
Oxidative and Nitrosative Stress in Age-Related Macular Degeneration: A Review of Their Role in Different Stages of Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Role of Choriocapillaris (CC)/Bruch’s Membrane (BrM)/Retinal Pigment Epithelium (RPE)/Photoreceptor Complex in Different Forms of AMD
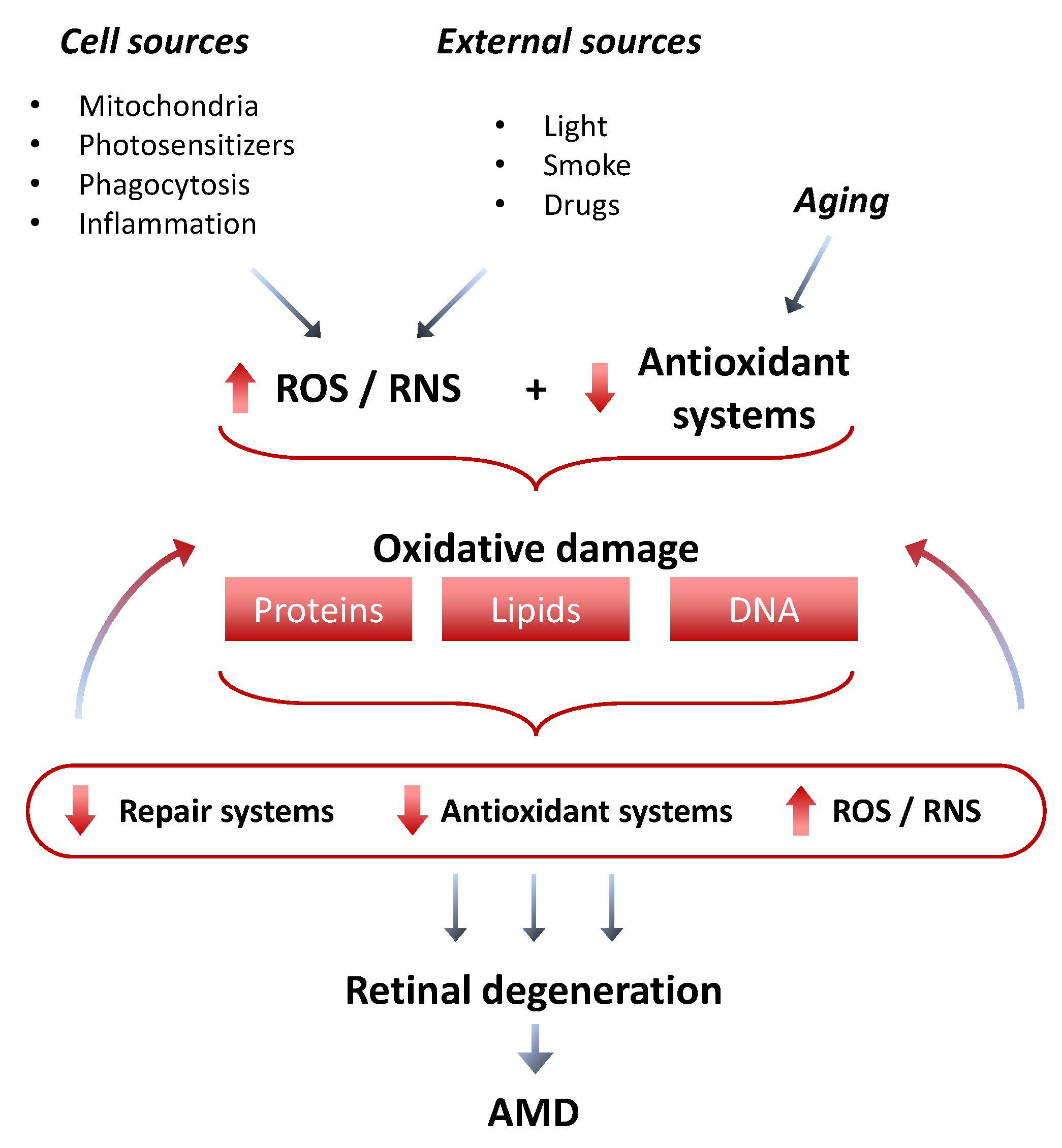
3. Oxidative Stress as the First Trigger for AMD Initiation
3.1. The Role of Mitochondria in ROS Production and Oxidative Damage in the Early Phases of AMD
3.2. Impairment of Autophagy and Oxidative Damage
4. Antioxidant Mechanisms in the Retina
The p62/Keap1/Nrf2 and PGC-1 Pathways
5. Oxidative Stress and Late Neovascular AMD
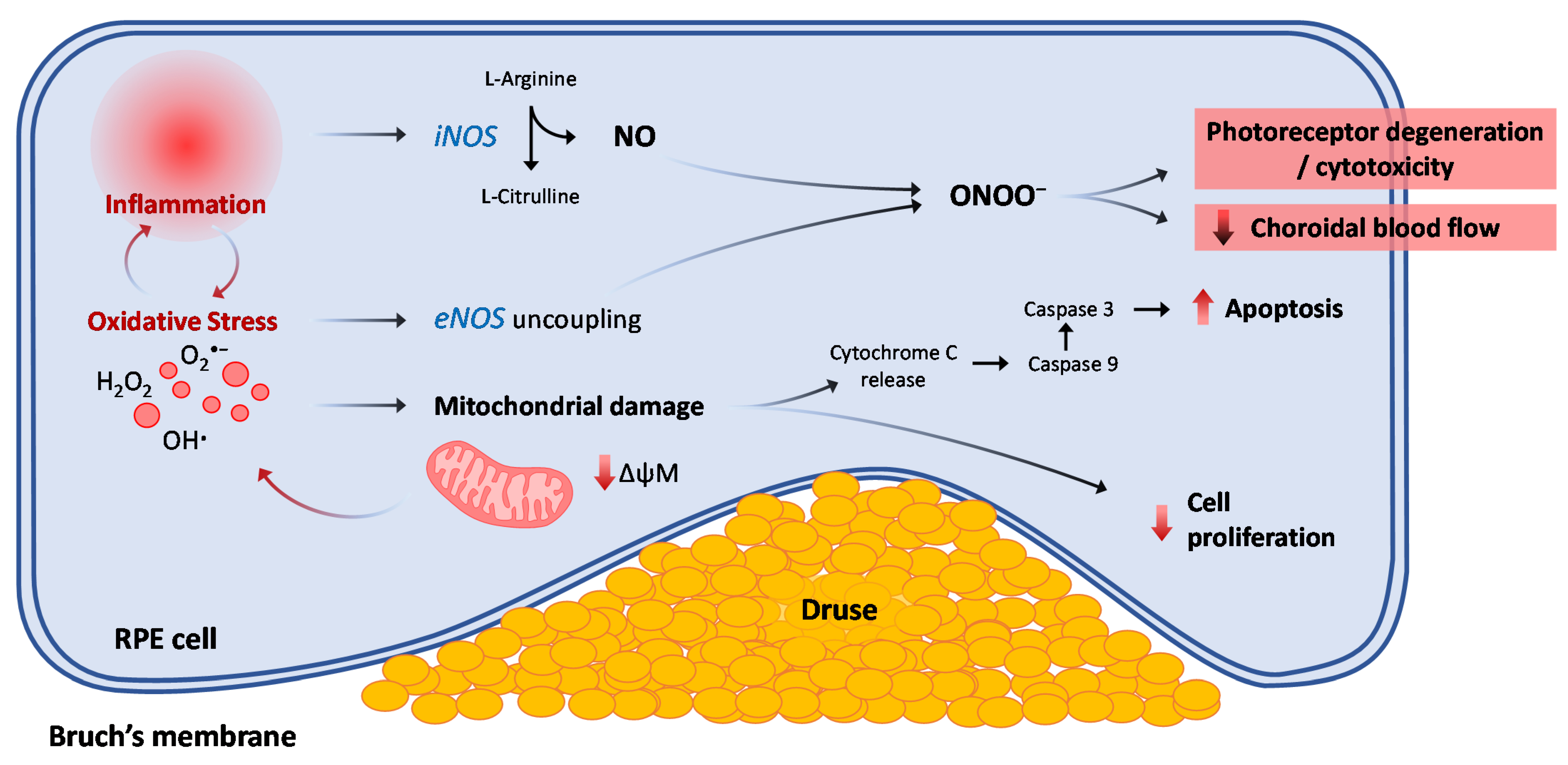
6. Nitric Oxide and Nitrosative Stress Role in AMD
Nitric Oxide and Nitrosative Stress in Late Neovascular AMD
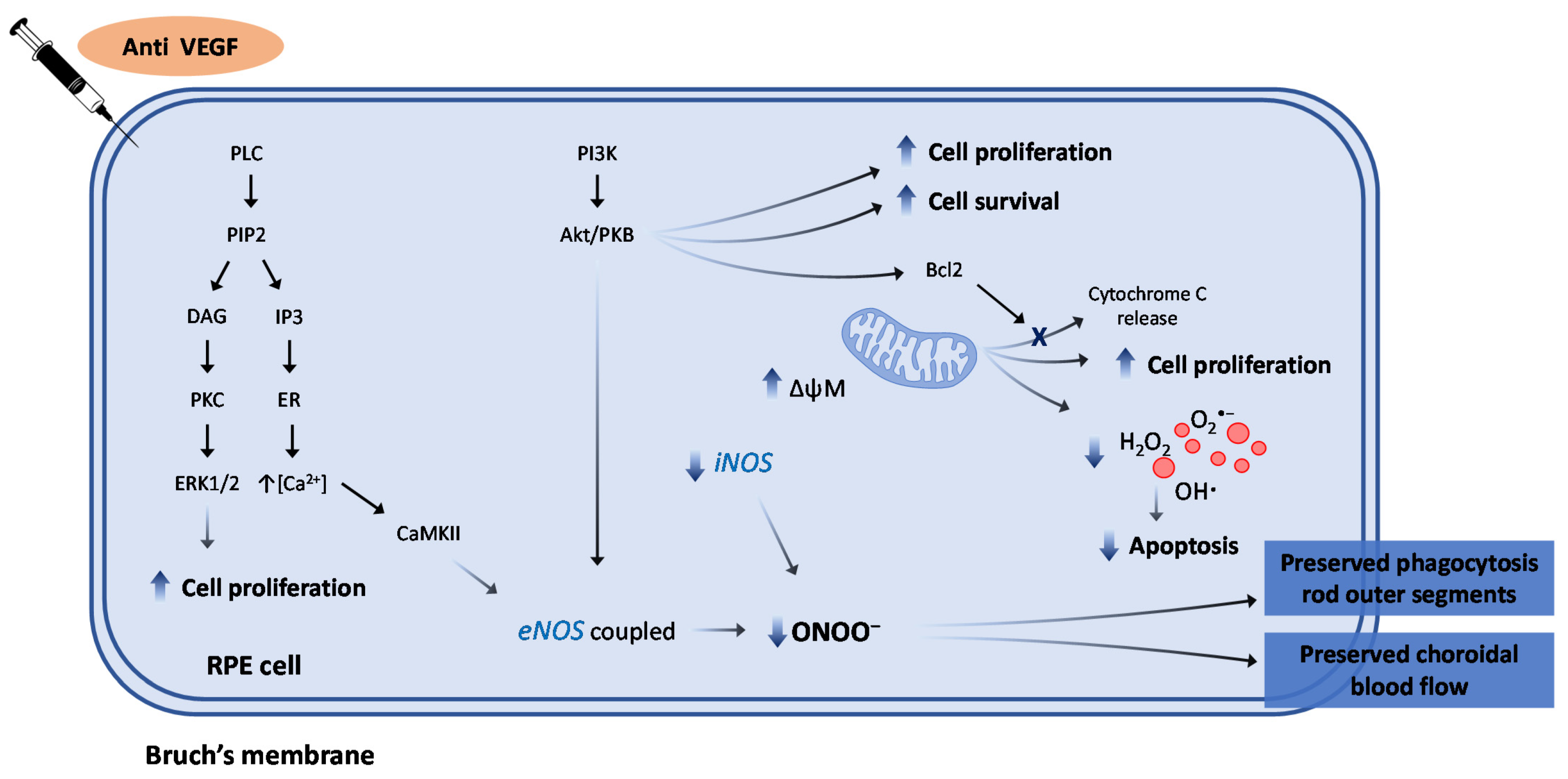
7. The Role of Anti-VEGF Agents against Oxidative/Nitrosative Stress
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, 106–116. [Google Scholar] [CrossRef] [Green Version]
- GBD 2019 Blindness and Vision Impairment Collaborators; Vision Loss Expert Group of the Global Burden of Disease Study. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: The Right to Sight: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Ferris, F.L.; Wilkinson, C.P.; Bird, A.; Chakravarthy, U.; Chew, E.; Csaky, K.; Sadda, S.R. Clinical classification of age-related macular degeneration. Ophthalmology 2013, 120, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Spaide, R.F.; Jaffe, G.J.; Sarraf, D.; Freund, K.B.; Sadda, S.R.; Staurenghi, G.; Waheed, N.K.; Chakravarthy, U.; Rosenfeld, P.J.; Holz, F.G.; et al. Consensus Nomenclature for Reporting Neovascular Age-Related Macular Degeneration Data: Consensus on Neovascular Age-Related Macular Degeneration Nomenclature Study Group. Ophthalmology 2020, 127, 616–636. [Google Scholar] [CrossRef] [PubMed]
- Heesterbeek, T.J.; Lorés-Motta, L.; Hoyng, C.B.; Lechanteur, Y.T.E.; den Hollander, A.I. Risk factors for progression of age-related macular degeneration. Ophthalmic Physiol. Opt. 2020, 40, 140–170. [Google Scholar] [CrossRef] [Green Version]
- Colijn, J.M.; Meester, M.; Verzijden, T.; de Breuk, A.; Silva, R.; Merle, B.M.J.; Cougnard-Grégoire, A.; Hoyng, C.B.; Fauser, S.; Coolen, T.; et al. Genetic risk, lifestyle, and AMD in Europe. The EYE-RISK consortium. Ophthalmology 2020. [Google Scholar] [CrossRef]
- Lambert, N.G.; ElShelmani, H.; Singh, M.K.; Mansergh, F.C.; Wride, M.A.; Padilla, M.; Keegan, D.; Hogg, R.E.; Ambati, B.K. Risk factors and biomarkers of age-related macular degeneration. Prog. Retin. Eye Res. 2016, 54, 64–102. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.; Assink, J.; Klein, R.; Mitchell, P.; Klaver, C.C.; Klein, B.E.; Hofman, A.; Jensen, S.; Wang, J.J.; De Jong, P.T. Risk factors for age-related macular degeneration: Pooled findings from three continents. Ophthalmology 2001, 108, 697–704. [Google Scholar] [CrossRef]
- Mitchell, P.; Wang, J.J.; Smith, W.; Leeder, S.R. Smoking and the 5-year incidence of age-related maculopathy: The Blue Mountains Eye Study. Arch. Ophthalmol. 2002, 120, 1357–1363. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.K.; Chong, E.W.; Williamson, E.; Aung, K.Z.; Makeyeva, G.A.; Giles, G.G.; English, D.R.; Hopper, J.; Guymer, R.H.; Baird, P.N.; et al. 20/20--Alcohol and age-related macular degeneration. Am. J. Epidemiol. 2012, 176, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-related macular degeneration: Genetics and biology coming together. Annu. Rev. Genom. Hum. Genet. 2014, 15, 151–171. [Google Scholar] [CrossRef] [Green Version]
- Tisi, A.; Feligioni, M.; Passacantando, M.; Ciancaglini, M.; Maccarono, R. The impact of oxidative stress on blood-retinal barrier physiology in age-related macular degeneration. Cells 2021, 10, 64. [Google Scholar] [CrossRef]
- Park, G.B.; Kim, D. Cigarette smoke-induced EGFR activation promotes Epithelial mesenchymal migration of human retinal pigment Epithelial cells through regulation of the fak-mediated Syk/Src pathway. Mol. Med. Rep. 2018, 17, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Kunchithapautham, K.; Atkinson, C.; Rohrer, B. Smoke exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J. Biol. Chem. 2014, 289, 14534–14546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, S.; Datta, S.; Wang, L.; Pegany, R.; Cano, M.; Handa, J.T. The impact of lipids, lipid oxidation, and inflammation on AMD, and the potential role of miRNAs on lipid metabolism in the RPE. Exp. Eye Res. 2019, 181, 346–355. [Google Scholar] [CrossRef]
- Schick, T.; Ersoy, L.; Lechanteur, Y.T.E.; Saksens, N.T.M.; Hoyng, C.B.; den Hollander, A.I.; Kirchhof, B.; Fauser, S. History of sunlight exposure is a risk factor for age-related macular degeneration. Retina 2016, 36, 787–790. [Google Scholar] [CrossRef]
- Sui, G.Y.; Liu, G.C.; Liu, G.Y.; Gao, Y.Y.; Deng, Y.; Wang, W.Y.; Tong, S.H.; Wang, L. Is sunlight exposure a risk factor for age-related macular degeneration? A systematic review and meta-analysis. Br. J. Ophthalmol. 2013, 97, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borras, C.; Canonica, J.; Jorieux, S.; Abache, T.; El Sanharawi, M.; Klein, C.; Delaunay, K.; Jonet, L.; Salvodelli, M.; Naud, M.C.; et al. CFH exerts anti-oxidant effects on retinal pigment epithelial cells independently from protecting against membrane attack complex. Sci. Rep. 2019, 9, 13873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weismann, D.; Hartvigsen, K.; Lauer, N.; Bennett, K.L.; Scholl, H.P.N.; Issa, P.C.; Cano, M.; Brandstätter, H.; Tsimikas, S.; Skerka, C.; et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 2011, 478, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.X.; Zhang, L.; Zhang, M.; Du, H.; Zhao, L.; Lee, C.; Grob, S.; Lim, S.L.; Hughes, G.; Lee, J.; et al. Complement factor H genotypes impact risk of age-related macular degeneration by interaction with oxidized phospholipids. Proc. Natl. Acad. Sci. USA 2012, 109, 13757–13762. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Redox theory of aging. Redox Biol. 2015, 5, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handa, J.T. How does the macula protect itself from oxidative stress? Mol. Asp. Med. 2012, 33, 418–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miceli, M.V.; Liles, M.R.; Newsome, D.A. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp. Cell Res. 1994, 214, 242–249. [Google Scholar] [CrossRef]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Asp. Med. 2012, 33, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangsa-Wirawan, N.D.; Linsenmeier, R.A. Retinal oxygen. Fundamental and clinical aspects. Arch. Ophthalmol. 2003, 21, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, J. The ageing retina: Physiology or pathology. Eye 1987, 1, 282–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, D.S.; Taomoto, M.; Otsuji, T.; Green, W.R.; Sunness, J.S.; Lutty, G.A. Quantifying changes in RPE and choriocapillaris in eyes with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1986–1993. [Google Scholar]
- Korte, G.E.; Repucci, V.; Henkind, P. RPE destruction causes choriocapillary atrophy. Investig. Ophthalmol. Vis. Sci. 1984, 25, 1135–1145. [Google Scholar]
- McLeod, D.S.; Grebe, R.; Bhutto, I.; Merges, C.; Baba, T.; Lutty, G.A. Relationship between RPE and choriocapillaris in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci 2009, 50, 4982–4991. [Google Scholar] [CrossRef] [Green Version]
- Zarbin, M.A.; Rosenfeld, P.J. Pathway-based therapies for age-related macular degeneration: An integrated survey of emerging treatment alternatives. Retina 2010, 30, 1350–1367. [Google Scholar] [CrossRef]
- Querques, G.; Rosenfeld, P.J.; Cavallero, E.; Borrelli, E.; Corvi, F.; Querques, L.; Bandello, F.M.; Zarbin, M.A. Treatment of dry age-related macular degeneration. Ophthalmic Res. 2014, 52, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, E.; Abdelfattah, N.S.; Uji, A.; Nittala, M.G.; Boyer, D.S.; Sadda, S.R. Postreceptor neuronal loss in intermediate age-related macular degeneration. Am. J. Ophthalmol. 2017, 181, 1–11. [Google Scholar] [CrossRef]
- Vujosevic, S.; Toma, C.; Villani, E.; Muraca, A.; Torti, E.; Florimbi, G.; Pezzotti, M.; Nucci, P.; De Cillà, S. Quantitative choriocapillaris evaluation in intermediate age-related macular degeneration by swept-source optical coherence tomography angiography. Acta Ophthalmol. 2019, 97, e919–e926. [Google Scholar] [CrossRef]
- Grunwald, J.E.; Metelitsina, T.I.; Dupont, J.C.; Ying, G.S.; Maguire, M.G. Reduced foveolar choroidal blood flow in eyes with increasing AMD severity. Investig. Ophthalmol. Vis. Sci 2005, 46, 1033–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutty, G.A.; McLeod, D.S.; Bhutto, I.A.; Edwards, M.M.; Seddon, J.M. Choriocapillaris dropout in early age-related macular degeneration. Exp. Eye Res. 2020, 192, 107939. [Google Scholar] [CrossRef]
- Seddon, J.M.; McLeod, D.S.; Bhutto, I.A.; Villalonga, M.B.; Silver, R.E.; Wenick, A.S.; Edwards, M.M.; Lutty, G.A. Histopathological Insights into Choroidal Vascular Loss in Clinically Documented Cases of Age-Related Macular Degeneration. JAMA Ophthalmol. 2016, 134, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, I.A.; McLeod, D.S.; Hasegawa, T.; Kim, S.Y.; Merges, C.; Tong, P.; Lutty, G.A. Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degeneration. Exp. Eye Res. 2006, 82, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellezza, I. Oxidative Stress in Age-Related Macular Degeneration: Nrf2 as Therapeutic Target. Front. Pharmacol. 2018, 9, 1280. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Luo, C.; Zhao, J.; Devarajan, G.; Xu, H. Immune regulation in the aging retina. Prog. Retin. Eye Res. 2019, 69, 159–172. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.C.; Hu, D.N.; Gao, H.X.; Chen, M.; Wang, D.; Rosen, R.; McCormick, S.A. Subtoxic levels hydrogen peroxide-induced production of interleukin-6 by retinal pigment epithelial cells. Mol. Vis. 2010, 16, 1864–1873. [Google Scholar]
- Glotin, A.L.; Debacq-Chainiaux, F.; Brossas, J.Y.; Faussat, A.M.; Treton, J.; Zubielewicz, A.; Toussaint, O.; Mascarelli, F. Prematurely senescent ARPE-19 cells display features of age-related macular degeneration. Free Radic. Biol. Med. 2008, 44, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Chaum, E.; Yin, J.; Lang, J.C. Molecular responses transduced by serial oxidative stress in the retinal pigment epithelium: Feedback control modeling of gene expression. Neurochem. Res. 2011, 36, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Strunnikova, N.; Zhang, C.; Teichberg, D.; Cousins, S.W.; Baffi, J.; Becker, K.G.; Csaky, K.G. Survival of retinal pigment epithelium after exposure to prolonged oxidative injury: A detailed gene expression and cellular analysis. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3767–3777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigel, A.L.; Handa, J.T.; Hjelmeland, L.M. Microarray analysis of H2O2-, HNE-, or tBH-treated ARPE-19 cells. Free Radic. Biol. Med. 2002, 33, 1419–1432. [Google Scholar] [CrossRef]
- Weigel Andrea, L.; Ida, H.; Boylan, S.A.; Hjelmeland, L.M. Acute hyperoxia-induced transcriptional response in the mouse RPE/choroid. Free Radic. Biol. Med. 2003, 35, 465–474. [Google Scholar] [CrossRef]
- Lu, L.; Hackett, S.F.; Mincey, A.; Lai, H.; Campochiaro, P.A. Effects of different types of oxidative stress in RPE cells. J. Cell Physiol. 2006, 206, 119–125. [Google Scholar] [CrossRef]
- Rabin, D.M.; Rabin, R.L.; Blenkinsop, T.A.; Temple, S.; Stern, J.H. Chronic oxidative stress upregulates Drusen-related protein expression in adult human RPE stem cell-derived RPE cells: A novel culture model for dry AMD. Aging 2013, 5, 51–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justilien, V.; Pang, J.J.; Renganathan, K.; Zhan, X.; Crabb, J.W.; Kim, S.R.; Sparrow, J.R.; Hauswirth, W.W.; Lewin, A.S. SOD2 knockdown mouse model of early AMD. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4407–4420. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-related retinopathy in NRF2-deficient mice. PLoS ONE 2011, 6, e19456. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Herrmann, R.; Bednar, A.; Saloupis, P.; Dwyer, M.A.; Yang, P.; Qi, X.; Thomas, R.S.; Jaffe, G.J.; Boulton, M.E.; et al. Aryl hydrocarbon receptor deficiency causes dysregulated cellular matrix metabolism and age-related macular degeneration-like pathology. Proc. Natl. Acad. Sci. USA 2013, 110, E4069–E4078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.Y.; Yang, H.J.; Chang, Y.S.; Kim, J.W.; Brooks, M.; Chew, E.Y.; Wong, W.T.; Fariss, R.N.; Rachel, R.A.; Cogliati, T.; et al. Deletion of aryl hydrocarbon receptor AHR in mice leads to subretinal accumulation of microglia and RPE atrophy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6031–6040. [Google Scholar] [CrossRef] [PubMed]
- Sickel, W. Retinal metabolism in dark and light. In Handbook of Sensory Physiology, 1st ed.; Fuortes, M.G.F., Ed.; Springer: Berlin/Heidelberg, Germany, 1972; Volume 7/2, pp. 667–727. [Google Scholar]
- Kortuem, K.; Geiger, L.K.; Levin, L.A. Differential susceptibility of retinal ganglion cells to reactive oxygen species. Investig. Ophtalmol. Vis. Sci. 2000, 41, 3176–3183. [Google Scholar]
- Beatty, S.; Koh, H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Marquioni-Ramella, M.D.; Suburo, A.M. Photo-damage, photo-protection and age-related macular degeneration. Photochem. Photobiol. Sci. 2015, 14, 1560–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefánsson, E.; Geirsdóttir, A.; Sigurdsson, H. Metabolic physiology in age related macular degeneration. Prog. Retin. Eye Res. 2011, 30, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Stefánsson, E.; Olafsdottir, O.B.; Eliasdottir, T.S.; Vehmeijer, W.; Einarsdottir, A.B.; Bek, T.; Torp, T.L.; Grauslund, J.; Eysteinsson, T.; Karlsson, R.A.; et al. Retinal oximetry: Metabolic imaging for diseases of the retina and brain. Prog. Retin. Eye Res. 2019, 70, 1–22. [Google Scholar] [CrossRef]
- Eells, J.T. Mitochondrial dysfunction in the aging retina. Biology 2019, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Blasiak, J.; Petrovski, G.; Veréb, Z.; Facskó, A.; Kaarniranta, K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. Biomed. Res. Int. 2014, 2014, 768026. [Google Scholar] [CrossRef] [PubMed]
- Roth, F.; Bindewald, A.; Holz, F.G. Keypathophysiologic pathways in age-related macular disease. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 710–716. [Google Scholar] [CrossRef]
- Stone, W.L.; Farnsworth, C.C.; Dratz, E.A. A reinvestigation of the fatty acid content of bovine, rat and frog retinal rod outer segments. Exp. Eye Res. 1979, 28, 387–397. [Google Scholar] [CrossRef]
- Arstila, A.U.; Smith, M.A.; Trump, B.F. Microsomal lipid peroxidation: Morphological characterization. Science 1972, 175, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef]
- Tate, D.J., Jr.; Miceli, M.V.; Newsome, D.A. Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1271–1279. [Google Scholar]
- Rózanowska, M.; Jarvis-Evans, J.; Korytowski, W.; Boulton, M.E.; Burke, J.M.; Sarna, T. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J. Biol. Chem. 1995, 270, 18825–18830. [Google Scholar] [CrossRef] [Green Version]
- Sparrow, J.R.; Nakanishi, K.; Parish, C.A. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1981–1989. [Google Scholar]
- Shamsi, F.A.; Boulton, M. Inhibition of RPE lysosomal and antioxidant activity by the age pigment lipofuscin. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3041–3046. [Google Scholar]
- Kopitz, J.; Holz, F.G.; Kaemmerer, E.; Schutt, F. Lipids and lipid peroxidation products in the pathogenesis of age-related macular degeneration. Biochimie 2004, 86, 825–831. [Google Scholar] [CrossRef]
- Kaidzu, S.; Tanito, M.; Ohira, A.; Umeda, S.; Suzuki, M.; Yoshikawa, Y.; Iwata, T. Immunohistochemical analysis of aldehyde-modified proteins in drusen in cynomolgus monkeys (Macaca fascicularis). Exp. Eye Res. 2008, 86, 856–859. [Google Scholar] [CrossRef]
- Ethen, C.M.; Reilly, C.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Age-related macular degeneration and retinal protein modification by 4-hydroxy-2-nonenal. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3469–3479. [Google Scholar] [CrossRef] [Green Version]
- Tanito, M.; Elliott, M.H.; Kotake, Y.; Anderson, R.E. Protein modifications by 4-hydroxynonenal and 4-hydroxyhexenal in light-exposed rat retina. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3859–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskol, G.; Karakucuk, S.; Oner, A.O.; Baskol, M.; Kocer, D.; Mirza, E.; Saraymen, R.; Ustdal, M. Serum paraoxonase 1 activity and lipid peroxidation levels in patients with age-related macular degeneration. Ophthalmologica 2006, 220, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Schutt, F.; Bergmann, M.; Holz, F.G.; Kopitz, J. Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3663–3668. [Google Scholar] [CrossRef] [PubMed]
- Totan, Y.; Yagci, R.; Bardak, Y.; Ozyurt, H.; Kendir, F.; Yilmaz, G.; Sahin, S.; Sahin Tig, U. Oxidative macromolecular damage in age-related macular degeneration. Curr. Eye Res. 2009, 34, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med. 2008, 14, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Kaneko, H.; Hayashi, Y.; Takayama, K.; Hwang, S.J.; Nishizawa, Y.; Kimoto, R.; Nagasaka, Y.; Tsunekawa, T.; Matsuura, T. Malondialdehyde induces autophagy dysfunction and VEGF secretion in the retinal pigment epithelium in age-related macular degeneration. Free Radic. Biol. Med. 2016, 94, 121–134. [Google Scholar] [CrossRef]
- Yadav, U.C.; Ramana, K.V. Regulation of NF-κB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid Med. Cell Longev. 2013, 2013, 690545. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Kambhampati, S.P.; Bhutto, I.A.; McLeod, D.S.; Lutty, G.A.; Kannan, R.M. Evolution of oxidative stress, inflammation and neovascularization in the choroid and retina in a subretinal lipid induced age-related macular degeneration model. Exp. Eye Res. 2021, 203, 108391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, L.; Weinreb, R.N. Ophthalmic drug discovery: Novel targets and mechanisms for retinal diseases and glaucoma. Nat. Rev. Drug Discov. 2012, 11, 541–559. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Tsujikawa, M.; Itabe, H.; Du, Z.J.; Xie, P.; Matsumura, N.; Fu, X.; Zhang, R.; Sonoda, K.H.; Egashira, K.; et al. Chronic photo-oxidative stress and subsequent MCP-1 activation as causative factors for age-related macular degeneration. J. Cell Sci. 2012, 125, 2407–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, M.; Thimmalappula, R.; Fujihara, M.; Nagai, N.; Sporn, M.; Wang, A.L.; Neufeld, A.H.; Biswal, S.; Handa, J.T. Cigarette smoking, oxidative stress, the anti-oxidant response through Nrf2 signaling, and Age-related Macular Degeneration. Vis. Res. 2010, 50, 652–664. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.C.; Wilkinson Berka, J.L.; Deliyanti, D.; Hunter, D.; Fung, A.; Liew, G.; White, A. The role of reactive oxygen species in the pathogenesis and treatment of retinal diseases. Exp. Eye Res. 2020, 201, 108255. [Google Scholar] [CrossRef]
- Gao, J.; Liu, R.T.; Cao, S.; Cui, J.Z.; Wang, A.; To, E.; Matsubara, J.A. NLRP3 inflammasome: Activation and regulation in age-related macular degeneration. Mediat. Inflamm. 2015, 2015, 690243. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Salminen, A. Age-related macular degeneration: Activation of innate immunity system via pattern recognition receptors. J. Mol. Med. (Berl.) 2009, 87, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Piippo, N.; Korhonen, E.; Hytti, M.; Kinnunen, K.; Kaarniranta, K.; Kauppinen, A. Oxidative Stress is the Principal Contributor to Inflammasome Activation in Retinal Pigment Epithelium Cells with Defunct Proteasomes and Autophagy. Cell Physiol. Biochem. 2018, 49, 359–367. [Google Scholar] [CrossRef]
- Wang, K.; Yao, Y.; Zhu, X.; Zhang, K.; Zhou, F.; Zhu, L. Amyloid β induces NLRP3 inflammasome activation in retinal pigment epithelial cells via NADPH oxidase- and mitochondria-dependent ROS production. J. Biochem. Mol. Toxicol. 2017, 31, e21887. [Google Scholar] [CrossRef]
- Madeira, M.H.; Rashid, K.; Ambrósio, A.F.; Santiago, A.R.; Langmann, T. Blockade of microglial adenosine A2A receptor impacts inflammatory mechanisms, reduces ARPE-19 cell dysfunction and prevents photoreceptor loss in vitro. Sci. Rep. 2018, 8, 2272. [Google Scholar] [CrossRef] [Green Version]
- Totsuka, K.; Ueta, T.; Uchida, T.; Roggia, M.F.; Nakagawa, S.; Vavvas, D.G.; Honjo, M.; Aihara, M. Oxidative stress induces ferroptotic cell death in retinal pigment epithelial cells. Exp. Eye Res. 2019, 181, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Sharma, R.; Chaudhary, P.; Vatsyayan, R.; Pearce, V.; Jeyabal, P.V.; Zimniak, P.; Awasthi, S.; Awasthi, Y.C. 4-Hydroxynonenal induces p53-mediated apoptosis in retinal pigment epithelial cells. Arch. Biochem. Biophys. 2008, 480, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Hanus, J.; Zhang, H.; Wang, Z.; Liu, Q.; Zhou, Q.; Wang, S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis. 2013, 4, e965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanus, J.; Anderson, C.; Sarraf, D.; Ma, J.; Wang, S. Retinal pigment epithelial cell necroptosis in response to sodium iodate. Cell Death Dis. 2016, 2, 16054. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y.; Fan, B.; Zheng, Y.C. Calcium overload is a critical step in programmed necrosis of ARPE-19 cells induced by high-concentration H2O2. Biomed. Environ. Sci. 2010, 23, 371–377. [Google Scholar] [CrossRef]
- Murakami, Y.; Matsumoto, H.; Roh, M.; Giani, A.; Kataoka, K.; Morizane, Y.; Kayama, M.; Thanos, A.; Nakatake, S.; Notomi, S.; et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ. 2014, 21, 270–277. [Google Scholar] [CrossRef]
- Yang, M.; So, K.F.; Lam, W.C.; Lo, A.C.Y. Novel Programmed Cell Death as Therapeutic Targets in Age-Related Macular Degeneration? Int. J. Mol. Sci. 2020, 21, 7279. [Google Scholar] [CrossRef]
- Juel, H.B.; Faber, C.; Svendsen, S.G.; Vallejo, A.N.; Nissen, M.H. Inflammatory cytokines protect retinal pigment epithelial cells from oxidative stress-induced death. PLoS ONE 2013, 8, e64619. [Google Scholar] [CrossRef]
- Malek, G.; Dwyer, M.; McDonnell, D. Exploring the potential role of the oxidant-activated transcription factor aryl hydrocarbon receptor in the pathogenesis of AMD. Adv. Exp. Med. Biol. 2012, 723, 51–59. [Google Scholar]
- Blasiak, J.; Pawlowska, E.; Sobczuk, A.; Szczepanska, J.; Kaarniranta, K. The Aging Stress Response and Its Implication for AMD Pathogenesis. Int. J. Mol. Sci. 2020, 21, 8840. [Google Scholar] [CrossRef] [PubMed]
- Barot, M.; Gokulgandhi, M.R.; Mitra, A.K. Mitochondrial dysfunction in retinal diseases. Curr. Eye Res. 2011, 36, 1069–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, J.S. Mitochondrial membrane permeabilization: The sine qua non for cell death. Bioessays 2006, 28, 253–260. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, Y.; Juhaszova, M.; Sollott, S.J. Age-related changes of myocardial ATP supply and demand mechanisms. Trends Endocrinol. Metab. 2013, 24, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Bannikova, S.Y.; Belousov, V.V.; Vyssokikh, M.Y.; Zorova, L.D.; Isaev, N.K.; Krasnikov, B.F.; Plotnikov, E.Y. Reactive oxygen and nitrogen species: Friends or foes? Biochemistry 2005, 70, 215–221. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218. [Google Scholar] [CrossRef]
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Balacco Gabrieli, C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar] [CrossRef]
- Nordgaard, C.L.; Karunadharma, P.P.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2848–2855. [Google Scholar] [CrossRef]
- Blasiak, J.; Glowacki, S.; Kauppinen, A.; Kaarniranta, K. Mitochondrial and nuclear DNA damage and repair in age-related macular degeneration. Int. J. Mol. Sci. 2013, 14, 2996–3010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allio, R.; Donega, S.; Galtier, N.; Nabholz, B. Large variation in the ratio of mitochondrial to nuclear mutation rate across animals: Implications for genetic diversity and the use of mitochondrial DNA as a molecular marker. Mol. Biol. Evol. 2017, 34, 2762–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badia, A.; Salas, A.; Duarri, A.; Ferreira-de-Souza, B.; Zapata, M.Á.; Fontrodona, L.; García-Arumí, J. Transcriptomics analysis of Ccl2/Cx3cr1/Crb1rd8 deficient mice provides new insights into the pathophysiology of progressive retinal degeneration. Exp. Eye Res. 2021, 203, 108424. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Lin, H.; Godley, B.F.; Boulton, M.E. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog. Retin. Eye Res. 2008, 27, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.Q.; Godley, B.F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res. 2003, 76, 397–403. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Mitter, S.K.; Song, C.; Qi, X.; Mao, H.; Rao, H.; Akin, D.; Lewin, A.; Grant, M.; Dunn, W., Jr.; Ding, J.; et al. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy 2014, 10, 1989–2005. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Schaaf, M.B.; Keulers, T.G.; Vooijs, M.A.; Rouschop, K.M. LC3/GABARAP family proteins: Autophagy-(un)related functions. FASEB J. 2016, 30, 3961–3978. [Google Scholar] [CrossRef] [Green Version]
- Kaarniranta, K.; Tokarz, P.; Koskela, A.; Paterno, J.; Blasiak, J. Autophagy regulates death of retinal pigment epithelium cells in age-related macular degeneration. Cell Biol. Toxicol. 2017, 33, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Sukseree, S.; Chen, Y.T.; Laggner, M.; Gruber, F.; Petit, V.; Nagelreiter, I.M.; Mlitz, V.; Rossiter, H.; Pollreisz, A.; Schmidt-Erfurth, U.; et al. Tyrosinase-Cre-Mediated Deletion of the Autophagy Gene Atg7 Leads to Accumulation of the RPE65 Variant M450 in the Retinal Pigment Epithelium of C57BL/6 Mice. PLoS ONE 2016, 11, e0161640. [Google Scholar] [CrossRef]
- Zhang, Y.; Cross, S.D.; Stanton, J.B.; Marmorstein, A.D.; Le, Y.Z.; Marmorstein, L.Y. Early AMD-like defects in the RPE and retinal degeneration in aged mice with RPE-specific deletion of Atg5 or Atg7. Mol. Vis. 2017, 23, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Jia, L.; Khan, N.; Lin, C.; Mitter, S.K.; Boulton, M.E.; Dunaief, J.L.; Klionsky, D.J.; Guan, J.L.; Thompson, D.A.; et al. Deletion of autophagy inducer RB1CC1 results in degeneration of the retinal pigment epithelium. Autophagy 2015, 11, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, M.; Frennesson, C.; Gustafsson, T.; Brunk, U.T.; Nilsson, S.E.; Kurz, T. Autophagy of iron-binding proteins may contribute to the oxidative stress resistance of ARPE-19 cells. Exp. Eye Res. 2013, 116, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2018, 8, e2537. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.T.; Błasiak, J.; Niittykoski, M.; Kinnunen, K.; Kauppinen, A.; Salminen, A. DNA damage response and autophagy in the degeneration of retinal pigment epithelial cells-Implications for age-related macular degeneration (AMD). Ageing Res. Rev. 2017, 36, 64–77. [Google Scholar] [CrossRef]
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef]
- Bienert, G.P.; Møller, A.L.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St Clair, D.K. Manganese superoxide dismutase: Guardian of the powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef]
- Decanini, A.; Nordgaard, C.L.; Feng, X.; Ferrington, D.A.; Olsen, T.W. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am. J. Ophthalmol. 2007, 143, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 233–260. [Google Scholar] [CrossRef]
- Rangasamy, T.; Cho, C.Y.; Thimmulappa, R.K.; Zhen, L.; Srisuma, S.S.; Kensler, T.W.; Yamamoto, M.; Petrache, I.; Tuder, R.M.; Biswal, S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Investig. 2004, 114, 1248–1259. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Mai, K.H.; Srisuma, S.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [Google Scholar] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Kensler, T.W. The role of Keap1 in cellular protective responses. Chem. Res. Toxicol. 2005, 18, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yamamoto, M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, N.; Itoh, K.; Wakabayashi, J.; Motohashi, H.; Noda, S.; Takahashi, S.; Imakado, S.; Kotsuji, T.; Otsuka, F.; Roop, D.R.; et al. Keap1-null mutation leads tompostnatal lethality due to constitutive Nrf2 activation. Nat. Genet. 2003, 35, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kondo, N.; Cano, M.; Ebrahimi, K.; Yoshida, T.; Barnett, B.P.; Biswal, S.; Handa, J.T. Nrf2 signaling modulates cigarette smoke-induced complement activation in retinal pigmented epithelial cells. Free Radic. Biol. Med. 2014, 70, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Bao, X.L.; Cong, Y.Y.; Fan, B.; Li, G.Y. Autophagy in Age-Related Macular Degeneration: A Regulatory Mechanism of Oxidative Stress. Oxid. Med. Cell. Longev. 2020, 8, 2896036. [Google Scholar] [CrossRef] [PubMed]
- Myeku, N.; Figueiredo-Pereira, M.E. Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: Association with sequestosome 1/p62. J. Biol. Chem. 2011, 286, 22426–22440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Chu, Y.; Mowery, J.; Konkel, B.; Galli, S.; Theos, A.C.; Golestaneh, N. Pgc-1α repression and high-fat diet induce age-related macular degeneration-like phenotypes in mice. Dis. Model. Mech. 2018, 11, dmm032698. [Google Scholar] [CrossRef] [Green Version]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Dong, A.; Xie, B.; Shen, J.; Yoshida, T.; Yokoi, K.; Hackett, S.F.; Campochiaro, P.A. Oxidative stress promotes ocular neovascularization. J. Cell Physiol. 2009, 219, 544–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klettner, A.; Roider, J. Constitutive and oxidative-stress-induced expression of VEGF in the RPE are differently regulated by different Mitogen-activated protein kinases. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 1487–1492. [Google Scholar] [CrossRef] [PubMed]
- Ellis, E.A.; Guberski, D.L.; Somogyi-Mann, M.; Grant, M.B. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/Wor diabetic rat. Free Radic. Biol. Med. 2000, 28, 91–101. [Google Scholar] [CrossRef]
- Kannan, R.; Zhang, N.; Sreekumar, P.G.; Spee, C.K.; Rodriguez, A.; Barron, E.; Hinton, D.R. Stimulation of apical and basolateral VEGF-A and VEGF-C secretion by oxidative stress in polarized retinal pigment epithelial cells. Mol. Vis. 2006, 12, 1649–1659. [Google Scholar] [PubMed]
- Wang, H.; Geisen, P. The role of rpe cell-associated vegf189 in choroidal endothelial cell transmigration across the rpe. Investig. Ophthalmol. Vis. Sci. 2011, 52, 570–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.E.; Lewin, A.S.; Ash, J.D. Mitochondria: Potential Targets for Protection in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2018, 1074, 11–17. [Google Scholar] [PubMed]
- Monaghan-Benson, E.; Hartmann, J.; Vendrov, A.E.; Budd, S.; Byfield, G.; Parker, A.; Ahmad, F.; Huang, W.; Runge, M.; Burridge, K.; et al. The role of vascular endothelial growth factor-induced activation of NADPH oxidase in choroidal endothelial cells and choroidal neovascularization. Am. J. Pathol. 2010, 177, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, Z.; Jiang, Y.; Hartnett, M.E. Endothelial NADPH oxidase 4 mediates vascular endothelial growth factor receptor 2-induced intravitreal neovascularization in a rat model of retinopathy of prematurity. Mol. Vis. 2014, 20, 231–241. [Google Scholar] [PubMed]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of nadph oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Chow, M.P. Flavonoids inhibit tumor necrosis factor-alpha-induced up-regulation of intercellular adhesion molecule-1 (icam-1) in respiratory epithelial cells through activator protein-1 and nuclear factor-kappab: Structure-activity relationships. Mol. Pharm. 2004, 66, 683–693. [Google Scholar]
- Nagata, M. Inflammatory cells and oxygen radicals. Curr. Drug Targets Inflamm. Allergy 2005, 4, 503–504. [Google Scholar] [CrossRef]
- l-Shabrawey, M.; Rojas, M. Role of nadph oxidase in retinal vascular inflammation. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3239–3244. [Google Scholar] [CrossRef]
- Ushio–Fukai, M. Vegf signaling through nadph oxidase-derived ros. Antioxid. Redox Signal. 2007, 9, 731–739. [Google Scholar] [CrossRef]
- Ruan, Y.; Jiang, S.; Gericke, A. Age-Related Macular Degeneration: Role of Oxidative Stress and Blood Vessels. Int. J. Mol. Sci. 2021, 22, 1296. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.L.; Jia, J.H.; Zhao, P.; Fan, R.; Pan, X.Y.; Yang, H.M.; Liu, L. Changes in blood oxidative and antioxidant parameters in a group of Chinese patients with age-related macular degeneration. J. Nutr. Health Aging 2012, 16, 201–204. [Google Scholar] [CrossRef]
- Othman, A.; Ahmad, S.; Megyerdi, S.; Mussell, R.; Choksi, K.; Maddipati, K.R.; Elmarakby, A.; Rizk, N.; Al-Shabrawey, M. 12/15-Lipoxygenase-derived lipid metabolites induce retinal endothelial cell barrier dysfunction: Contribution of NADPH oxidase. PLoS ONE 2013, 8, e57254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zor, R.K.; Erşan, S.; Küçük, E.; Yıldırım, G.; Sarı, İ. Serum malondialdehyde, monocyte chemoattractant protein-1, and vitamin C levels in wet type age-related macular degeneration patients. Ther. Adv. Ophthalmol. 2020, 12, 2515841420951682. [Google Scholar] [CrossRef]
- Bergmann, M.; Holz, F.; Kopitz, J. Lysosomal stress and lipid peroxidation products induce VEGF-121 and VEGF-165 expression in ARPE-19 cells. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T.; Takayama, K.; Kaneko, H.; Ye, F.; Fukukita, H.; Tsunekawa, T.; Kataoka, K.; Hwang, S.J.; Nagasaka, Y.; Ito, Y.; et al. Nutritional Supplementation Inhibits the Increase in Serum Malondialdehyde in Patients with Wet Age-Related Macular Degeneration. Oxid Med. Cell Longev. 2017, 2017, 9548767. [Google Scholar] [CrossRef]
- Demontis, G.C.; Longoni, B.; Marchiafava, P.L. Molecular steps involved in light-induced oxidative damage to retinal rods. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2421–2427. [Google Scholar]
- Tisi, A.; Parete, G.; Flati, V.; Maccarone, R. Up-regulation of pro-angiogenic pathways and induction of neovascularization by an acute retinal light damage. Sci. Rep. 2020, 10, 6376. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Nakabeppu, Y.; Sonoda, K.H. Oxidative Stress and Microglial Response in Retinitis Pigmentosa. Int. J. Mol. Sci. 2020, 21, 7170. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.R.; Baptista, F.I.; Santos, P.F.; Cristóvão, G.; Ambrósio, A.F.; Cunha, R.A.; Gomes, C.A. Role of microglia adenosine A(2A) receptors in retinal and brain neurodegenerative diseases. Mediat. Inflamm. 2014, 2014, 465694. [Google Scholar] [CrossRef]
- Hurst, J.; Kuehn, S.; Jashari, A.; Tsai, T.; Bartz-Schmidt, K.U.; Schnichels, S.; Joachim, S.C. A novel porcine ex vivo retina culture model for oxidative stress induced by H2O2. Altern. Lab. Anim. 2017, 45, 11–25. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Vielma, A.H.; Retamal, M.A.; Schmachtenberg, O. Nitric oxide signaling in the retina: What have we learned in two decades? Brain Res. 2012, 1430, 112–125. [Google Scholar] [CrossRef]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [CrossRef]
- Goldstein, I.M.; Ostwald, P.; Roth, S. Nitric oxide: A review of its role in retinal function and disease. Vis. Res. 1996, 36, 2979–2994. [Google Scholar] [CrossRef] [Green Version]
- Cantó, A.; Olivar, T.; Romero, F.J.; Miranda, M. Nitrosative Stress in Retinal Pathologies: Review. Antioxidants 2019, 8, 543. [Google Scholar] [CrossRef] [Green Version]
- Sripathi, S.R.; He, W.; Um, J.Y.; Moser, T.; Dehnbostel, S.; Kindt, K.; Goldman, J.; Frost, M.C.; Jahng, W.J. Nitric oxide leads to cytoskeletal reorganization in the retinal pigment epithelium under oxidative stress. Adv. Biosci. Biotechnol. 2012, 3, 1167–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutto, I.A.; Baba, T.; Merges, C.; McLeod, D.S.; Lutty, G.A. Low nitric oxide synthases (NOSs) in eyes with age-related macular degeneration (AMD). Exp. Eye Res. 2010, 90, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Friedman, E. A hemodynamic model of the pathogenesis of age-related macular degeneration. Am. J. Ophthalmol. 1997, 124, 677–682. [Google Scholar] [CrossRef]
- Grunwald, J.E.; Hariprasad, S.M.; DuPont, J. Effect of aging on foveolar choroidal circulation. Arch. Ophthalmol. 1998, 116, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Ruiz, A.; Cadenas, S.; Lamas, S. Nitric oxide signaling: Classical, less classical, and nonclassical mechanisms. Free Radic. Biol. Med. 2011, 51, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Bossy-Wetzel, E. Nitric oxide in health and disease of the nervous system. Antioxid. Redox Signal. 2009, 11, 541–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totan, Y.; Koca, C.; Erdurmuş, M.; Keskin, U.; Yiğitoğlu, R. Endothelin-1 and Nitric Oxide Levels in Exudative Age-Related Macular Degeneration. J. Ophthalmic. Vis. Res. 2015, 10, 151–154. [Google Scholar]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crabtree, M.J.; Brixey, R.; Batchelor, H.; Hale, A.B.; Channon, K.M. Integrated redox sensor and effector functions for tetrahydrobiopterin- and glutathionylation-dependent endothelial nitric-oxide synthase uncoupling. J. Biol. Chem. 2013, 288, 561–569. [Google Scholar] [CrossRef] [Green Version]
- McLeod, D.S.; Baba, T.; Bhutto, I.A.; Lutty, G.A. Co-expression of endothelial and neuronal nitric oxide synthases in the developing vasculatures of the human fetal eye. Graefe’s Arch. Clin. Exp. Ophthalmol. 2012, 250, 839–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekmen-Clark, M.; Gleason, E. Nitric oxide production and the expression of two nitric oxide synthases in the avian retina. Vis. Neurosci. 2013, 30, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Haverkamp, S.; Eldred, W.D. Localization of nNOS in photoreceptor, bipolar and horizontal cells in turtle and rat retinas. Neuroreport 1998, 9, 2231–2235. [Google Scholar] [CrossRef] [PubMed]
- Giove, T.J.; Deshpande, M.M.; Eldred, W.D. Identification of alternate transcripts of neuronal nitric oxide synthase in the mouse retina. J. Neurosci. Res. 2009, 87, 3134–3142. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.J.; Gao, F.; Wu, S.M. Light responses and morphology of bNOS-immunoreactive neurons in the mouse retina. J. Comp. Neurol. 2010, 518, 2456–2474. [Google Scholar] [CrossRef] [Green Version]
- Blom, J.; Give, T.; Deshpande, M.; Eldred, W.D. Characterization of nitric oxide signaling pathways in the mouse retina. J. Comp. Neurol. 2012, 520, 4204–4217. [Google Scholar] [CrossRef]
- Waldman, S.A.; Murad, F. Cyclic GMP synthesis and function. Pharmacol. Rev. 1987, 39, 163–196. [Google Scholar]
- Schmetterer, L.; Polak, K. Role of nitric oxide in the control of ocular blood flow. Prog. Retin. Eye Res. 2001, 20, 823–847. [Google Scholar] [CrossRef]
- Stringham, J.M.; Stringham, N.T. Nitric oxide and lutein: Function, performance, and protection of neural tissue. Foods 2015, 4, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Sennlaub, F.; Courtois, Y.; Goureau, O. Inducible nitric oxide synthase mediates the change from retinal to vitreal neovascularization in ischemic retinopathy. J. Clin. Investig. 2001, 107, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhou, J.; Marshall, B.; Xin, H.; Atherton, S.S. Lack of iNOS facilitates MCMV spread in the retina. Investig. Ophthalmol. Vis. Sci. 2007, 48, 285–292. [Google Scholar] [CrossRef]
- Yang, L.P.; Li, Y.; Zhu, X.A.; Tso, M.O. Minocycline delayed photoreceptor death in rds mice through iNOS-dependent mechanism. Mol. Vis. 2007, 13, 1073–1082. [Google Scholar] [PubMed]
- Papapetropoulos, A.; García-Cardeña, G.; Madri, J.A.; Sessa, W.C. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J. Clin. Investig. 1997, 100, 3131–3139. [Google Scholar] [CrossRef]
- McLaren, A.T.; Marsden, P.A.; Mazer, C.D.; Baker, A.J.; Stewart, D.J.; Tsui, A.K.; Li, X.; Yucel, Y.; Robb, M.; Boyd, S.R.; et al. Increased expression of HIF-1alpha, nNOS, and VEGF in the cerebral cortex of anemic rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R403–R414. [Google Scholar] [CrossRef] [Green Version]
- Bernatchez, P.N.; Bauer, P.M.; Yu, J.; Prendergast, J.S.; He, P.; Sessa, W.C. Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc. Natl. Acad. Sci. USA 2005, 102, 761–766. [Google Scholar] [CrossRef] [Green Version]
- Duda, D.G.; Fukumura, D.; Jain, R.K. Role of eNOS in neovascularization: NO for endothelial progenitor cells. Trends Mol. Med. 2004, 10, 143–145. [Google Scholar] [CrossRef]
- Hattenbach, L.O.; Falk, B.; Nürnberger, F.; Koch, F.H.; Ohrloff, C. Detection of inducible nitric oxide synthase and vascular endothelial growth factor in choroidal neovascular membranes. Ophthalmologica 2002, 216, 209–214. [Google Scholar] [CrossRef]
- Ando, A.; Yang, A.; Nambu, H.; Campochiaro, P.A. Blockade of nitric-oxide synthase reduces choroidal neovascularization. Mol. Pharmacol. 2002, 62, 539–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Wu, M.; Liu, Y.; Song, L.; Li, S.; Wang, X.; Zhang, Y.F.; Fang, J.; Wu, S. Serine racemase deficiency attenuates choroidal neovascularization and reduces nitric oxide and VEGF levels by retinal pigment epithelial cells. J. Neurochem. 2017, 143, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, F.; Feder, R.S.; McLeod, S.D.; Parke, D.W., 2nd. The Preferred Practice Pattern Guidelines in Ophthalmology. Ophthalmology 2016, 123, 928–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cillà, S.; Farruggio, S.; Vujosevic, S.; Raina, G.; Filippini, D.; Gatti, V.; Clemente, N.; Mary, D.; Vezzola, D.; Casini, G.; et al. Anti-Vascular Endothelial Growth Factors Protect Retinal Pigment Epithelium Cells Against Oxidation by Modulating Nitric Oxide Release and Autophagy. Cell Physiol. Biochem. 2017, 42, 1725–1738. [Google Scholar] [CrossRef]
- Malik, D.; Tarek, M.; Caceres del Carpio, J.; Ramirez, C.; Boyer, D.; Kenney, M.C.; Kuppermann, B.D. Safety profiles of anti-VEGF drugs: Bevacizumab, ranibizumab, aflibercept and ziv-aflibercept on human retinal pigment epithelium cells in culture. Br. J. Ophthalmol. 2014, 98 (Suppl. 1), i11–i16. [Google Scholar] [CrossRef] [Green Version]
- Sheu, S.J.; Chao, Y.M.; Liu, N.C.; Chan, J.Y. Differential effects of bevacizumab, ranibizumab and aflibercept on cell viability, phagocytosis and mitochondrial bioenergetics of retinal pigment epithelial cell. Acta Ophthalmol. 2015, 93, e631–e643. [Google Scholar] [CrossRef] [PubMed]
- Gemenetzi, M.; Lotery, A.J.; Patel, P.J. Risk of geographic atrophy in age-related macular degeneration patients treated with intravitreal anti-VEGF agents. Eye (Lond.) 2017, 31, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lois, N.; McBain, V.; Abdelkader, E.; Scott, N.W.; Kumari, R. Retinal pigment epithelial atrophy in patients with exudative age-related macular degeneration undergoing anti-vascular endothelial growth factor therapy. Retina 2013, 33, 13–22. [Google Scholar] [CrossRef]
- Grunwald, J.E.; Pistilli, M.; Daniel, E.; Ying, G.S.; Pan, W.; Jaffe, G.J.; Toth, C.A.; Hagstrom, S.A.; Maguire, M.G.; Martin, D.F. Comparison of Age-Related Macular Degeneration Treatments Trials Research Group. Incidence and Growth of Geographic Atrophy during 5 Years of Comparison of Age-Related Macular Degeneration Treatments Trials. Ophthalmology 2017, 124, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, U.; Harding, S.P.; Rogers, C.A.; Downes, S.M.; Lotery, A.J.; Culliford, L.A.; Reeves, B.C. IVAN study investigators: Alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet 2013, 382, 1258–1267. [Google Scholar] [CrossRef]
- Bhisitkul, R.B.; Mendes, T.S.; Rofagha, S.; Enanoria, W.; Boyer, D.S.; Sadda, S.R.; Zhang, K. Macular atrophy progression and 7-year vision outcomes in subjects from the ANCHOR, MARINA, and HORIZON studies: The SEVEN-UP study. Am. J. Ophthalmol. 2015, 159, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Wons, J.; Wirth, M.A.; Graf, N.; Becker, M.D.; Michels, S. Comparison of progression rate of retinal pigment epithelium loss in patients with neovascular age-related macular degeneration treated with Ranibizumab and Aflibercept. J. Ophthalmol. 2017, 2017, 7432739. [Google Scholar] [CrossRef] [PubMed]
- De Cillà, S.; Farruggio, S.; Cocomazzi, G.; Mary, D.; Alkabes, M.; Rossetti, L.; Vujosevic, S.; Grossini, E. Aflibercept and Ranibizumab Modulate Retinal Pigment Epithelial Cells Function by Acting on Their Cross Talk with Vascular Endothelial Cells. Cell Physiol. Biochem. 2020, 54, 161–179. [Google Scholar] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toma, C.; De Cillà, S.; Palumbo, A.; Garhwal, D.P.; Grossini, E. Oxidative and Nitrosative Stress in Age-Related Macular Degeneration: A Review of Their Role in Different Stages of Disease. Antioxidants 2021, 10, 653. https://doi.org/10.3390/antiox10050653
Toma C, De Cillà S, Palumbo A, Garhwal DP, Grossini E. Oxidative and Nitrosative Stress in Age-Related Macular Degeneration: A Review of Their Role in Different Stages of Disease. Antioxidants. 2021; 10(5):653. https://doi.org/10.3390/antiox10050653
Chicago/Turabian StyleToma, Caterina, Stefano De Cillà, Aurelio Palumbo, Divya Praveen Garhwal, and Elena Grossini. 2021. "Oxidative and Nitrosative Stress in Age-Related Macular Degeneration: A Review of Their Role in Different Stages of Disease" Antioxidants 10, no. 5: 653. https://doi.org/10.3390/antiox10050653