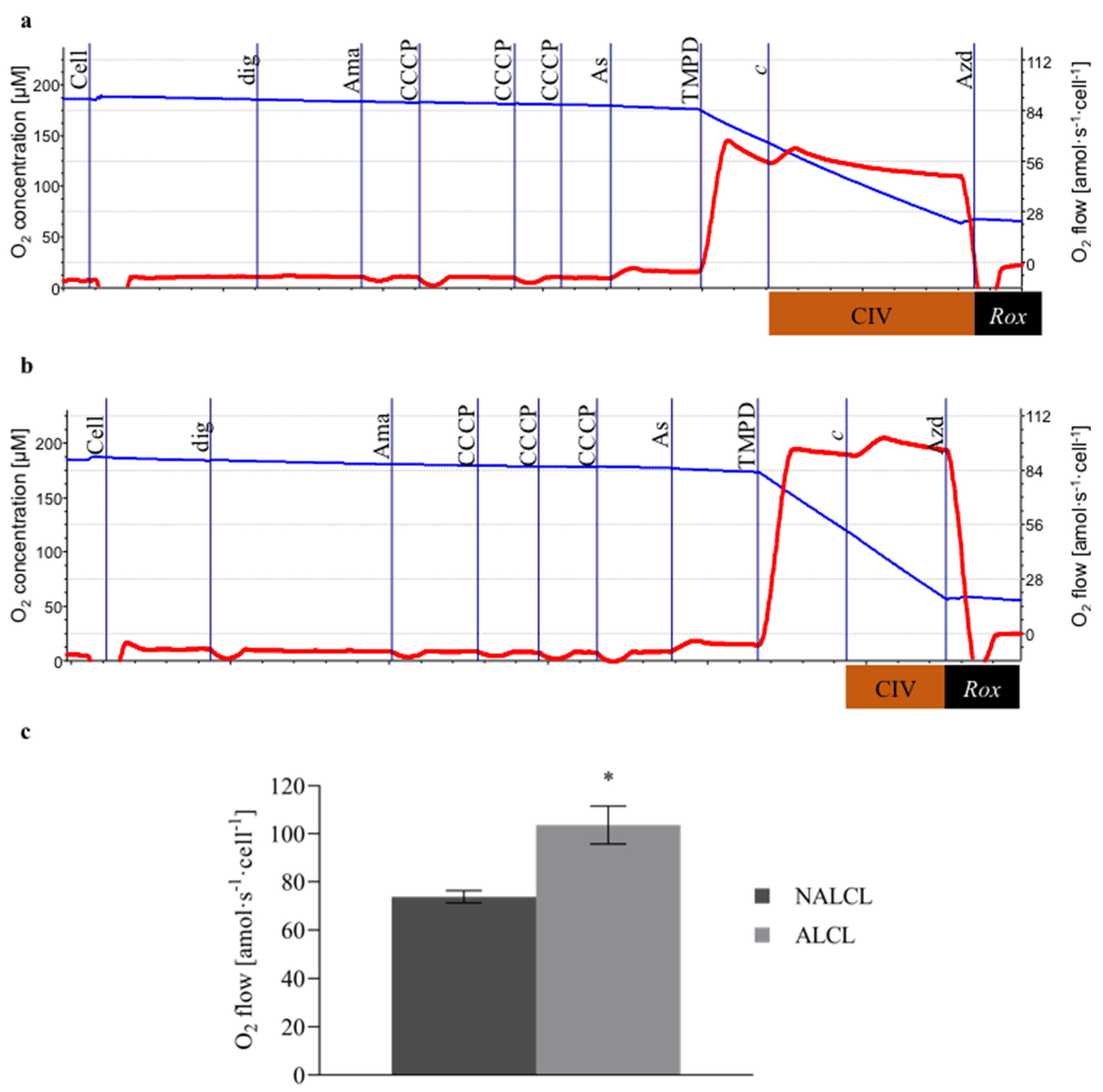
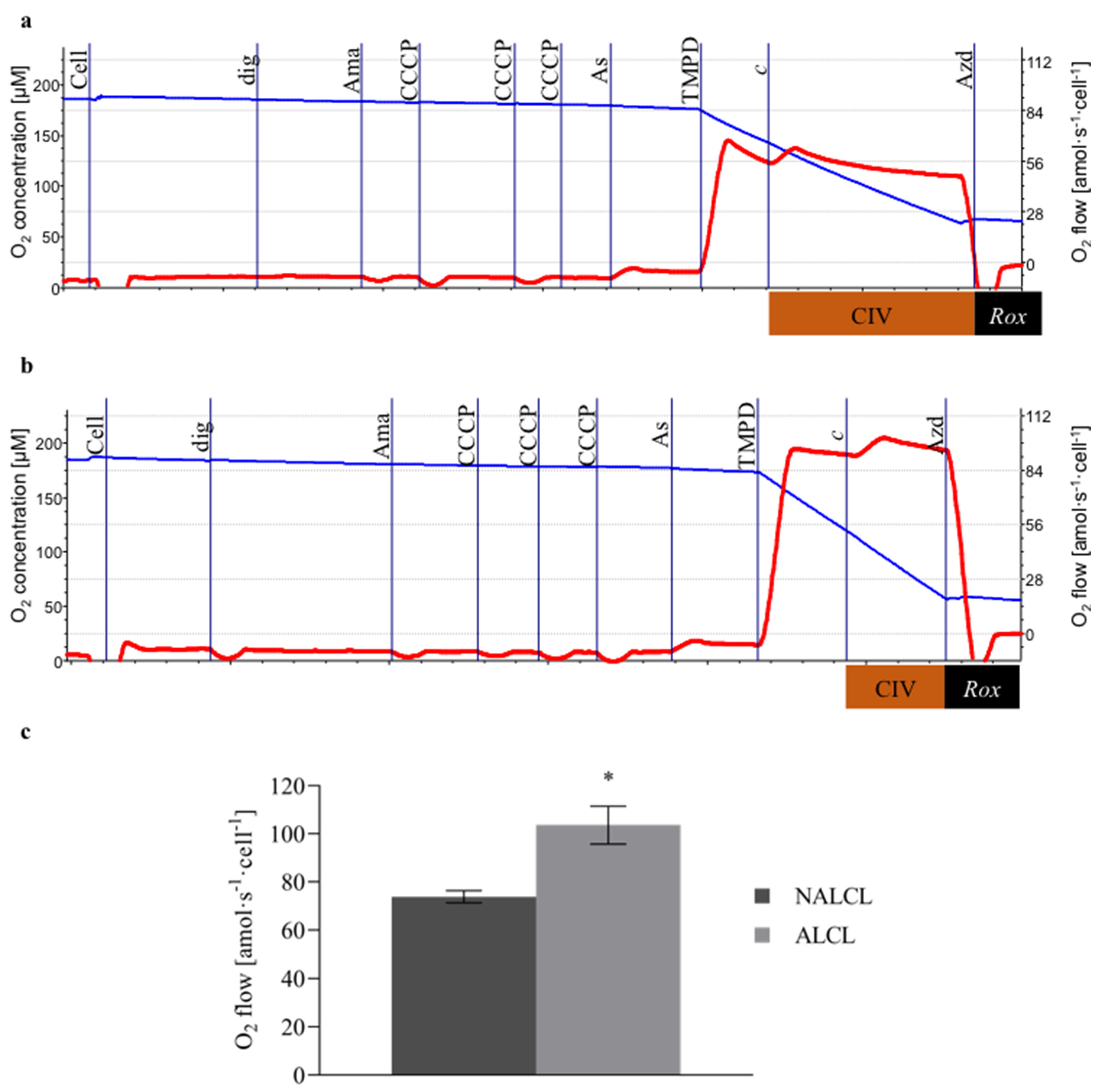
3.1. Complex IV Activity
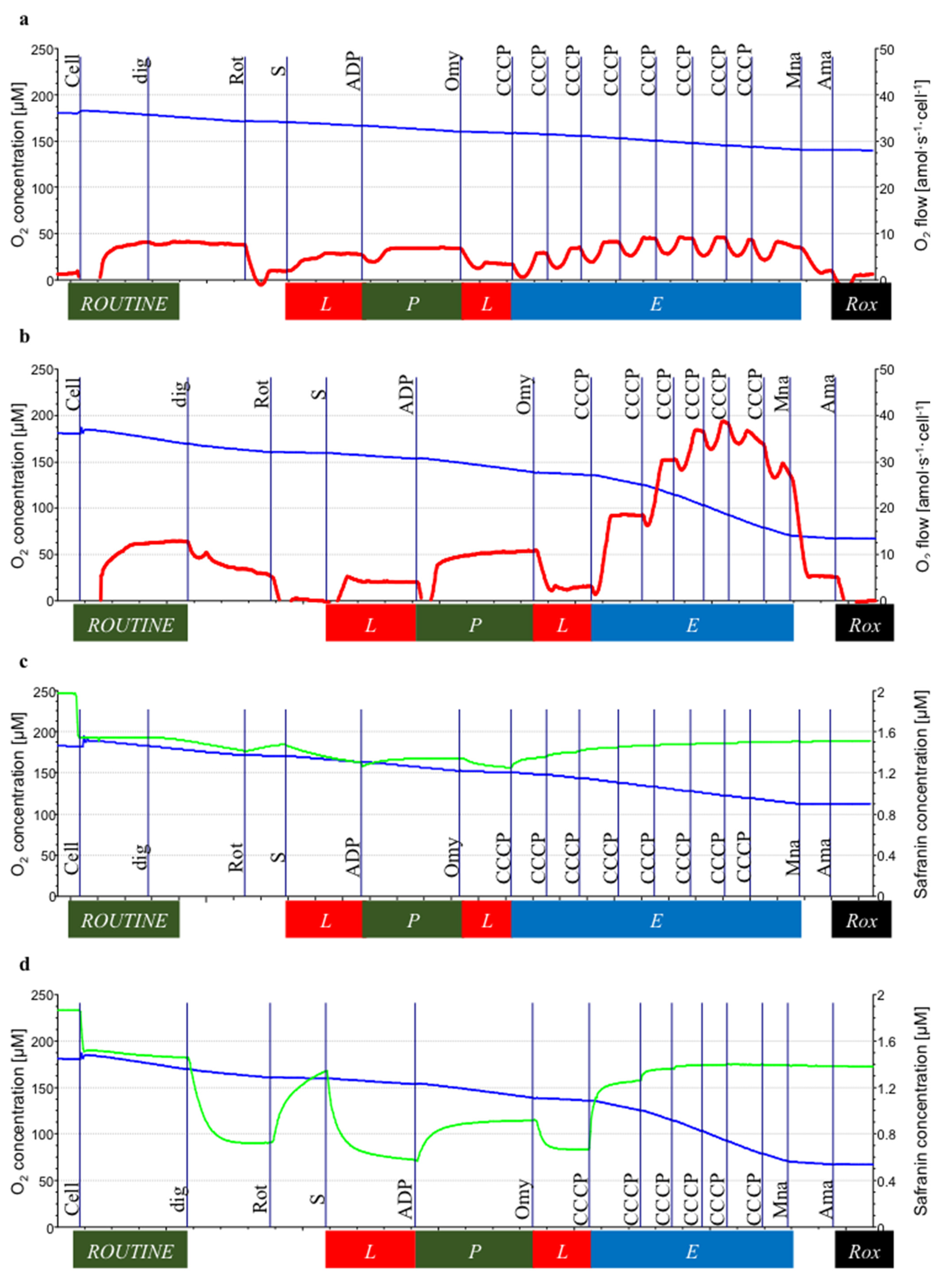
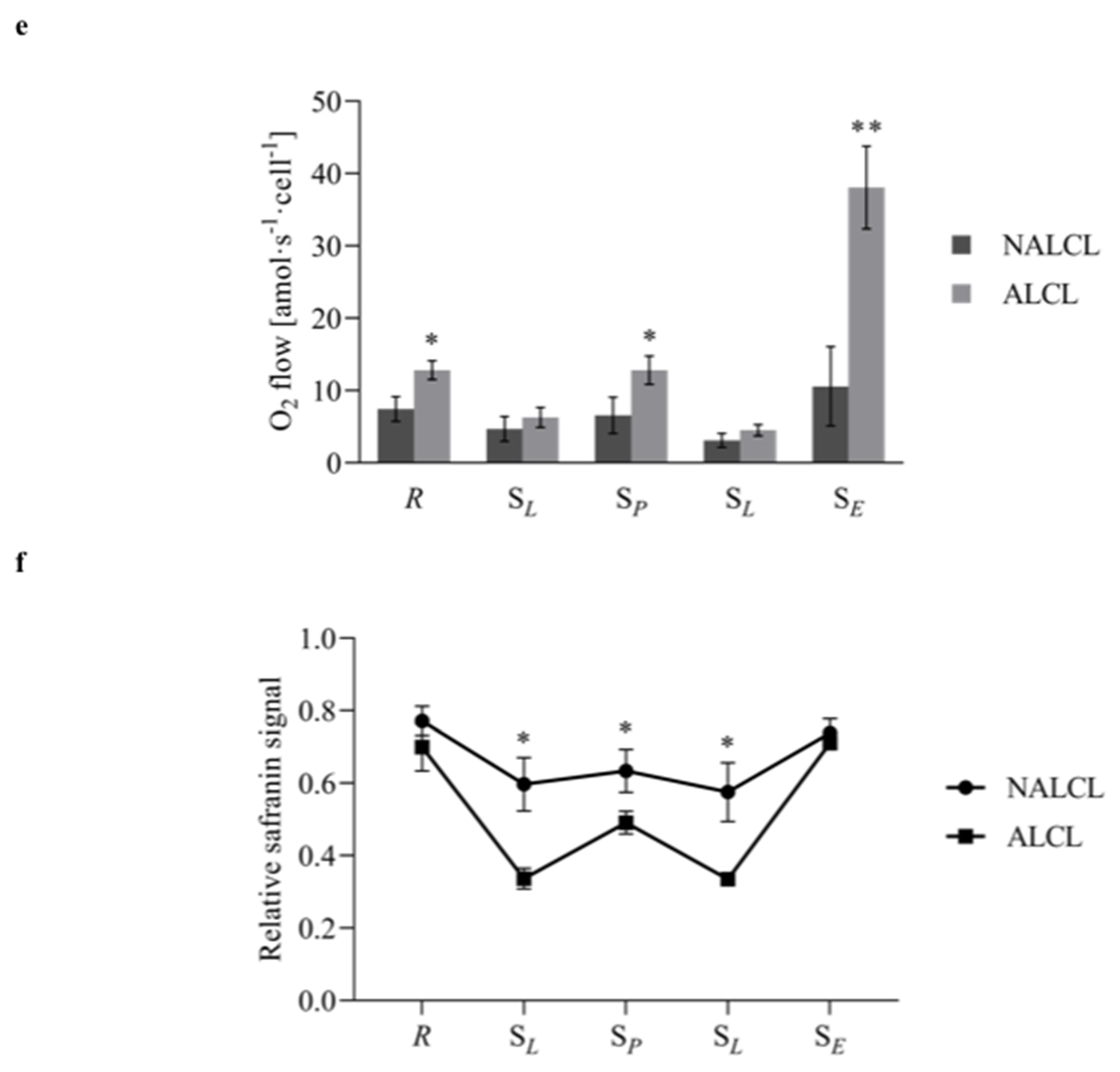
3.2. Mitochondrial Respiration
3.3. Mitochondrial Membrane Potential
4. Discussion
5. Conclusions
6. Limitations & Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elsabbagh, M.; Divan, G.; Koh, Y.J.; Kim, Y.S.; Kauchali, S.; Marcin, C.; Montiel-Nava, C.; Patel, V.; Paula, C.S.; Wang, C.; et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef] [Green Version]
- WHO. Autism Spectrum Disorders. Available online: https://www.who.int/news-room/fact-sheets/detail/autism-spectrum-disorders (accessed on 28 May 2019).
- Lima Antão, J.Y.F.D.; Oliveira, A.S.B.; Almeida Barbosa, R.T.D.; Crocetta, T.B.; Guarnieri, R.; Arab, C.; Massetti, T.; Antunes, T.P.C.; Silva, A.P.D.; Bezerra, Ĺ.M.P.; et al. Instruments for augmentative and alternative communication for children with autism spectrum disorder: A systematic review. Clinics 2018, 73, e497. [Google Scholar]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Manivasagam, T.; Arunadevi, S.; Essa, M.M.; SaravanaBabu, C.; Borah, A.; Thenmozhi, A.J.; Qoronfleh, M.W. Role of oxidative stress and antioxidants in autism. Adv. Neurobiol. 2020, 24, 193–206. [Google Scholar]
- Pahrudin Arrozi, A.; Wan Ngah, W.Z.; Mohd Yusof, Y.A.; Ahmad Damanhuri, M.H.; Makpol, S. Antioxidant modulation in restoring mitochondrial function in neurodegeneration. Int. J. Neurosci. 2017, 127, 218–235. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Scorza, F.A.; Menezes-Rodrigues, F.S.; Olszewer, E.; Errante, P.R.; Tavares, J.G.P.; Scorza, C.A.; Ferraz, H.B.; Finsterer, J.; Caricati-Neto, A. The mitochondrial calcium uniporter: A new therapeutic target for parkinson’s disease-related cardiac dysfunctions? Clinics 2020, 75, e1299. [Google Scholar] [CrossRef]
- Makpol, S.; Abdul Rahim, N.; Hui, C.K.; Ngah, W.Z. Inhibition of mitochondrial cytochrome c release and suppression of caspases by gamma-tocotrienol prevent apoptosis and delay aging in stress-induced premature senescence of skin fibroblasts. Oxid. Med. Cell. Longev. 2012, 2012, 785743. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, L.; Persico, A.M. Mitochondrial dysfunction in autism spectrum disorders: Cause or effect? Biochim. Biophys. Acta 2010, 1797, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Goldani, A.A.; Downs, S.R.; Widjaja, F.; Lawton, B.; Hendren, R.L. Biomarkers in autism. Front. Psychiatry 2014, 5, 100. [Google Scholar] [CrossRef]
- Fernandez-Checa, J.C.; Kaplowitz, N.; Garcia-Ruiz, C.; Colell, A.; Miranda, M.; Mari, M.; Ardite, E.; Morales, A. Gsh transport in mitochondria: Defense against tnf-induced oxidative stress and alcohol-induced defect. Am. J. Physiol. 1997, 273, G7–G17. [Google Scholar] [CrossRef]
- Gargus, J.J.; Imtiaz, F. Mitochondrial energy-deficient endophenotype in autism. Am. J. Biochem. Biotechnol. 2008, 4, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Obata, K. Synaptic inhibition and γ-aminobutyric acid in the mammalian central nervous system. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2013, 89, 139–156. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.P.; Hooker, B.S.; Herbert, M.R. Bridging from cells to cognition in autism pathophysiology: Biological pathways to defective brain function and plasticity. Am. J. Biochem. Biotechnol. 2008, 4, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the mitochondrial medicine society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [Green Version]
- Ghanizadeh, A.; Berk, M.; Farrashbandi, H.; Alavi Shoushtari, A.; Villagonzalo, K.A. Targeting the mitochondrial electron transport chain in autism, a systematic review and synthesis of a novel therapeutic approach. Mitochondrion 2013, 13, 515–519. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Djordjevic, J.; Albensi, B.C.; Fernyhough, P. Simultaneous evaluation of substrate-dependent oxygen consumption rates and mitochondrial membrane potential by tmrm and safranin in cortical mitochondria. Biosci. Rep. 2015, 36, e00286. [Google Scholar] [CrossRef]
- Krumschnabel, G.; Eigentler, A.; Fasching, M.; Gnaiger, E. Use of safranin for the assessment of mitochondrial membrane potential by high-resolution respirometry and fluorometry. Methods Enzymol. 2014, 542, 163–181. [Google Scholar]
- Doerrier, C.; Garcia-Souza, L.F.; Krumschnabel, G.; Wohlfarter, Y.; Mészáros, A.T.; Gnaiger, E. High-resolution fluorespirometry and oxphos protocols for human cells, permeabilized fibers from small biopsies of muscle, and isolated mitochondria. Methods Mol. Biol. 2018, 1782, 31–70. [Google Scholar]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control. An Introduction to Oxphos Analysis; Oroboros MiPNet Publications: Innsbruck, Austria, 2014; p. 88. [Google Scholar]
- Gnaiger, E. Mitochondrial respiratory states and rates. MitoFit Prep. Arch. 2019, 1, 1–40. [Google Scholar]
- Lemieux, H.; Blier, P.U.; Gnaiger, E. Remodeling pathway control of mitochondrial respiratory capacity by temperature in mouse heart: Electron flow through the q-junction in permeabilized fibers. Sci. Rep. 2017, 7, 2840. [Google Scholar] [CrossRef] [Green Version]
- Perevoshchikova, I.V.; Sorochkina, A.I.; Zorov, D.B.; Antonenko, Y.N. Safranine o as a fluorescent probe for mitochondrial membrane potential studied on the single particle level and in suspension. Biochem. Biokhimiia 2009, 74, 663–671. [Google Scholar] [CrossRef]
- Akerman, K.E.; Wikstrom, M.K. Safranine as a probe of the mitochondrial membrane potential. FEBS Lett. 1976, 68, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Figueira, T.R.; Melo, D.R.; Vercesi, A.E.; Castilho, R.F. Safranine as a fluorescent probe for the evaluation of mitochondrial membrane potential in isolated organelles and permeabilized cells. Methods Mol. Biol. 2012, 810, 103–117. [Google Scholar]
- Chauhan, A.; Essa, M.M.; Muthaiyah, B.; Brown, W.T.; Chauhan, V. Mitochondrial abnormalities in lymphoblasts from autism. J. Neurochem. 2009, 109, 273. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Gu, F.; Essa, M.M.; Wegiel, J.; Kaur, K.; Brown, W.T.; Chauhan, V. Brain region-specific deficit in mitochondrial electron transport chain complexes in children with autism. J. Neurochem. 2011, 117, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.S.; Tsai, C.S.; Kuo, C.L.; Chen, H.W.; Lii, C.K.; Ma, Y.S.; Wei, Y.H. Oxidative stress-related alteration of the copy number of mitochondrial DNA in human leukocytes. Free Radic. Res. 2003, 37, 1307–1317. [Google Scholar] [CrossRef]
- Soon, B.H.; Abdul Murad, N.A.; Then, S.M.; Abu Bakar, A.; Fadzil, F.; Thanabalan, J.; Mohd Haspani, M.S.; Toh, C.J.; Mohd Tamil, A.; Harun, R.; et al. Mitochondrial DNA mutations in grade ii and iii glioma cell lines are associated with significant mitochondrial dysfunction and higher oxidative stress. Front. Physiol. 2017, 8, 231. [Google Scholar] [CrossRef]
- Giulivi, C.; Zhang, Y.F.; Omanska-Klusek, A.; Ross-Inta, C.; Wong, S.; Hertz-Picciotto, I.; Tassone, F.; Pessah, I.N. Mitochondrial dysfunction in autism. JAMA 2010, 304, 2389–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzecry, J.M.; Deth, R.; Holtzman, D. Abnormalities in respiratory properties of lymphoblasts in autistic spectrum disorders. Acta Paediatr. 2008, 97, 859–860. [Google Scholar]
- Poling, J.S.; Frye, R.E.; Shoffner, J.; Zimmerman, A.W. Developmental regression and mitochondrial dysfunction in a child with autism. J. Child Neurol. 2006, 21, 170–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Gutierrez Rios, P.; Kuo, S.H.; Akman, H.O.; Rosoklija, G.; Tanji, K.; Dwork, A.; Schon, E.A.; Dimauro, S.; Goldman, J.; et al. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol. Dis. 2013, 54, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Guevara-Campos, J.; Gonzalez-Guevara, L.; Puig-Alcaraz, C.; Cauli, O. Autism spectrum disorders associated to a deficiency of the enzymes of the mitochondrial respiratory chain. Metab. Brain Dis. 2013, 28, 605–612. [Google Scholar] [CrossRef]
- Palmieri, L.; Papaleo, V.; Porcelli, V.; Scarcia, P.; Gaita, L.; Sacco, R.; Hager, J.; Rousseau, F.; Curatolo, P.; Manzi, B.; et al. Altered calcium homeostasis in autism-spectrum disorders: Evidence from biochemical and genetic studies of the mitochondrial aspartate/glutamate carrier agc1. Mol. Psychiatry 2010, 15, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E.; Naviaux, R.K. Autistic disorder with complex iv overactivity: A new mitochondrial syndrome. J. Pediatr. Neurol. 2011, 9, 427–434. [Google Scholar]
- Chauhan, A.; Essa, M.M.; Merz, G.; Muthaiyah, B.; Ted Brown, W.; Chauhan, V. 81 mitochondrial abnormalities in lymphoblasts from autism. Mitochondrion 2010, 10, 223. [Google Scholar] [CrossRef]
- James, S.J.; Rose, S.; Melnyk, S.; Jernigan, S.; Blossom, S.; Pavliv, O.; Gaylor, D.W. Cellular and mitochondrial glutathione redox imbalance in lymphoblastoid cells derived from children with autism. FASEB J. 2009, 23, 2374–2383. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between mitochondrial membrane potential and ros formation. Methods Mol. Biol. 2012, 810, 183–205. [Google Scholar] [PubMed]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Role of uncoupled and non-coupled oxidations in maintenance of safely low levels of oxygen and its one-electron reductants. Q. Rev. Biophys. 1996, 29, 169–202. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrates | Function | Concentration |
|---|---|---|
| Digitonin (Dig) | Plasma membrane permeabilization | 7.5 µg∙mL−1 (NALCL) 27.5 µg∙mL−1 (ALCL) |
| Pyruvate (P) | NADH-generating substrate (Substrates for CI) | 5 mM |
| Glutamate (G) | 10 mM | |
| Malate (M) | 2 mM | |
| Succinate (S) | CII substrate | 10 mM |
| Ascorbate (As) | Maintains TMPD in a reduced state | 2 mM |
| N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride (TMPD) | Substrate for reducing cytochrome c (Substrate for CIV) | 0.5 mM |
| Cytochrome c (c) | Mitochondrial outer membrane permeability test | 10 µM |
| ADP (D) | Substrate of ANT, F1F0-ATPase (Substrate for CV) | 2.5 mM |
| Carbonyl cyanide m-chlorophenyl hydrazone, CCCP (U) | Uncoupler, protonophore | 0.5 µM steps |
| Rotenone (Rot) | CI inhibitor | 0.5 µM |
| Malonate (Mna) | CII inhibitor | 5 mM |
| Antimycin A (Ama) | CIII inhibitor | 2.5 µM |
| Sodium azide (Azd) | CIV inhibitor | 100 mM |
| Oligomycin (Omy) | ATP synthase inhibitor | 2.5 µM |
| Safranin (Saf) | Fluorophore, dye for measuring mitochondrial membrane potential | 2 µM |
| States and Ratios | Definition and Rate |
|---|---|
| N-pathway (CI-linked pathway) | Respiration induced by the addition of NADH-generating substrates. Electrons are transferred from CI to CIII and then to CIV. |
| S-pathway (CII-linked pathway) | Respiration induced by the addition of succinate and Rot. CI is inhibited by Rot while electrons can only be generated by CII, transferred to CIII and then to CIV. |
| NS-pathway (CI- and CII-linked pathways) | Respiration induced by the addition of NADH-generating substrates and succinate without Rot. Combination of both pathways (usual pathway) whereby electrons move from both CI and CII, to CIII and then to CIV. |
| ROUTINE, R | ROUTINE respiration controlled by intrinsic energy demand. This represents energy demand under steady-state conditions. |
| LEAK, L | LEAK respiration caused by proton leak, proton slip, cation cycling, and electron leak. L is measured in the presence of reducing substrate(s) but absence of ADP or after enzymatic inhibition of the phosphorylation system by Omy. |
| OXPHOS, P | Respiration in the ADP-stimulated state of oxidative phosphorylation; OXPHOS capacity. |
| ET, E | Oxygen consumption in the non-coupled state at optimum uncoupler concentration, ET capacity. |
| ROX, Rox | Residual oxygen consumption, measured after inhibition of the ETS. |
| (E − P)/E | Relative E − P excess capacity, defines the limitation of OXPHOS capacity exerted by the phosphorylation system. |
| (P − L)/P | OXPHOS coupling efficiency, combining the effects of coupling and limitation by the phosphorylation system. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, H.; Zakaria, F.; Makpol, S.; Karim, N.A. A Link between Mitochondrial Dysregulation and Idiopathic Autism Spectrum Disorder (ASD): Alterations in Mitochondrial Respiratory Capacity and Membrane Potential. Curr. Issues Mol. Biol. 2021, 43, 2238-2252. https://doi.org/10.3390/cimb43030157
Hassan H, Zakaria F, Makpol S, Karim NA. A Link between Mitochondrial Dysregulation and Idiopathic Autism Spectrum Disorder (ASD): Alterations in Mitochondrial Respiratory Capacity and Membrane Potential. Current Issues in Molecular Biology. 2021; 43(3):2238-2252. https://doi.org/10.3390/cimb43030157
Chicago/Turabian StyleHassan, Hazirah, Fazaine Zakaria, Suzana Makpol, and Norwahidah Abdul Karim. 2021. "A Link between Mitochondrial Dysregulation and Idiopathic Autism Spectrum Disorder (ASD): Alterations in Mitochondrial Respiratory Capacity and Membrane Potential" Current Issues in Molecular Biology 43, no. 3: 2238-2252. https://doi.org/10.3390/cimb43030157
APA StyleHassan, H., Zakaria, F., Makpol, S., & Karim, N. A. (2021). A Link between Mitochondrial Dysregulation and Idiopathic Autism Spectrum Disorder (ASD): Alterations in Mitochondrial Respiratory Capacity and Membrane Potential. Current Issues in Molecular Biology, 43(3), 2238-2252. https://doi.org/10.3390/cimb43030157