
(5-Chloroquinolin-8-yl)-2-fluorobenzoate. The Halogen Bond as a Structure Director

 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
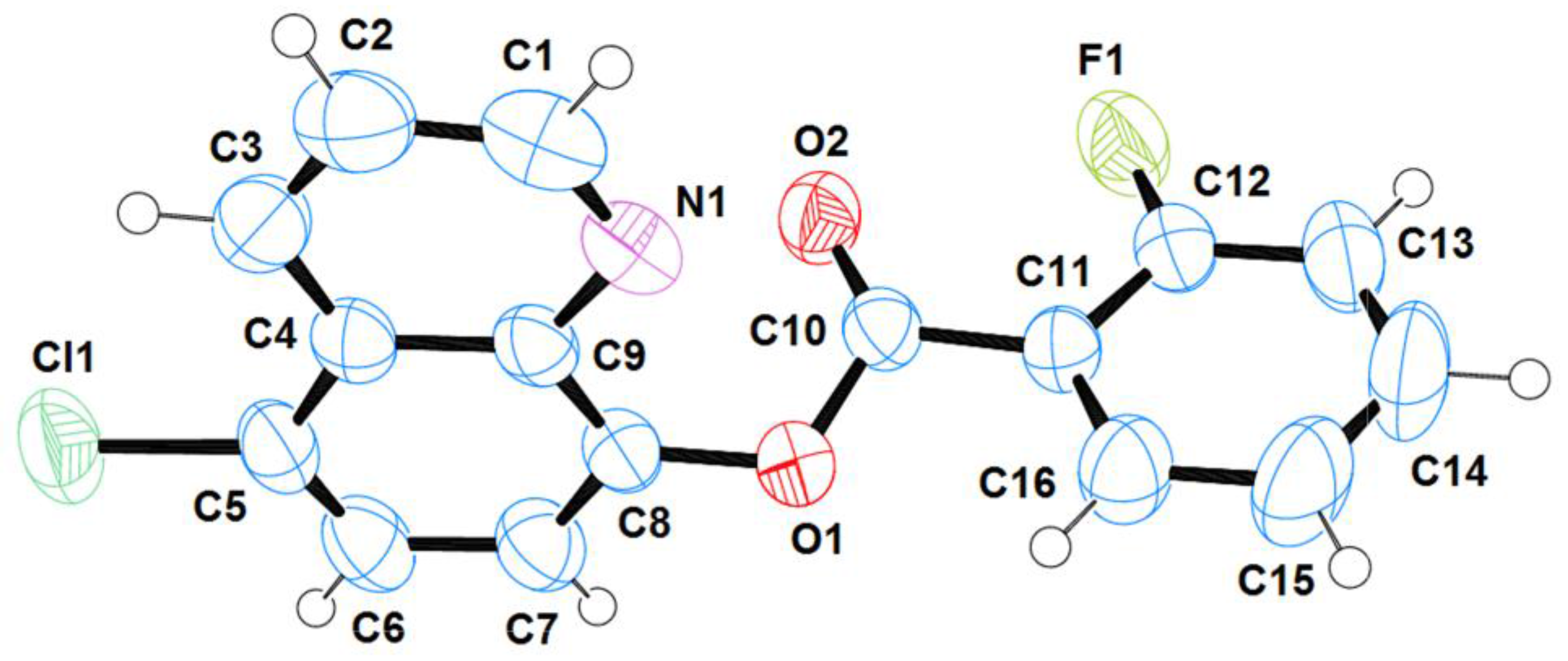
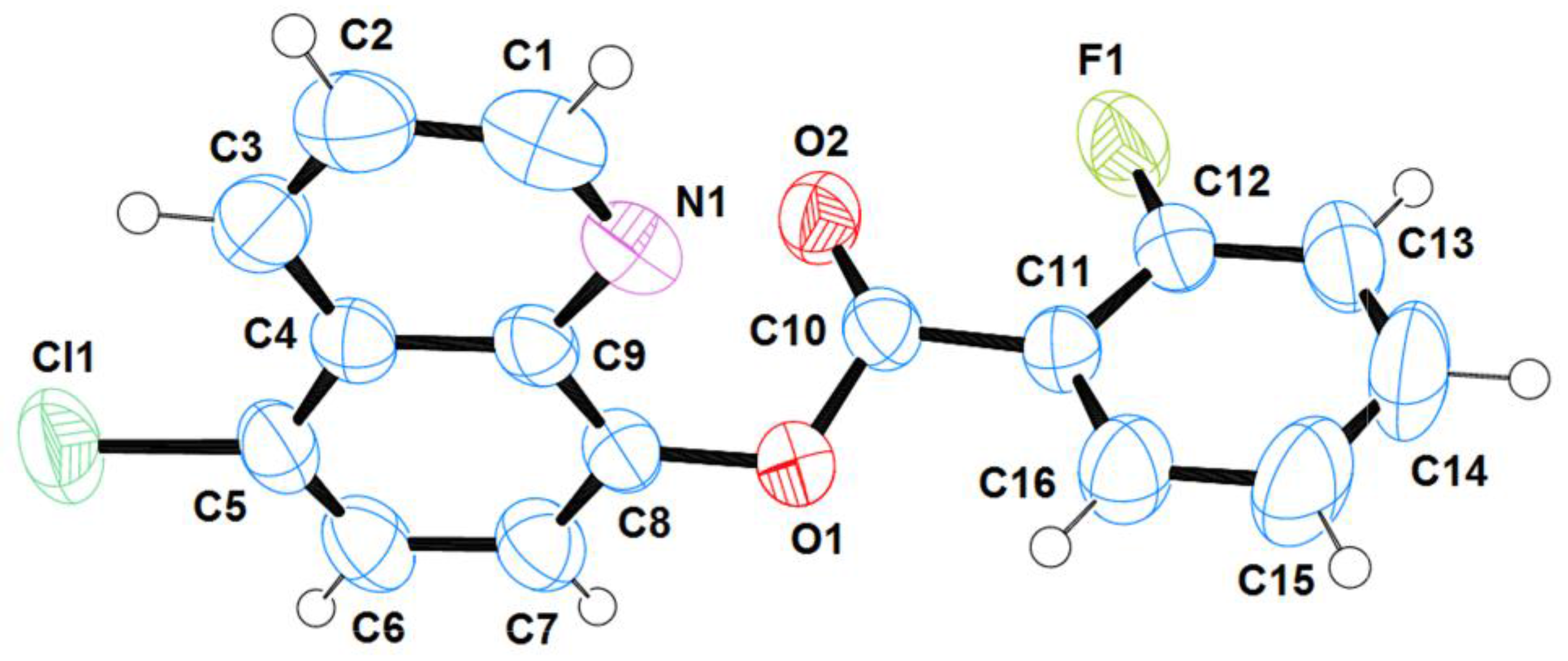
2.1. Structural Analysis
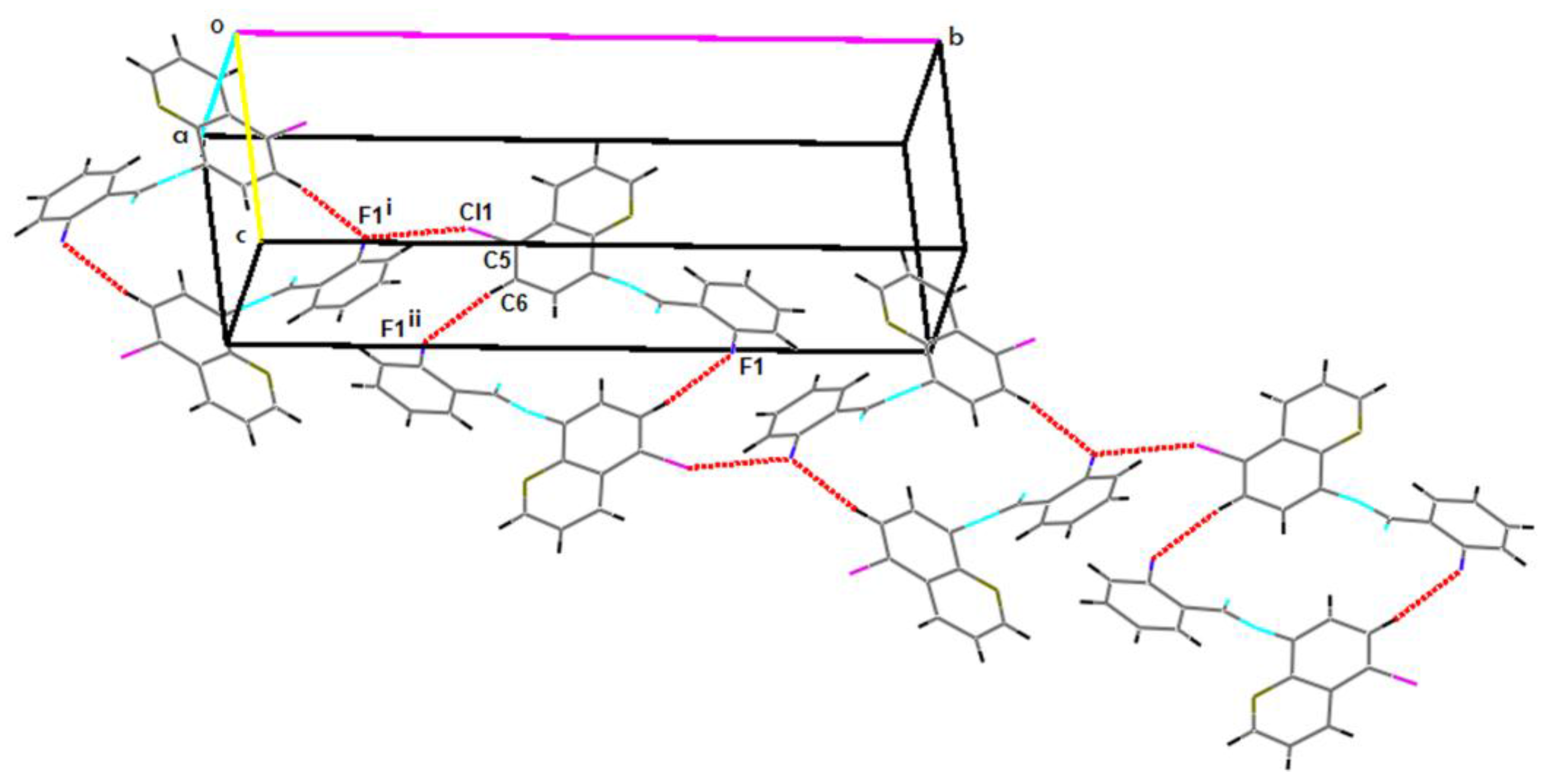
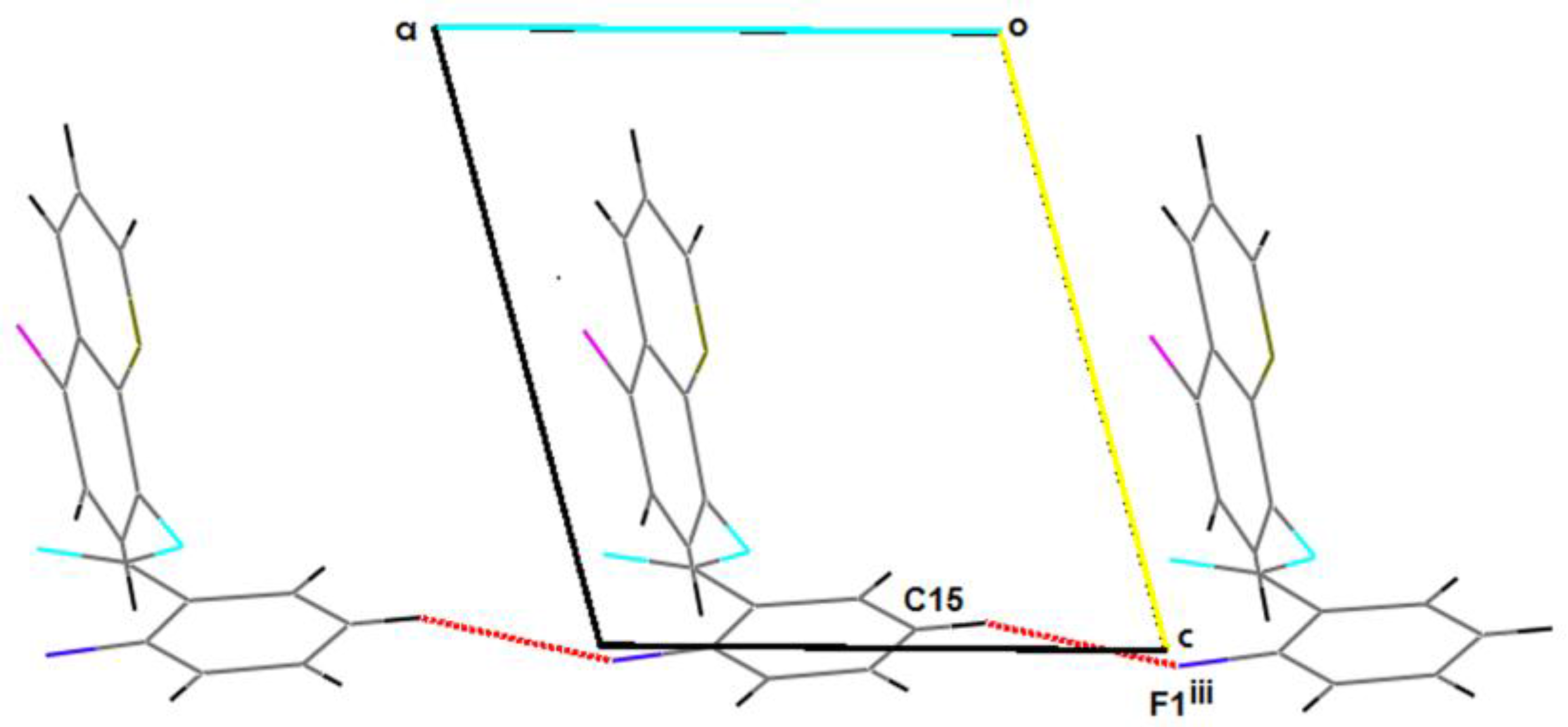
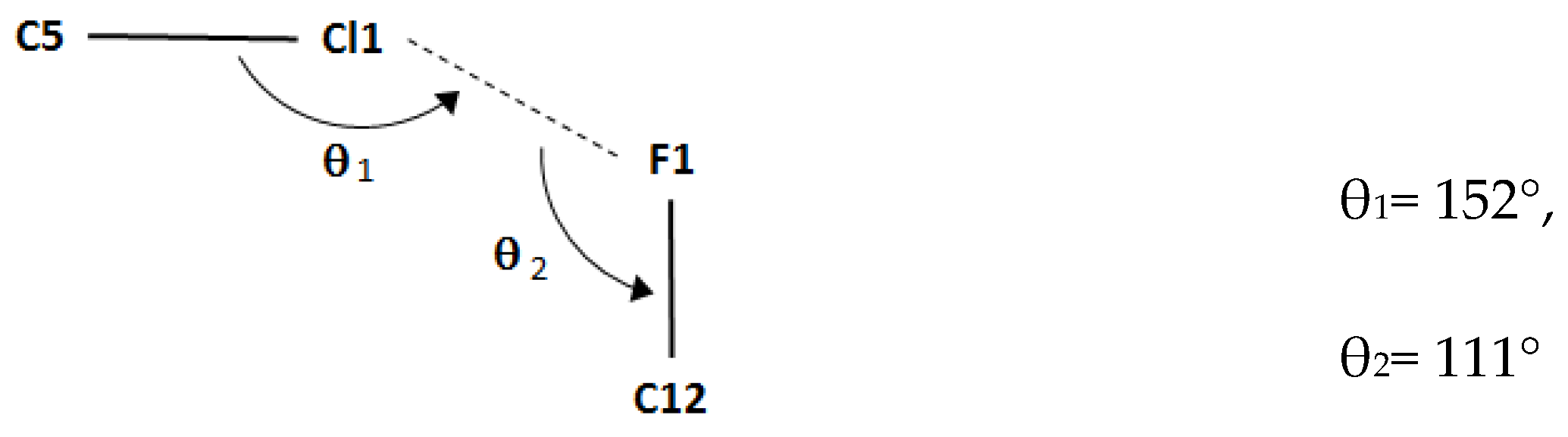
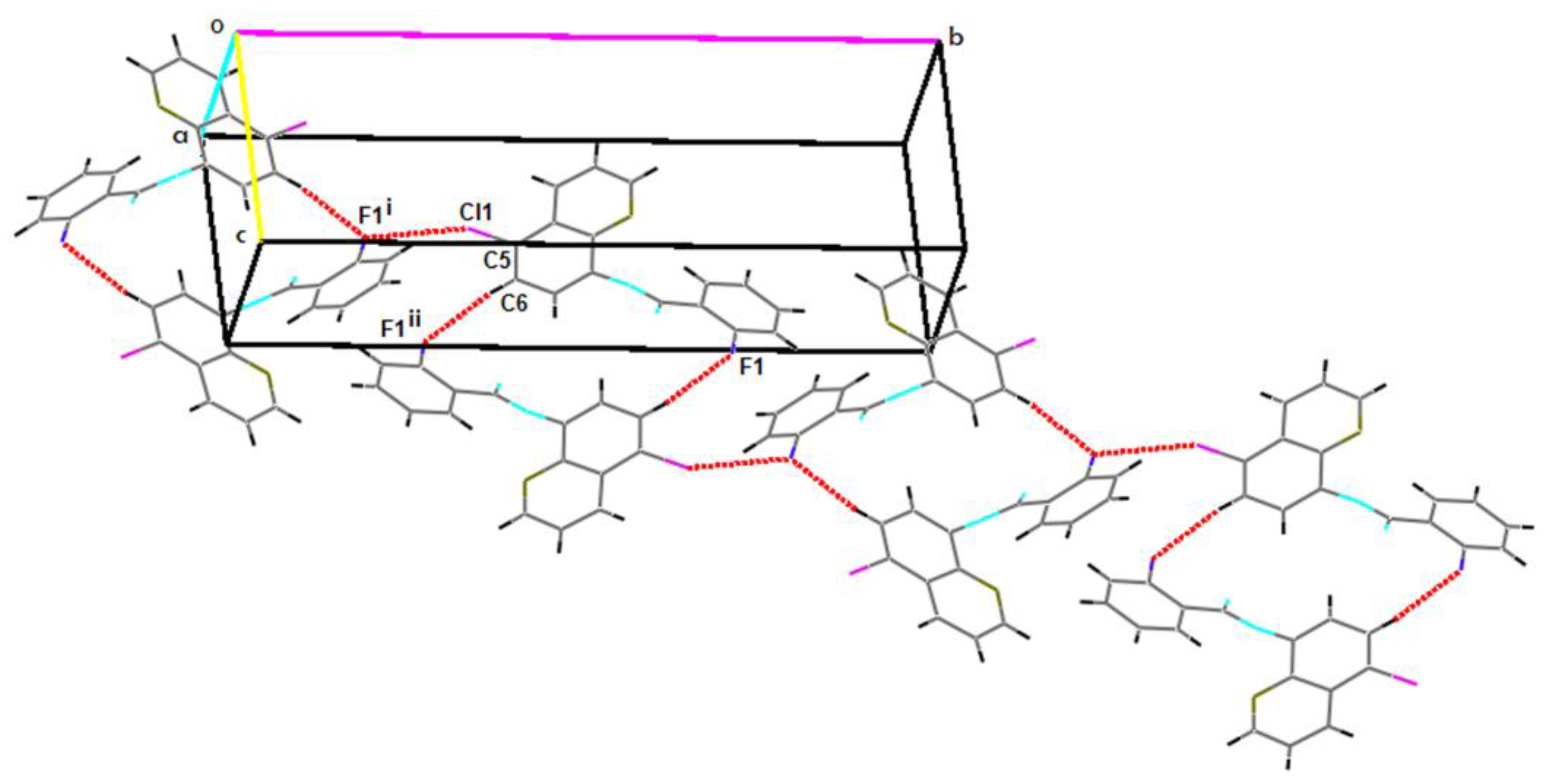
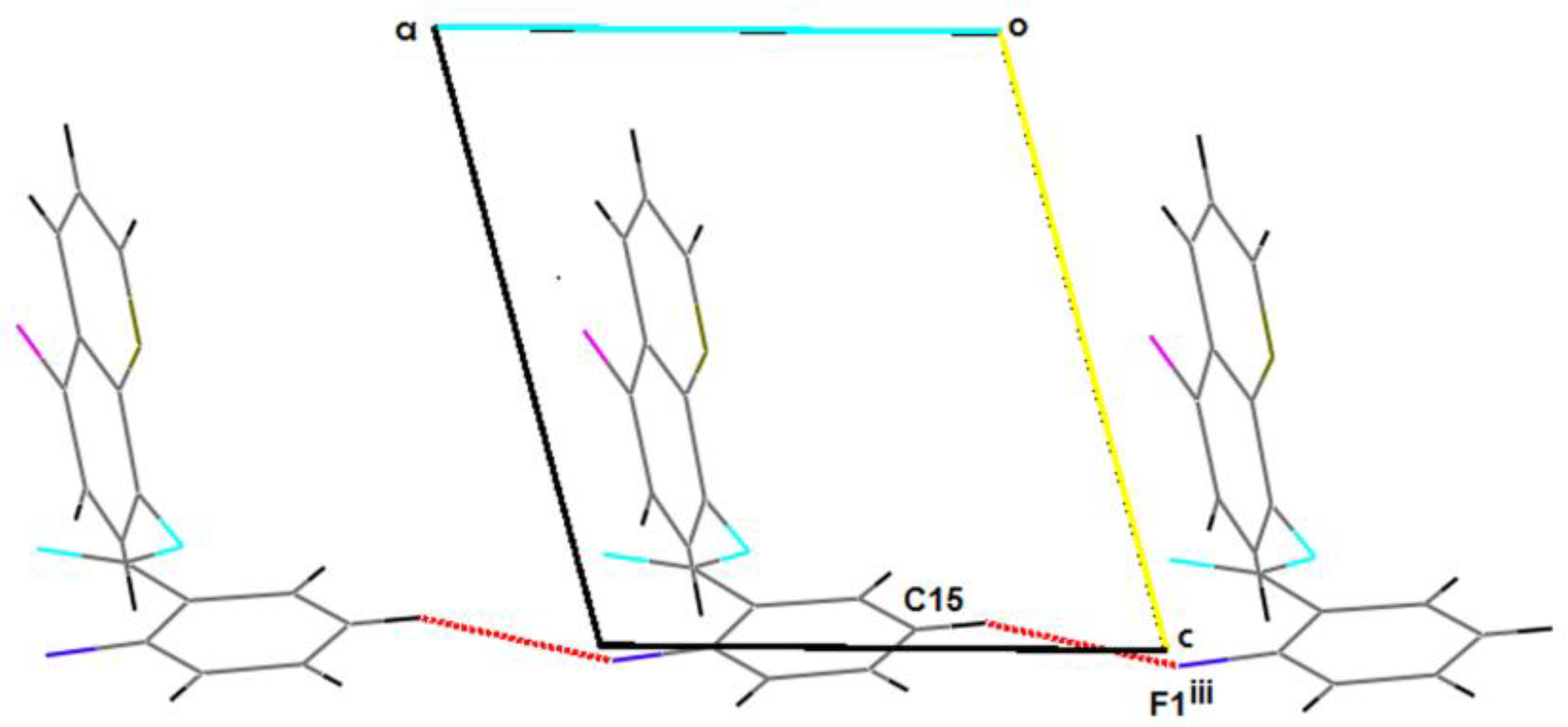
2.2. Supramolecular Features
3. Materials and Methods
3.1. General Information
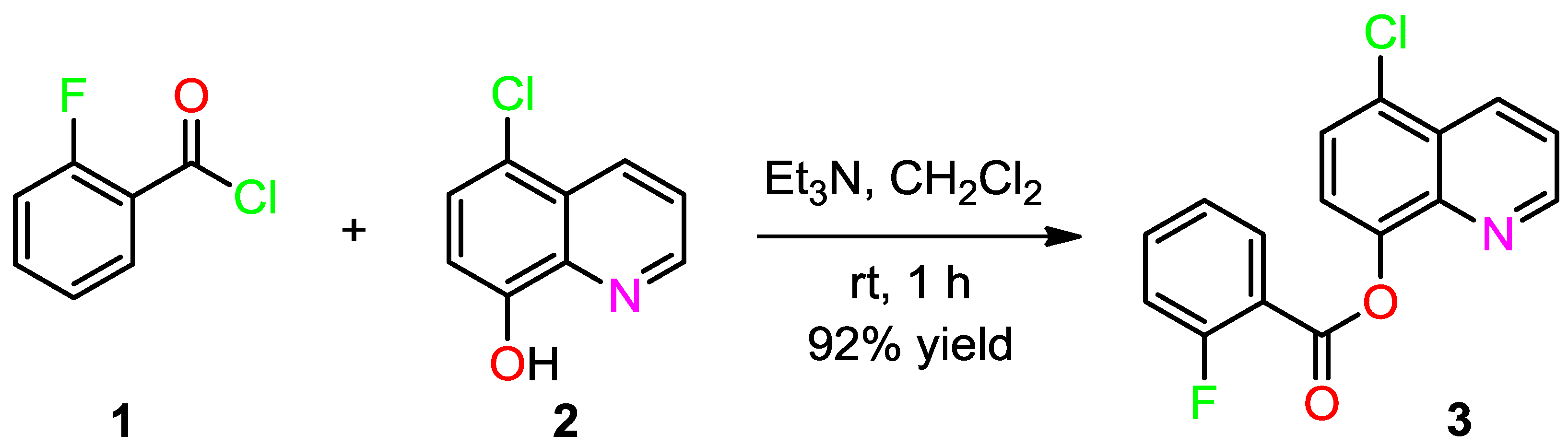
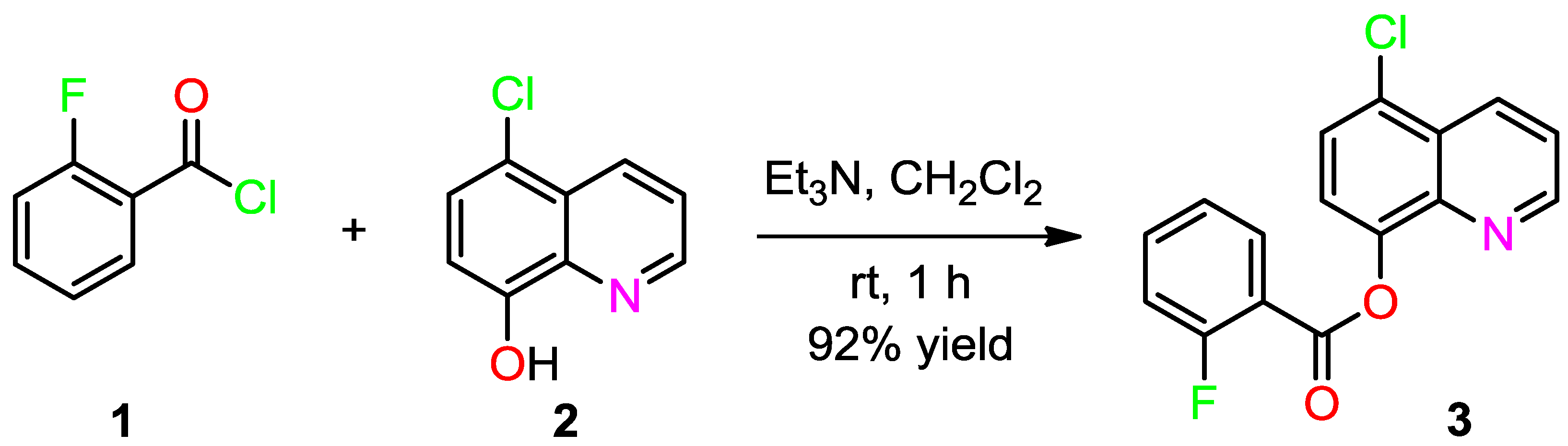
3.2. Synthesis of (5-Chloroquinolin-8-yl)-2-fluorobenzoate (3)
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Klingenstein, R.; Melnyk, P.; Leliveld, S.R.; Ryckebusch, A.; Korth, C. Similar structure-activity relationships of quinoline derivatives for antiprion and antimalarial effects. J. Med. Chem. 2006, 49, 5300–5308. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Deng, X.; Zhang, L.; Roth, M.G.; Fontoura, B.M.A.; Phillips, M.A.; De Brabander, J.K. SAR-based optimization of a 4-quinoline carboxylic acid analogue with potent antiviral activity. ACS Med. Chem. Lett. 2013, 4, 571–521. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, S.; Gosetto, F.; Manfroni, G.; Tabarrini, O.; Kaatz, G.W.; Patel, D.; Cecchetti, V. Evolution from a natural nucleus to obtain 2-(4-propoxyphenyl)quinoline derivatives as potent inhibitors of the S. aureus NorA efflux pump. J. Med. Chem. 2011, 54, 5722–5736. [Google Scholar] [PubMed]
- Chioua, M.; Sucunza, D.; Soriano, E.; Hadjipavlou-Litina, D.; Alcázar, A.; Ayuso, I.; Oset-Gasque, M.J.; González, M.P.; Monjas, L.; Rodríguez-Franco, M.I.; et al. α-Aryl-N-alkyl nitrones, as potential agents for stroke treatment: Synthesis, theoretical calculations, antioxidant, anti-inflammatory, neuroprotective, and brain-blood barrier permeability properties. J. Med. Chem. 2012, 55, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Hammarström, L.G.J.; Harmel, R.K.; Granath, M.; Ringom, R.; Gravenfors, Y.; Färnegardh, K.; Svensson, P.H.; Wennman, D.; Lundin, G.; Roddis, Y.; et al. The oncolytic efficacy and in vivo pharmacokinetics of [2-(4-chlorophenyl)quinolin-4-yl](piperidine-2-yl)methanol (vacquinol-1) are governed by distinct stereochemical features. J. Med. Chem. 2016, 59, 8577–8592. [Google Scholar] [CrossRef] [PubMed]
- Degorce, S.L.; Barlaam, B.; Cadogan, E.; Dishington, A.; Ducray, R.; Glossop, S.C.; Hassall, L.A.; Lau, A.; McGuire, T.M.; Nowak, T.; et al. Discovery of novel 3-quinoline carboxamides as potent, selective, and orally bioavailable inhibitors of ataxia telangiectasia mutated (ATM) kinase. J. Med. Chem. 2016, 59, 6281–6292. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Barilli, A.; Bassanetti, I.; Tegoni, M.; Bussolati, O.; Franchi-Gazzola, R.; Mucchino, C.; Marchiò, L. Copper-dependent cytotoxicity of 8-hydroxyquinoline derivatives correlates with their hydrophobicity and does not require caspase activation. J. Med. Chem. 2012, 55, 10448–10459. [Google Scholar] [CrossRef] [PubMed]
- For a review, see Andersson, M.I.; MacGowan, A.P. J. Antimicrob. Chemother. 2003, 51 (Suppl. S1), 1–11. [PubMed]
- Rabbani, M.G.; Islam, Md.R.; Ahmad, M.; Hossion, A.M.L. Synthesis of some NH-derivatives of ciprofloxacin as antibacterial and antifungal agents. Bangladesh J. Pharmacol. 2011, 6, 8–13. [Google Scholar] [CrossRef]
- Adsule, S.; Barve, V.; Chen, D.; Ahmed, F.; Dou, Q.P.; Padhye, S.; Sarkar, F.H. Novel Schiff base copper complexes of quinoline 2-carboxaldehyde as proteasome inhibitors in human prostate cancer cells. J. Med. Chem. 2006, 49, 7242–7246. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fuquen, R.; Mosquera, F.; Kennedy, A.R. 2,4,6-Trinitrophenyl furan-2-carboxylate. Acta Cryst. 2013, E69, o1682. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fuquen, R.; Hernández, G.; Ellena, J.; De Simone, C.; Tenorio, J.C. 5-Chloroquinolin-8-yl furan-2-carboxylate. Acta Cryst. 2013, E69, o501. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fuquen, R.; Hernández, G.; Kennedy, A.R. 4-Formyl-2-nitrophenyl benzoate. Acta Cryst. 2014, E70, o268. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fuquen, R.; Mosquera, F.; Ellena, J.; Tenorio, J.C. 2,4,6-Trinitrophenyl 3-bromobenzoate. Acta Cryst. 2014, E70, o689. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fuquen, R.; Hernández, G.; Ellena, J.; De Simone, C.; Tenorio, J.C. Crystal structure of 4-formyl-2-nitrophenyl-4-chloro-2-nitrobenzoate. Acta Cryst. 2015, E71, o940. [Google Scholar] [CrossRef] [PubMed]
- This compound is supplied by MCULE, among others: https://mcule.com/mcule-5727823188/ under the CAS Number 5727823188.
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen bonds in crystal engineering: Like hydrogen bonds yet different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole bond vs π-hole bond: A comparison based on halogen bond. Chem. Rev. [CrossRef] [PubMed]
- Lei, G. 8-Quinolyl benzoate. Acta Cryst. 2006, E62, o4666–o4667. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Parthasarathy, R. The nature of halogen…halogen interactions- Are short halogen contacts due to specific attractive forces or due close packing of nonspherical atoms. J. Am. Chem. Soc. 1989, 111, 8725–8726. [Google Scholar] [CrossRef]
- Rigaku. CrystalClear; Rigaku Corporation: Tokyo, Japan, 2008. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond lengths | |
| C(5)-Cl(1); 1.7310(19) | O(1)-C(10); 1.355(2) |
| C(9)-N(1); 1.361(2) | C(10)-O(2); 1.191(2) |
| C(1)-N(1); 1.300(3) | C(10)-C(11); 1.473(2) |
| C(8)-O(1); 1.389(2) | C(12)-F(1); 1.337(3) |
| Angles | |
| C(8)-O(1)-C(10); 117.06(14) | O(1)-C(10)-C(11); 110.26(15) |
| Torsion angles | |
| O(2)-C(10)-C(11)-C(12); −11.7(3) | O(2)-C(10)-C(11)-C(12); −11.7(3) |
| F(1)-C(12)-C(11)-C(10); −5.5(3) | O(1)-C(10)-C(11)-C(16); −16.1(3) |
| C(9)-C(8)-O(1)-C(10); −70.6(2) | |
| D–H…A | D–H | H…A | D…A | D–H…A |
|---|---|---|---|---|
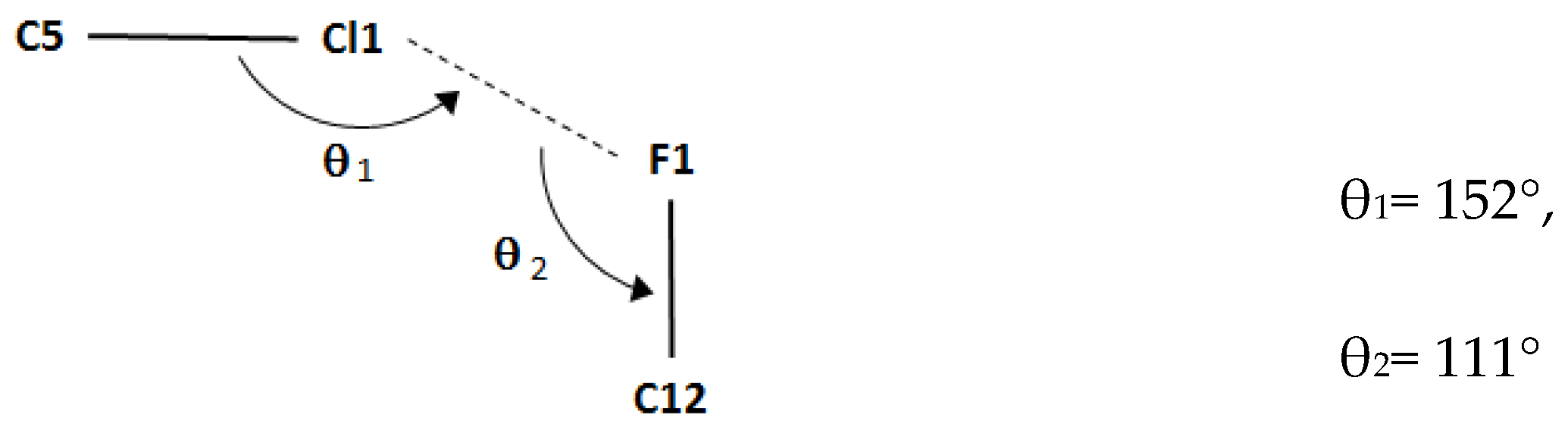
| C5–Cl1…F1i | 1.7310(19) | 3.2171(15) | 4.816(2) | 152 |
| C6–H6…F1ii | 0.93 | 2.57 | 3.491(3) | 169.8 |
| C15–H15…F1iii | 0.93 | 2.62 | 3.515(3) | 161.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Fuquen, R.; Castillo, J.C.; Abonía, R.; Portilla, J.; Henao, J.A. (5-Chloroquinolin-8-yl)-2-fluorobenzoate. The Halogen Bond as a Structure Director. Molbank 2017, 2017, M934. https://doi.org/10.3390/M934
Moreno-Fuquen R, Castillo JC, Abonía R, Portilla J, Henao JA. (5-Chloroquinolin-8-yl)-2-fluorobenzoate. The Halogen Bond as a Structure Director. Molbank. 2017; 2017(1):M934. https://doi.org/10.3390/M934
Chicago/Turabian StyleMoreno-Fuquen, Rodolfo, Juan Carlos Castillo, Rodrigo Abonía, Jaime Portilla, and José Antonio Henao. 2017. "(5-Chloroquinolin-8-yl)-2-fluorobenzoate. The Halogen Bond as a Structure Director" Molbank 2017, no. 1: M934. https://doi.org/10.3390/M934
APA StyleMoreno-Fuquen, R., Castillo, J. C., Abonía, R., Portilla, J., & Henao, J. A. (2017). (5-Chloroquinolin-8-yl)-2-fluorobenzoate. The Halogen Bond as a Structure Director. Molbank, 2017(1), M934. https://doi.org/10.3390/M934