
The ATM Gene in Breast Cancer: Its Relevance in Clinical Practice
by
 , , , , and
, , , , and
Luigia Stefania Stucci
1,* ,
,
Valeria Internò
1 ,
,
Marco Tucci
1,2,
Martina Perrone
1,
Francesco Mannavola
1,
Raffaele Palmirotta
3 and
and
Camillo Porta
1 1
Division of Medical Oncology, Department of Biomedical Sciences and Human Oncology, University of Bari ‘Aldo Moro’, A.O.U. Consorziale Policlinico di Bari, 70121 Bari, Italy
2
National Cancer Research Center, Tumori Institute IRCCS Giovanni Paolo II, 70121 Bari, Italy
3
Interdisciplinary Department of Medicine, Section of Sciences and Technologies of Laboratory Medicine, University of Bari, 70121 Bari, Italy
*
Author to whom correspondence should be addressed.
Genes 2021, 12(5), 727; https://doi.org/10.3390/genes12050727
Submission received: 19 April 2021
/
Revised: 9 May 2021
/
Accepted: 10 May 2021
/
Published: 13 May 2021
(This article belongs to the Special Issue Breast Cancer Genetics: Diagnostic and Treatment)
Abstract
:Molecular alterations of the Ataxia-telangiectasia (AT) gene are frequently detected in breast cancer (BC), with an incidence ranging up to 40%. The mutated form, the Ataxia-telangiectasia mutated (ATM) gene, is involved in cell cycle control, apoptosis, oxidative stress, and telomere maintenance, and its role as a risk factor for cancer development is well established. Recent studies have confirmed that some variants of ATM are associated with an increased risk of BC development and a worse prognosis. Thus, many patients harboring ATM mutations develop intermediate- and high-grade disease, and there is a higher rate of lymph node metastatic involvement. The evidence concerning a correlation of ATM gene mutations and the efficacy of therapeutic strategies in BC management are controversial. In fact, ATM mutations may sensitize cancer cells to platinum-derived drugs, as BRCA1/2 mutations do, whereas their implications in objective responses to hormonal therapy or target-based agents are not well defined. Herein, we conducted a review of the role of ATM gene mutations in BC development, prognosis, and different treatment strategies.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The Ataxia-telangiectasia mutated (ATM) gene is an oncosuppressor, located on chromosome 11q23, that encodes a 350-KDa protein consisting of 3056 amino acids [1]. It belongs to the superfamily of phosphatidylinositol 3-kinase-related protein kinases (PIKKs). The PIKK superfamily includes six serine/threonine kinases showing a sequence similarity to phosphatidylinositol 3-kinases (PI3Ks), including ATR (ATM- and RAD3-related), DNA-PKcs (DNA-dependent protein kinase catalytic subunit), and mTOR (mammalian target of rapamycin). The ATM protein is involved in DNA repair and activates DNA damage response pathways [2]; indeed, upon DNA damage, it is recruited to double-strand breaks where it holds the two ends together. ATM mutations cause Ataxia-telangiectasia (AT), an autosomal recessive neurodegenerative disorder characterized by a progressive neuromotor dysfunction resulting from several neuropathological processes dominated by gradual cerebellar cortical atrophy, telangiectasia in the eyes and sometimes on the facial skin, thymic degeneration, immune deficiency, recurrent sinopulmonary infections (at least in some patients), retarded somatic growth, premature aging, gonadal dysgenesis, predisposition to lymphoreticular malignancies, and acute sensitivity to ionizing radiation [3,4,5].
In general, the ATM gene is involved in cell cycle control, apoptosis, gene regulation, oxidative stress, and telomere maintenance and is deregulated in many malignancies such as breast cancer (BC) [6]. Many ATM mutations have been described and associated with a moderate risk of BC development [7]. Furthermore, epidemiological studies based on relatives affected by both AT and BC suggested that heterozygous carriers of ATM mutations have a two- to thirteen-fold increased risk of BC development, with a higher relative risk under 50 years old [8,9,10,11].
Previous studies emphasized the evidence of a strong association between ATM variants and the risk of BC development. The V2424G variant confers the highest risk of BC development, while the D1853V, L546, and S707P isoforms are associated with the lowest risk [12]. Moreover, next generation sequencing (NGS) analysis revealed that ATM is among the most aberrant gene in sporadic cancer (as shown by the COSMIC database), and that loss of heterozygosity in the region of the ATM has been detected in approximately 40% of human sporadic BC [13,14,15].
In wider large-scale studies including solid cancers, 5% of patients showed ATM aberrations (either mutation or loss). As described, 8% of lung cancer patients showed ATM mutations that were largely mutually exclusive with those of TP53. More recently, ATM alterations have been found in colorectal cancer (CRC) both in patients bearing both microsatellite stable and unstable tumors. In prostate cancer, targeted next-generation sequencing has revealed an 8% incidence of ATM mutations. In a range from 1% to 5%, ATM mutations were reported in endometrial, kidney, liver, esophageal, ovarian, salivary gland, gastric, thyroid, and urinary tract cancers [12].
Clinical and pathologic characteristics of ATM-associated BC have not been well defined, but it is known that ATM-mutated BCs are mostly endocrine-positive, dedifferentiated, and more aggressive, and thus have poor prognosis [11].
As regards therapeutic implications, ATM aberrations may sensitize cancer cells to platinum-derived drugs, similarly to the effect of BRCA1 mutations, but have a worse effect in case of radiotherapy (RT). In fact, ATM mutations increase the risk for development of a second tumor after RT [16,17,18,19].
2. The ATM Gene and Its Role in Cancer
Since the ATM protein plays an important role in DNA repair through the activation of enzymes that fix the broken strands, its biology is of interest in cancer research [20]. The physiological structure of the ATM protein is characterized by an N-terminal half that is largely unique and a C-terminal half showing homology with other PI3K-like kinases such as ATR, mTOR, and DNA-PKcs. ATM contains at least five autophosphorylation sites. The N-terminal portion interacts with substrates and cofactors such as NBS1, p53, BRCA1, LKB1, and BLM [12]. Once activated, ATM phosphorylates many downstream effectors, as illustrated in Figure 1.
Pathogenic variants in ATM are common. In particular, around 0.35% of people carry an ATM mutation, and there is a strong association between mutations in ATM and cancers. Some researchers found a four-fold increased risk for pancreatic cancer, a three-fold increase for stomach cancer, and a two- to three-fold increase for prostate cancer and confirmed the previously known two-fold invasive ductal BC in patients with an ATM mutation. They also found a low to moderate increase in risk for male breast cancer, ovarian cancer, colorectal cancer, and melanoma. ATM is a large gene with many thousands of locations where a mutation can occur. One common mutation, known as c.7271T>G, is associated with a significantly higher risk of BC (about four-fold) than other ATM mutations [21].
In BC, the physiological function of ATM is downregulated by malignant cells. In fact, ATM phosphorylates DBC1 (deleted in BC) and promotes apoptosis by the activation of p53 and caspase-2 [22,23,24,25]. Moreover, ATM signaling can also be upregulated in cancer cells that have already evaded cell apoptosis through other mechanisms, as seen in melanoma through upregulation of melanoma-associated antigen-encoding (MAGE) genes as well as in prostate cancer by activation of the Androgen receptor (AR) [1,2]. Regarding the role of ATM mutations on the efficacy of therapeutic strategies, it is well known that activation of the ATM-dependent pathway in tumor cells can promote chemoresistance and radiotherapy resistance through the activation of p38 MAPK and enzyme transglutaminase 2 [3,26], as well as by inducing enzymes involved in DNA double-strand break (DSB) repair [27]. Although ATM is considered as an oncosuppressor gene owing to its role in enhancing chemo-radioresistance of tumor cells, this aberrant protein could potentially be explored as a target for cancer treatment. Moreover, ATM signaling induces tumor progression via the NF-kb-dependent pathway that promotes the release of pro-tumorigenic cytokines, as well as the epithelial–mesenchymal transition [28]. Furthermore, in some tumors, ATM signaling upregulates the alphavbeta3 integrin pathway [29], leading to tumor progression and downregulation of immune-mediated cell responses [30]. The tumor-specific alphavbeta3 integrin expression targeted dendritic cells, facilitating their ability to phagocytose viable therapy-resistant tumor cells, and thereby impaired their ability to cross-prime antigen-specific T lymphocyte, and it has been clearly demonstrated that the integrin plays a critical role in triggering invasive and metastatic activities of tumor cells.
It is well known that ATM germline mutations have different effects on tumor cells [31]. Particularly, the increased incidence of BC in families harboring AT has been clearly demonstrated [13], but roles for specific mutations of ATM are now emerging. Heterozygous ATM mutations are associated with a five-fold higher risk of BC in subjects under 50 years of age [32] and are well classified in the COSMIC (Catalogue of Somatic Mutations in Cancer) database [33]. In addition to conventional BC subtypes, ATM heterozygous mutations were recently associated to the predisposition of familial ductal pancreatic adenocarcinoma [34]. ATM-silencing mutations or deletions have also been found in other types of tumors, such as lung adenocarcinoma and colon cancer [35,36]. Moreover, ATM inactivating mutations are more frequently described in solid tumors with areas of hypoxia, such as gliomas, thus contributing to radiotherapy resistance [37].
3. Role of ATM Gene Mutations in BC Susceptibility and Prognosis
ATM mutations increase the BC risk, as demonstrated in a recent systematic review and meta-analysis [17]. It is well known that BRCA1/2 mutations have been associated to a hereditary BC in 5% of patients, but also the incidence of BC in AT families was found to be increased two- to five-fold [12,13,38,39,40,41,42]. The data collected by Moslemi et al. confirmed that ATM missense variants increase the risk of BC. The risk of BC is enhanced to a degree ranging from 2.8 to 3.04 [43,44]. Among the different variants explored, the V2424G (c. 7271 T>G) missense variant had the highest association with BC incidence in all subgroups [10,43,44,45,46,47]. While the ATM V2424G variant was one of the forms associated with an increased risk of cancer, the ATM D1853V missense variant has the least association with BC risk [48]. Moreover, a high association of ATM variants with BC is more frequently described in Asian than Caucasian patients, mostly due to racial differences, environmental conditions, or lifestyle. Recently, the Breast Cancer Association Consortium designed a panel of 34 putative susceptibility genes which were checked in 60,466 specimens from BC patients and 53,461 controls. It was demonstrated that protein-truncating variants in five genes (ATM, BRCA1, BRCA2, CHEK2, and PALB2) were associated with a risk of BC overall (p < 0.0001). For protein-truncating variants in ATM and CHEK2, the odds ratio was higher for estrogen receptor (ER)-positive than ER-negative disease. Rare missense variants in ATM, CHEK2, and TP53 were associated with an overall risk of BC (p < 0.001). The results of this study defined those genes that could most usefully be included in screening panels predictive of BC. Furthermore, ATM proved to be potentially useful for genetic counseling [49]. As previously mentioned, ATM mutations also occur in sporadic BC leading to ATM gene inactivation, but the mechanisms are still unclear. Different mutations have been described, such as allelic loss [50] and ATM epigenetic silencing mediated by CpG island methylation [51]. Post-transcriptional ATM regulation mediated by microRNAs has been reported in gliomas and BC [52,53,54]. The miR-18 was reported as a putative ATM regulatory miRNA in BC [23], but other studies showed no correlation with ATM transcript and miR-18a. A recent study integrated genomic, transcriptomic, and proteomic analyses in a large series of BC and identified tumor subtypes with different subsets of genetic and epigenetic abnormalities [55]. For example, ATM loss and MDM2 amplification proved to be more common in aggressive luminal B subtype BC. The clinical impact of ATM downregulation in defining the prognosis of BC patients is limited and not yet validated. Lower levels of ATM gene products have been discovered in high-grade BC [56,57,58], suggesting association of the ATM mutation with more aggressive disease. Analysis of a large cohort of patients with long-term follow-up showed a strong correlation between the absence of ATM protein expression and distant metastasis, resulting in a worse outcome of BC patients [11]. Survival analysis revealed that BC patients harboring inactivation of the ATM gene had a shorter disease-free survival (DFS) and overall survival (OS). Moreover, a multivariate analysis by Bueno et al. demonstrated that ATM is an independent prognostic factor, in association with clinical–pathological factors such as tumor size and lymph node involvement. Other reports described a strong correlation between ATM downregulation and poor survival in patients with p53 wild-type tumors [59,60]. Another study explored patients who underwent multigene panel testing (MGPT) between 2013–2019, identifying those harboring ATM mutations. Heterozygous germline ATM mutation carriers had an increased risk of developing cancer of the breast, pancreas, and other organs [61]. Thus, a decreased ATM expression is associated with a worse prognosis in BC, suggesting it may be a potential marker of disease outcome. The majority of patients had intermediate- to high-grade, hormone receptor-positive disease and a possibly higher rate of HER2 positivity and lymph node involvement. It was also reported that ATM expression promotes HER2-dependent tumorigenicity in vitro and in vivo. Stagni V et al. [62] demonstrated a correlation between ATM activation and a reduced time to recurrence in patients diagnosed with invasive HER2-positive BC. Moreover, ATM was identified as a novel modulator of HER2 protein integrity through a complex of HER2 with the chaperone HSP90, therefore preventing HER2 ubiquitination and degradation. Thus, since downstream activation of HER2 by ATM may modulate the response to therapeutic approaches, it is conceivable that assessing the ATM activity status may be useful in the treatment and prognosis of HER2-positive tumors [62].
4. Therapeutic Implications of ATM Gene Mutations in BC
Nowadays, it is well known that tumors with mutations in genes encoding proteins involved in DNA repair may be more sensitive to treatments that induce cytotoxicity by inducing DNA damage or inhibiting DNA repair mechanisms. As BRCA1-mutated tumors may be more sensitive to treatments with platinum derivatives and benefit from treatment with inhibitors of PARP, similar strategies could also be hypothesized for BC patients harboring ATM gene mutations [63]. Moreover, it is recognized that radiosensitivity is a hallmark of AT syndrome. As a consequence, heterozygous ATM mutations increase radiotherapy toxicity [64,65], probably due to defective DNA repair and genomic instability in normal tissues. In view of this evidence, adverse events occurring in BC patients during chemotherapy may be increased in those bearing germline ATM mutations. Indeed, some reports showed a high risk of myeloid suppression in ATM mutated patients compared to wild-type [65]. ATM mutations are predicted to result in an increased sensitivity to platinum-based chemotherapy used for BC treatment [66,67]. Similarly, PARP-inhibitors could also be more effective in ATM-deficient BC tumors, but they have not been specifically evaluated in ATM-mutated BC. PARP-inhibitors have shown promising results in tumor cells defective in DNA damage repair, in particular DSBs, as ATM-mutated tumor cells. Some studies have considered the efficacy of Olaparib in ATM-deficient leukemic cells from patients with leukemia [68] or in patients with gastric cancer [69], but no studies in BC tumor cells have yet been performed. The ATM-dependent pathway is also involved in resistance to treatment with CDK4/6 inhibitors, recently introduced in clinical practice for the treatment of advanced estrogen receptor-positive BC [70]. In this regard, a recent study showed that defects in single-strand break repair in luminal BC can drive endocrine therapy resistance and are closely associated with the ATM-CHK2-CDC25A pathway. ATM, as a DNA damage sensor, activates CHK2, which in turn phosphorylates CDC25A that could inhibit the phosphorylation of CDK4/6. Therefore, the cross talk between the CDK4/6-Rb and the ATM-CHK2-CDC25A axes is very important [71]. More recently, Haricharan et al. demonstrated that both ATM and CHK2 gene alterations are required to boost the efficacy of endocrine agents in luminal tumors. In fact, the inactivation of either of these negative cell cycle regulators prevents cell cycle arrest upon ER inhibition [72]. To date, ATM alone has not been associated with a high incidence of contralateral BC [73], but genetic variants in ATM have been demonstrated to play a clinically significant role in radiation-induced contralateral breast cancer. The Women’s Environmental, Cancer, and Radiation Epidemiology Study, an international population-based case–control study, collected patients with contralateral relapse and a cohort of survivors of unilateral BC. Among women who carried ATM missense mutations, those who were exposed to radiation had a statistically significantly higher risk of contralateral BC as compared to those with wild-type or subjects who did not undergo radiotherapy carrying the same predicted deleterious missense variant. Thus, the authors concluded that women who carry rare deleterious ATM missense variants and have been treated with radiation may have a more elevated risk of developing contralateral BC. This effect proved to be dose-dependent, and the risk of contralateral BC was greater in cases with ATM missense variants. The potential mechanism could be associated to the presence of rare missense variants effectively reducing the level of ATM activity, increasing the susceptibility to radiation-induced tumorigenesis [19].
5. Discussion: How Could the ATM Gene Mutation Influence BC Management?
Traditionally, gene testing or inherited BC genes has focused on women at high risk who have a family history of BC or who were diagnosed at an early age. In a recent study [49], mutations or variants in eight genes as BRCA1/2, PALB2, BARD1, RAD51C, ATM, and CHECK2 were found to be significantly associated with BC. To date, clinical practice also relies on the use of gene panel testing of unaffected women with a moderate risk of BC in the family history, in particular, counseling women with ATM gene mutations. The management of women with these mutations will consist of screening alone and magnetic resonance imaging (MRI) at the age of 40 years. Nowadays, clinicians are not ready to expand the gene panel test to the general population, and the ATM mutation is mostly diagnosed after the diagnosis of BC. Thus, the role of the ATM gene in predisposing to BC development seems to be limited. However, the present review aims to guide the clinician to use ATM in clinical practice during management of BC (Figure 2).
Firstly, we have learned that ATM gene mutations are correlated with specific clinical characteristics of BC such as high risk of ER-positive BC, grade two or three tumors, lymph node involvement, and HER2-positivity as well as the development of a contralateral breast tumor in patients resistant to radiotherapy. Thus, ATM mutations in BC patients are associated with a worse prognosis. These data could support clinicians in personalizing both treatments, as well as follow-up, in these patients. Moreover, since mutations in ATM encoding protein are involved in DNA repair mechanisms, ATM-aberrations may also sensitize BC cells to platinum drugs or PARP inhibitors in triple-negative BC, similarly to the effect of BRCA1 mutations. Some evidence suggests that ATM mutations could also be involved in resistance to CDK4/6 inhibitors in luminal positive BC. Relative to the triple-negative BC subtype, in the era of immunotherapy for early and advanced disease, the role of ATM mutations in predicting treatment efficacy and good or poor response to immune checkpoint inhibitors, such as PD1/PDL1 inhibitors, could be interesting. As in colon cancer, immunotherapy proved to be effective in patients with alterations of mismatch repair gene alterations; we could hypothesize that ATM mutations could enhance the genomic instability of DNA and enhance the immunotherapy response in triple-negative BC patients. ATM missense mutations among women diagnosed with BC before the age of 45 years could enhance the risk of contralateral breast cancer in radio-treated patients after a long latency period. In addition, although ATM is considered a tumor suppressor, ATM mutations can enhance chemo-radioresistance to tumor cells, and this could be useful to explore as a potential target for cancer treatment. In order to overcome the drug resistance in ATM-deficient tumors, some studies tested the ATR-checkpoint kinase 1 (Chk1) cascade as a potential target [74,75,76,77,78], showing an improved response to therapy of these tumors. Based on these findings, additional studies are needed to elucidate the unique characteristics of ATM-associated BC, which may have implications on personalized management, from diagnosis to treatment and follow-up of BC patients.
Author Contributions
L.S.S. formulated the idea of the work, V.I., M.P. and F.M. analyzed the literature and prepared the figures. M.T. and R.P. checked the manuscript. C.P. contributed to revision of the work. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moslemi, M.; Moradi, Y.; Dehghanbanadaki, H.; Afkhami, H.; Khaledi, M.; Sedighimehr, N.; Fathi, J.; Sohrabi, E. The association between ATM variants and risk of breast cancer: A systematic review and meta-analysis. BMC Cancer 2021, 21, 27. [Google Scholar] [CrossRef]
- Dörk, T.; Bendix, R.; Bremer, M.; Rades, D.; Klöpper, K.; Nicke, M.; Skawran, B.; Hector, A.; Yamini, P.; Steinmann, D.; et al. Spectrum of ATM Gene Mutations in a Hospital-Based Series of Unselected Breast Cancer Patients. Cancer Res. 2001, 61, 7608–7615. [Google Scholar]
- Bernstein, J.L.; Teraoka, S.; Southey, M.C.; Jenkins, M.A.; Andrulis, I.L.; Knight, J.A.; John, E.M.; Lapinski, R.; Wolitzer, A.L.; Whittemore, A.S. Population-Based Estimates of Breast Cancer Risks Associated with ATM Gene Variants c. 7271T> G and c. 1066–6T> G (IVS10–6T> G) from the Breast Cancer Family Registry. Hum. Mutat. 2006, 27, 1122–1128. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and Related Protein Kinases: Safeguarding Genome Integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S.; et al. A Single Ataxia Elangiectasia Gene with a Product Similar to PI-3 Kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef]
- Rotman, G.; Shiloh, Y. ATM: From Gene to Function. Hum. Mol. Genet. 1998, 7, 1555–1563. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Thompson, D.; Duedal, S.; Kirner, J.; McGuffog, L.; Last, J.; Reiman, A.; Byrd, P.; Taylor, M.; Easton, D.F. Cancer Risks and Mortality in Heterozygous ATM Mutation Carriers. J. Natl. Cancer Inst. 2005, 97, 813–822. [Google Scholar] [CrossRef]
- Goldgar, D.E.; Healey, S.; Dowty, J.G.; Da Silva, L.; Chen, X.; Spurdle, A.B.; Terry, M.B.; Daly, M.J.; Buys, S.M.; Southey, M.C.; et al. Rare Variants in the ATM Gene and Risk of Breast Cancer. Breast Cancer Res. 2011, 13, R73. [Google Scholar] [CrossRef] [Green Version]
- Angele, S.; Hall, J. The ATM Gene and Breast Cancer: Is It Really a Risk Factor? Mutat. Res. 2000, 462, 167–178. [Google Scholar] [CrossRef]
- Broeks, A.; Urbanus, J.H.; Floore, A.N.; Dahler, E.C.; Klijn, J.G.; Rutgers, E.J.T.; Devilee, P.; Russell, N.S.; van Leeuwen, F.E.; Veer, L.J.V. ATM-Heterozygous Germline Mutations Contribute to Breast Cancer-Susceptibility. Am. J. Hum. Genet. 2000, 66, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, O.; Johnson, N.; Silva, I.D.S.; Orr, N.; Ashworth, A.; Nevanlinna, H.; Heikkinen, T.; Aittomäki, K.; Blomqvist, C.; Burwinkel, B.; et al. Missense Variants in ATM in 26,101 Breast Cancer Cases and 29,842 Controls. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2143–2151. [Google Scholar] [CrossRef] [Green Version]
- Thorstenson, Y.R.; Roxas, A.; Kroiss, R.; Jenkins, M.A.; Yu, K.M.; Bachrich, T.; Muhr, D.; Wayne, T.L.; Chu, G.; Davis, R.W.; et al. Contributions of ATM Mutations to Familial Breast and Ovarian Cancer. Cancer Res. 2003, 63, 3325–3333. [Google Scholar]
- Cremona, C.A.; Behrens, A. ATM Signalling and Cancer. Oncogene 2014, 33, 3351–3360. [Google Scholar] [CrossRef] [Green Version]
- Negrini, M.; Rasio, D.; Hampton, G.M.; Sabbioni, S.; Rattan, S.; Carter, S.L.; Rosenberg, A.L.; Schwartz, G.F.; Shiloh, Y.; Cavenee, W.K. Definition and Refinement of Chromosome 11 Regions of Loss of Heterozygosity in Breast Cancer: Identification of a New Region at 11q23.3. Cancer Res. 1995, 55, 3003–3007. [Google Scholar]
- Laake, K.; Launonen, V.; Niederacher, D.; Gudlaugsdottir, S.; Seitz, S.; Rio, P.; Champème, M.H.; Bièche, I.; Birnbaum, D.; White, G.; et al. Loss of Heterozygosity at 11q23.1 and Survival in Breast Cancer: Results of a Large European Study. Breast Cancer Somatic Genet. Consort. Genes Chromosomes Cancer 1999, 25, 212–221. [Google Scholar] [CrossRef]
- Bueno, R.C.; Canevari, R.A.; Villacis, R.A.R.; Domingues, M.A.C.; Caldeira, J.R.F.; Rocha, R.M.; Drigo, S.A.; Rogatto, S.R. ATM Down-Regulation Is Associated with Poor Prognosis in Sporadic Breast Carcinomas. Ann. Oncol. 2014, 25, 69–75. [Google Scholar] [CrossRef]
- Berstein, J.L.; Haile, R.W.; Stovall, M.; Boice, J.D., Jr.; Shore, R.E.; Langholz, B.; Thomas, D.C.; Bernstein, L.; Charles, F.L.; Olsen, J.H.; et al. Radiation Exposure, the ATM Gene, and Contralateral Breast Cancer in the Women’s Environmental Cancer and Radiation Epidemiology Study. J. Natl. Cancer Inst. 2010, 102, 475–483. [Google Scholar] [CrossRef]
- Abraham, R.T. PI 3-Kinase Related Kinases: ‘Big’ Players in Stress-Induced Signaling Pathways. DNA Repair 2004, 3, 883–887. [Google Scholar] [CrossRef]
- Hall, M.J.; Bernhisel, R.; Hughes, E.; Larson, K.; Rosenthal, E.T.; Singh, N.A.; Lancaster, J.M.; Kurian, A.W. Germline Pathogenic Variants in the Ataxia telangiectasia Mutated (ATM) Gene Are Associated with High and Moderate Risks for Multiple Cancers. Cancer Prev. Res. 2021, 14, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Kernan, J.L.; Liu, P.H.; Sanda, T.; Logette, E.; Tschopp, J.; Look, A.T.; Wang, J.; Bouchier-Hayes, L.; Sidi, S. PIDD Death-Domain Phosphorylation by ATM Controls Prodeath versus Prosurvival PIDDosome Signaling. Mol. Cell 2012, 47, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Lin, C.; Wu, Z.; Gong, H.; Zeng, Y.; Wu, J.; Li, M.; Li, J. miR-18a Impairs DNA Damage Response Through Downregulation of Ataxia telangiectasia Mutated (ATM) Kinase. PLoS ONE 2011, 6, e25454. [Google Scholar] [CrossRef]
- Le Guezennec, X.; Bulavin, D.V. WIP1 Phosphatase at the Crossroads of Cancer and Aging. Trends Biochem. Sci. 2010, 35, 109–114. [Google Scholar] [CrossRef]
- Wang, L.; Mosel, A.J.; Oakley, G.G.; Peng, A. Deficient DNA Damage Signaling Leads to Chemoresistance to Cisplatin in Oral Cancer. Mol. Cancer Ther. 2012, 11, 2401–2409. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Xia, F.; Hermance, N.; Mabb, A.; Simonson, S.; Morrissey, S.; Gandhi, P.; Munson, M.; Miyamoto, S.; Kelliher, M.A. A Cytosolic ATM/NEMO/RIP1 Complex Recruits TAK1 to Mediate the NF-kappaB and p38 Mitogen-Activated Protein Kinase (MAPK)/MAPK-Activated Protein 2 Responses to DNA Damage. Mol. Cell. Biol. 2011, 31, 2774–2786. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Glass, J. The Phenotypic Radiation Resistance of CD44 þ/CD24(-or Low) Breast Cancer Cells Is Mediated Through the Enhanced Activation of ATM Signaling. PLoS ONE 2011, 6, e24080. [Google Scholar] [CrossRef]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin Remodeling Underlies the Senescence-Associated Secretory Phenotype of Tumor Stromal Fibroblasts That Supports Cancer Progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Stucci, L.S.; Tucci, M.; Passarelli, A.; Silvestris, F. Avβ3 Integrin: Pathogenetic Role in Osteotropic Tumors. Crit. Rev. Oncol. Hematol. 2015, 96, 183–193. [Google Scholar] [CrossRef]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Felici, C.; Stucci, L.S.; Cives, M.; Silvestris, F. Immune System Evasion as Hallmark of Melanoma Progression: The Role of Dendritic Cells. Front. Oncol. 2019, 9, 1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keimling, M.; Volcic, M.; Csernok, A.; Wieland, B.; Dörk, T.; Wiesmüller, L. Functional Characterization Connects Individual Patient Mutations in Ataxia telangiectasia Mutated (ATM) with Dysfunction of Specific DNA Double-Strand Break-Repair Signaling Pathways. FASEB J. 2011, 25, 3849–3860. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Wasielewski, M.; Martens, J.W.; Schutte, M. Discovering Moderate-Risk Breast Cancer Susceptibility Genes. Curr. Opin. Genet. Dev. 2010, 20, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of Somatic Mutation in Human Cancer Genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM Mutations in Patients with Hereditary Pancreatic Cancer. Cancer Discov. 2011, 2, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; et al. Somatic Mutations Affect Key Pathways in Lung Adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Sriramulu, S.; Ramachandran, M.; Subramanian, S.; Kannan, R.; Gopinath, M.; Sollano, J.; Bissi, L.; Banerjee, S.; Marotta, F.; Pathak, S. A Review on Role of ATM Gene in Hereditary Transfer of Colorectal Cancer. Acta Biomed. 2018, 89, 463–469. [Google Scholar]
- Squatrito, M.; Brennan, C.W.; Helmy, K.; Huse, J.T.; Petrini, J.H.; Holland, E.C. Loss of ATM/Chk2/p53 Pathway Components Accelerates Tumor Development and Contributes to Radiation Resistance in Gliomas. Cancer Cell 2010, 18, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Swift, M.; Reitnauer, P.J.; Morrell, D.; Chase, C.L. Breast and Other Cancers in Families with Ataxia-telangiectasia. N. Engl. J. Med. 1987, 316, 1289–1294. [Google Scholar] [CrossRef]
- Chen, J.; Birkholtz, G.G.; Lindblom, P.; Rubio, C.; Lindblom, A. The Role of Ataxia-telangiectasia Heterozygotes in Familial Breast Cancer. Cancer Res. 1998, 58, 1376–1379. [Google Scholar]
- Milne, R.L. Variants in the ATM Gene and Breast Cancer Susceptibility. Genome Med. 2009, 1, 12. [Google Scholar] [CrossRef] [Green Version]
- Athma, P.; Rappaport, R.; Swift, M. Molecular Genotyping Shows That Ataxia-telangiectasia Heterozygotes Are Predisposed to Breast Cancer. Cancer Genet. Cytogenet. 1996, 92, 130–134. [Google Scholar] [CrossRef]
- Inskip, H.M.; Kinlen, L.J.; Taylor, A.M.R.; Woods, C.G.; Arlett, C.F. Risk of Breast Cancer and Other Cancers in Heterozygotes for Ataxia-Telangiectasia. Br. J. Cancer 1999, 79, 1304–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Os, N.J.; Roeleveld, N.; Weemaes, C.M.; Jongmans, M.C.; Janssens, G.O.; Taylor, A.M.; Hoogerbrugge, N.; Willemsen, M.A. Health Risks for Ataxia-telangiectasia Mutated Heterozygotes: A Systematic Review, Meta-Analysis and Evidence-Based Guideline. Clin. Genet. 2016, 90, 105–117. [Google Scholar] [CrossRef]
- Southey, M.C.; Goldgar, D.E.; Winqvist, R.; Pylkäs, K.; Couch, F.; Tischkowitz, M.; Foulkes, W.D.; Dennis, J.; Michailidou, K.; van Rensburg, E.J.; et al. PALB2, CHEK2 and ATM Rare Variants and Cancer Risk: Data from COGS. J. Med. Genet. 2016, 53, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Marabelli, M.; Cheng, S.C.; Parmigiani, G. Penetrance of ATM Gene Mutations in Breast Cancer: A Meta-Analysis of Different Measures of Risk. Genet. Epidemiol. 2016, 40, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitui, M.; Nahas, S.; Du, L.; Yang, Z.; Lai, C.; Nakamura, K.; Arroyo, S.; Scott, S.; Purayidom, A.; Concannon, P.; et al. Functional and Computational Assessment of Missense Variants in the Ataxia-telangiectasia Mutated (ATM) Gene: Mutations with Increased Cancer Risk. Hum. Mutat. 2009, 30, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.-B.; Pan, X.-M.; Sun, H.; Wang, X.; Rao, L.; Li, L.-J.; Liang, W.-B.; Lv, M.-L.; Yang, W.-Z.; Zhang, L. The Association between ATM D1853N Polymorphism and Breast Cancer Susceptibility: A Meta-Analysis. J. Exp. Clin. Cancer Res. 2010, 29, 117. [Google Scholar] [CrossRef] [Green Version]
- Breast Cancer Association Consortium; Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; et al. Easton DF. Breast Cancer Risk Genes-Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar]
- Meng, Z.H.; Ben, Y.; Li, Z.; Chew, K.; Ljung, B.-M.; Lagios, M.D.; Dairkee, S.H. Aberrations of Breast Cancer Susceptibility Genes Occur Early in Sporadic Breast Tumors and in Acquisition of Breast Epithelial Immortalization. Genes Chromosomes Cancer 2004, 41, 214–222. [Google Scholar] [CrossRef]
- Vo, Q.N.; Kim, W.J.; Cvitanovic, L.; Boudreau, D.A.; Ginzinger, D.G.; Brown, K.D. The ATM Gene Is a Target for Epigenetic Silencing in Locally Advanced Breast Cancer. Oncogene 2004, 23, 9432–9437. [Google Scholar] [CrossRef] [Green Version]
- Ng, W.L.; Yan, D.; Zhang, X.; Mo, Y.-Y.; Wang, Y. Over-Expression of miR-100 Is Responsible for the Low-Expression of ATM in the Human Glioma Cell Line: M059J. DNA Repair 2010, 9, 1170–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannavola, F.; D’Oronzo, S.; Cives, M.; Stucci, L.S.; Ranieri, G.; Silvestris, F.; Tucci, M. Extracellular Vesicles and Epigenetic Modifications Are Hallmarks of Melanoma Progression. Int. J. Mol. Sci. 2019, 21, 52. [Google Scholar] [CrossRef] [Green Version]
- Pellerino, A.; Bruno, F.; Internò, V.; Rudà, R.; Soffietti, R. Current Clinical Management of Elderly Patients with Glioma. Expert Rev. Anticancer Ther. 2020, 20, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Cai, Q.; Dai, Q.; Shu, X.-O.; Shin, A.; Gao, Y.-T.; Zheng, W. Expression Patterns of the ATM Gene in Mammary Tissues and Their Associations with Breast Cancer Survival. Cancer 2007, 109, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Tommiska, J.; Bartkova, J.; Heinonen, M.; Hautala, L.; Kilpivaara, O.; Eerola, H.; Aittomäki, K.; Hofstetter, B.; Lukas, J.; von Smitten, K.; et al. The DNA Damage Signalling Kinase ATM Is Aberrantly Reduced or Lost in BRCA1/BRCA2-Deficient and ER/PR/ERBB2-Triple-Negative Breast Cancer. Oncogene 2008, 27, 2501–2506. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.L.; Sheu, L.F.; Yu, J.C.; Yang, T.L.; Chen, B.F.; Leu, F.J.; Shen, C.Y. Abnormality of the DNA Double-Strand-Break Checkpoint/Repair Genes, ATM, BRCA1 and TP53, in Breast Cancer Is Related to Tumour Grade. Br. J. Cancer 2004, 90, 1995–2001. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Reinhardt, H.C.; Bartkova, J.; Tommiska, J.; Blomqvist, C.; Nevanlinna, H.; Bartek, J.; Yaffe, M.B.; Hemann, M.T. The Combined Status of ATM and p53 Link Tumor Development with Therapeutic Response. Genes Dev. 2009, 23, 1895–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knappskog, S.; Chrisanthar, R.; Løkkevik, E.; Anker, G.; Østenstad, B.; Lundgren, S.; Risberg, T.; Mjaaland, I.; Leirvaag, B.; Miletic, H.; et al. Low Expression Levels of ATM May Substitute for CHEK2/TP53 Mutations Predicting Resistance Towards Anthracycline and Mitomycin Chemotherapy in Breast Cancer. Breast Cancer Res. 2012, 14, R47. [Google Scholar] [CrossRef] [Green Version]
- Konde, A.S.; Ivan, K.; Klavanian, J.; Rangarajan, T.; Jaiyesimi, I.A.; Zakalik, D. Heterozygous Germline ATM Mutations in Breast Cancer: A Single Academic Center Experience. J. Clin. Oncol. 2020, 38, 1537. [Google Scholar] [CrossRef]
- Stagni, V.; Manni, I.; Oropallo, V.; Mottolese, M.; Di Benedetto, A.; Piaggio, G.; Falcioni, R.; Giaccari, D.; Di Carlo, S.; Sperati, F.; et al. ATM Kinase Sustains HER2 Tumorigenicity in Breast Cancer. Nat. Commun. 2015, 6, 6886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, H.H.; Sun, X.; Nahas, S.A.; Teraoka, S.; Lai, C.-H.; Concannon, P.; Gatti, R.A. Improved Diagnostic Testing for Ataxia–telangiectasia by Immunoblotting of Nuclear Lysates for ATM Protein Expression. Mol. Genet. Metab. 2003, 80, 437–443. [Google Scholar] [CrossRef]
- Kabacik, S.; Ortega-Molina, A.; Efeyan, A.; Finnon, P.; Bouffler, S.; Serrano, M.; Badie, C. A Minimally Invasive Assay for Individual Assessment of the ATM/CHEK2/p53 Pathway Activity. Cell Cycle 2011, 10, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Rigakos, G.; Razis, E. BRCAness: Finding the Achilles Heel in Ovarian Cancer. Oncologist 2012, 17, 956–962. [Google Scholar] [CrossRef] [Green Version]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thorton, A.; Norquist, B.; Casadei, S.; Nord, A.S.; et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Williamson, C.T.; Kubota, E.; Hamill, J.D.; Klimowicz, A.; Ye, R.; Muzik, H.; Dean, M.; Tu, L.; Gilley, D.; Magliocco, A.M.; et al. Enhanced Cytotoxicity of PARP Inhibition in Mantle Cell Lymphoma Harbouring Mutations in Both ATM and p53. EMBO Mol. Med. 2012, 4, 515–527. [Google Scholar] [CrossRef]
- Kubota, E.; Williamson, C.T.; Ye, R.; Elegbede, A.; Peterson, L.; Lees-Miller, S.P.; Bebb, D.G. Low ATM Protein Expression and Depletion of p53 Correlates with Olaparib Sensitivity in Gastric Cancer Cell Lines. Cell Cycle 2014, 13, 2129–2137. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zou, W.; Zhang, J.; Zhang, Y.; Xu, Q.; Li, S.; Chen, C. Mechanisms of CDK4/6 Inhibitor Resistance in Luminal Breast Cancer. Front. Pharmacol. 2020, 11, 580251. [Google Scholar] [CrossRef]
- Anurag, M.; Punturi, N.; Hoog, J.; Bainbridge, M.N.; Ellis, M.J.; Haricharan, S. Comprehensive Profiling of DNA Repair Defects in Breast Cancer Identifies a Novel Class of Endocrine Therapy Resistance Drivers. Clin. Cancer Res. 2018, 24, 4887–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haricharan, S.; Punturi, N.; Singh, P.; Holloway, K.R.; Anurag, M.; Schmelz, J.; Schmidt, C.; Lei, J.T.; Suman, V.; Hunt, K.; et al. Loss of MutL Disrupts CHK2-Dependent Cell-Cycle Control Through CDK4/6 to Promote Intrinsic Endocrine Therapy Resistance in Primary Breast Cancer. Cancer Discov. 2017, 7, 1168–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.D.; Blades, K.; Foote, K.M.; Guichard, S.M.; Jewsbury, P.J.; McGuire, T. Discovery of AZD6738, a Potent and Selective Inhibitor with the Potential to Test the Clinical Efficacy of ATR Kinase Inhibition in Cancer Patients. Cancer Res. 2013, 73, 2348. [Google Scholar]
- Vendetti, F.P.; Lau, A.; Schamus, S.; Conrads, T.P.; O’Connor, M.J.; Bakkenist, C.J. The Orally Active and Bioavailable ATR Kinase Inhibitor AZD6738 Potentiates the Anti-Tumor Effects of Cisplatin to Resolve ATM-Deficient Non-Small Cell Lung Cancer In Vivo. Oncotarget 2015, 6, 44289–44305. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Fecteau, J.-F.; Brown, J.; Lau, A.; Kipps, T.J. Induction of Proliferation Sensitizes Chronic Lymphocytic Leukemic Cells to Apoptosis Mediated by the ATR Inhibitor AZD6738. Cancer Res. 2014, 74, 5485. [Google Scholar]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR Inhibition Induces Synthetic Lethality and Overcomes Chemore- Sistance in TP53- or ATM-Defective Chronic Lymphocytic Leukemia Cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Palii, S.S.; Innes, C.L.; Paules, R.S. Depletion of ATR Selectively Sensitizes ATM-Deficient Human Mammary Epithelial Cells to Ionizing Radiation and DNA-Damaging Agents. Cell Cycle 2014, 13, 3541–3550. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Structure of human ATM and function in DNA damage repair. (A) ATM is characterized by an N-terminal and a C-terminal half that shows homology to other phosphoinositide-3 kinase (PI3K)-like kinases and a portion that contains a FAT domain (named after the FRAP, ATR, and TRRAP proteins). The N-terminal portion interacts with substrates and cofactors such as NBS1, p53, and BRCA1. In addition, the N-terminal portion is characterized by a proposed chromatin- interaction domain, a nuclear localisation sequence (NLS), two caspase-3 cleavage sites, and a putative leucine zipper region. (B) Following DNA damage, ATM is recruited and is catalytically activated by autophosphorylation. Once activated, ATM serves as a transducer, phosphorylates, and activates other protein kinases such as checkpoint kinase 2 (CHK2), which in turn modulates its own substrates, resulting in cell cycle arrest, or ATM can also activate p53. ATM can activate mechanisms involved in DSB repair, such as BRCA proteins.
Figure 1.
Structure of human ATM and function in DNA damage repair. (A) ATM is characterized by an N-terminal and a C-terminal half that shows homology to other phosphoinositide-3 kinase (PI3K)-like kinases and a portion that contains a FAT domain (named after the FRAP, ATR, and TRRAP proteins). The N-terminal portion interacts with substrates and cofactors such as NBS1, p53, and BRCA1. In addition, the N-terminal portion is characterized by a proposed chromatin- interaction domain, a nuclear localisation sequence (NLS), two caspase-3 cleavage sites, and a putative leucine zipper region. (B) Following DNA damage, ATM is recruited and is catalytically activated by autophosphorylation. Once activated, ATM serves as a transducer, phosphorylates, and activates other protein kinases such as checkpoint kinase 2 (CHK2), which in turn modulates its own substrates, resulting in cell cycle arrest, or ATM can also activate p53. ATM can activate mechanisms involved in DSB repair, such as BRCA proteins.
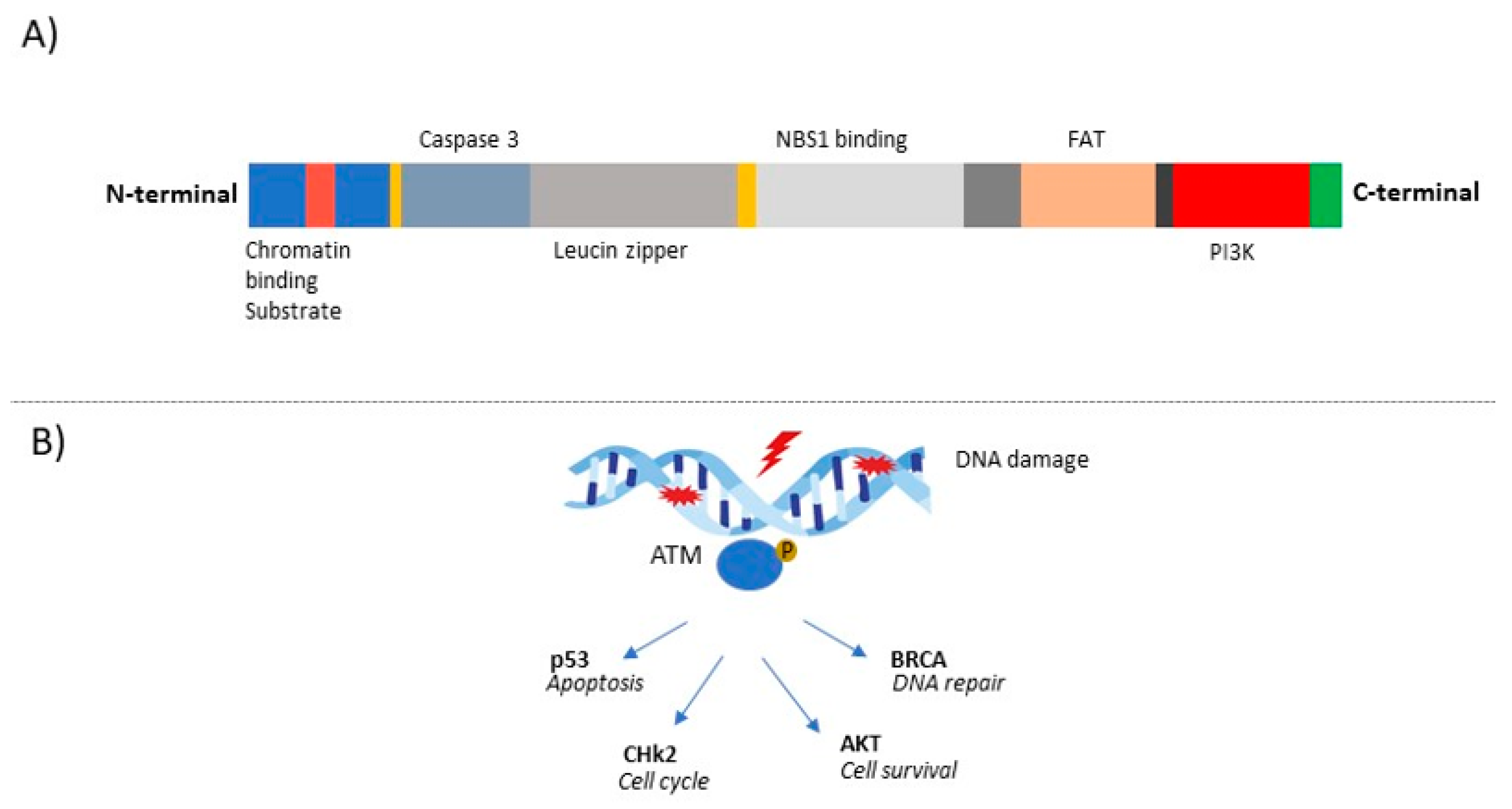
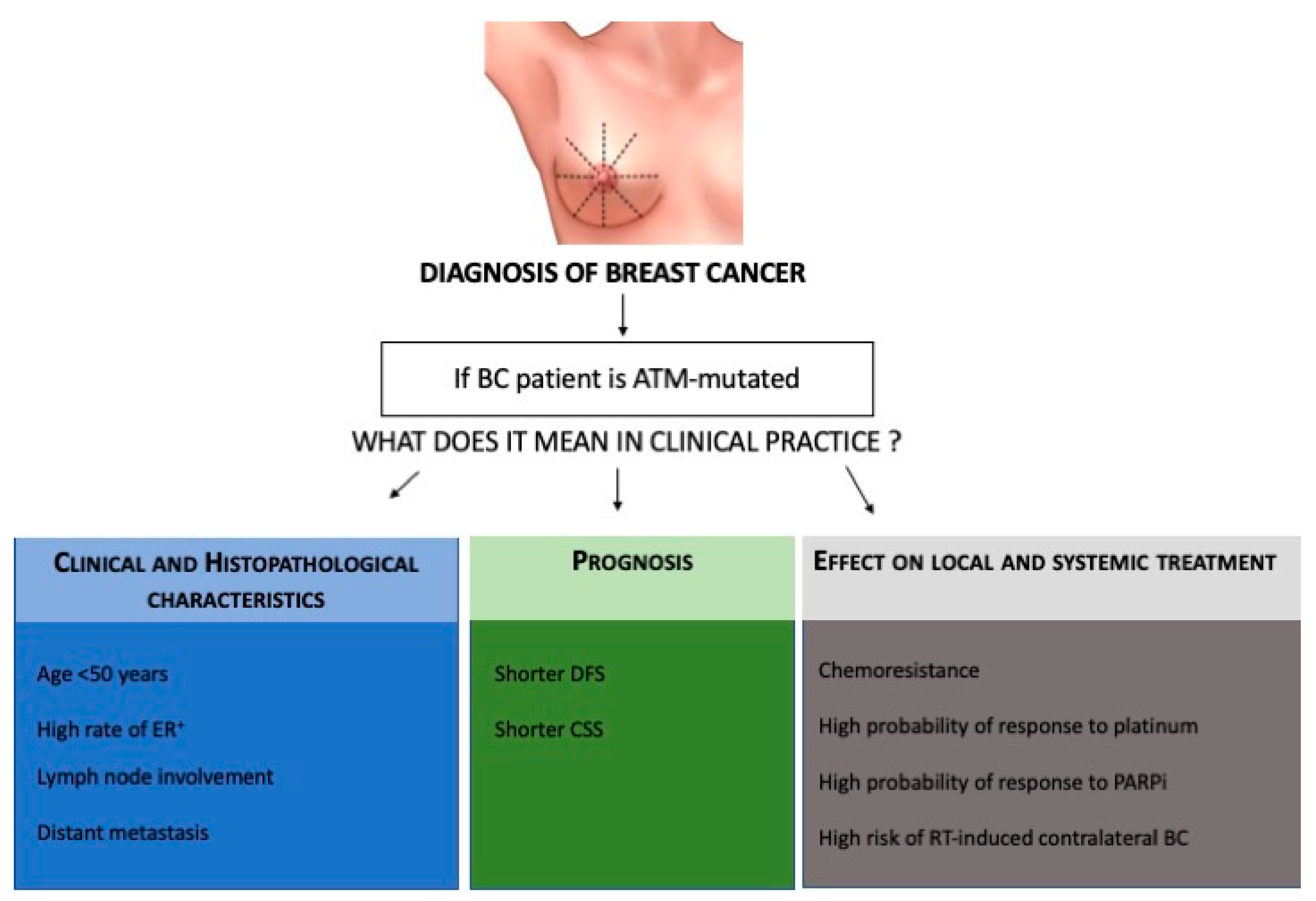
Figure 2.
Flow-chart of clinical management of ATM-mutated BC patients from diagnosis, treatment to follow-up. BC, breast cancer; ER, estrogen-receptor; DFS, disease-free survival; CSS, cancer-specific survival; PARPi, Poly (ADP-Ribose) Polymerase inhibitors.
Figure 2.
Flow-chart of clinical management of ATM-mutated BC patients from diagnosis, treatment to follow-up. BC, breast cancer; ER, estrogen-receptor; DFS, disease-free survival; CSS, cancer-specific survival; PARPi, Poly (ADP-Ribose) Polymerase inhibitors.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stucci, L.S.; Internò, V.; Tucci, M.; Perrone, M.; Mannavola, F.; Palmirotta, R.; Porta, C. The ATM Gene in Breast Cancer: Its Relevance in Clinical Practice. Genes 2021, 12, 727. https://doi.org/10.3390/genes12050727
AMA Style
Stucci LS, Internò V, Tucci M, Perrone M, Mannavola F, Palmirotta R, Porta C. The ATM Gene in Breast Cancer: Its Relevance in Clinical Practice. Genes. 2021; 12(5):727. https://doi.org/10.3390/genes12050727
Chicago/Turabian StyleStucci, Luigia Stefania, Valeria Internò, Marco Tucci, Martina Perrone, Francesco Mannavola, Raffaele Palmirotta, and Camillo Porta. 2021. "The ATM Gene in Breast Cancer: Its Relevance in Clinical Practice" Genes 12, no. 5: 727. https://doi.org/10.3390/genes12050727
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.