




Int. J. Mol. Sci. 2022, 23(11), 6111; https://doi.org/10.3390/ijms23116111 - 30 May 2022
Cited by 6 | Viewed by 2617
Abstract
►
Show Figures
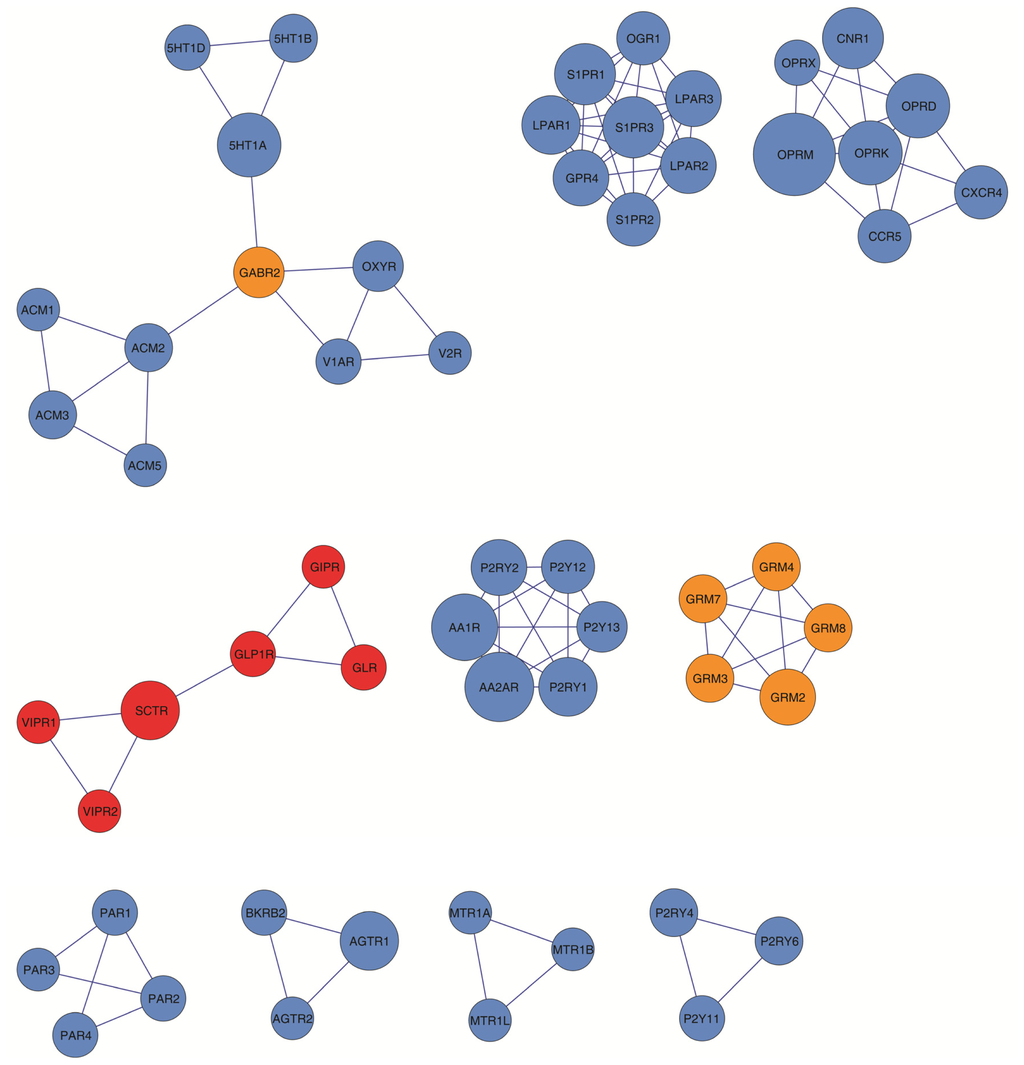
The 5-HT2A serotonin receptor (HTR2A) has been reported to be involved in the serotonin- or serotonin receptor 2A agonist-induced melanogenesis in human melanoma cells. However, the molecular mechanisms underlying HTR2A in regulating melanogenesis remain poorly understood. In this research, cultured mouse melanoma cell
[...] Read more.
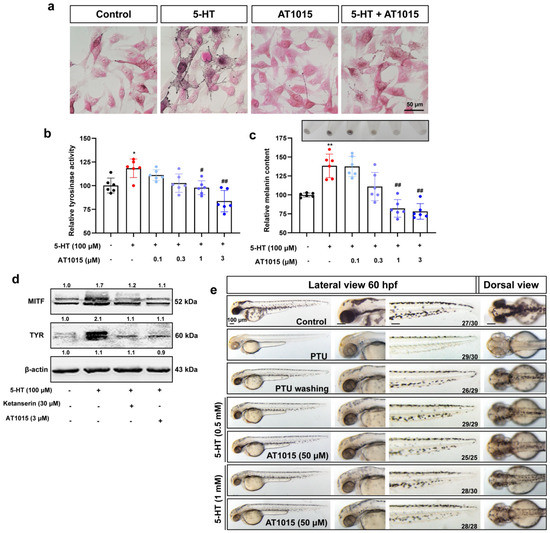
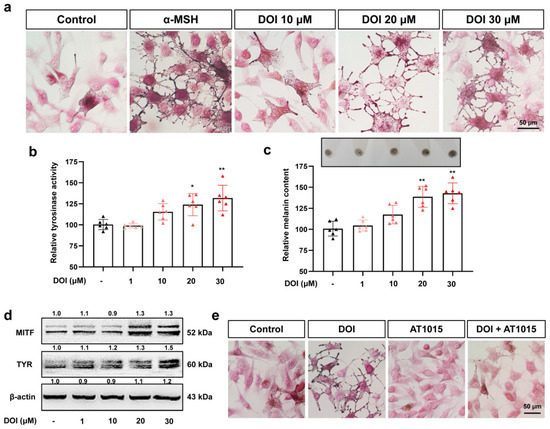
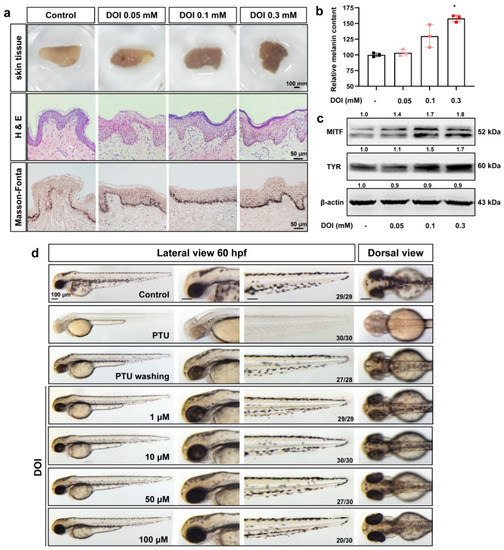
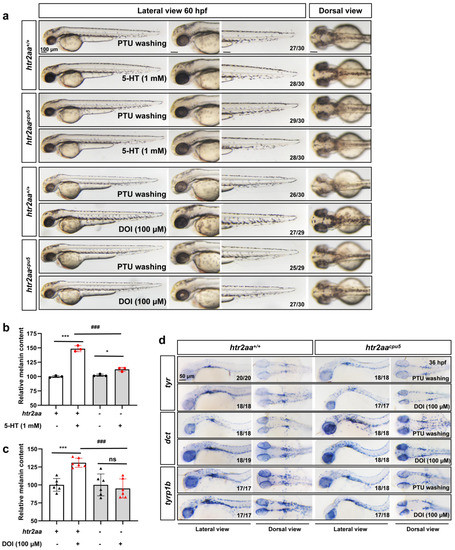
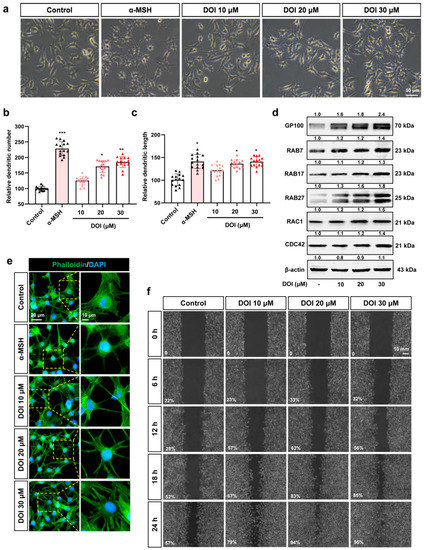
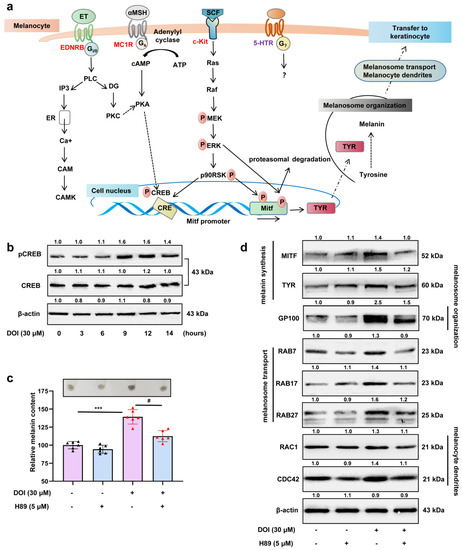
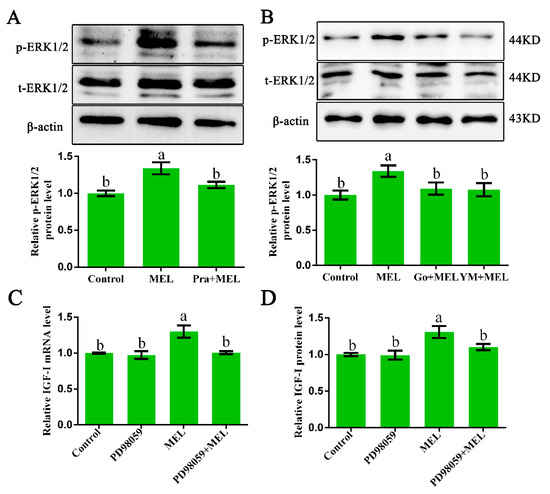
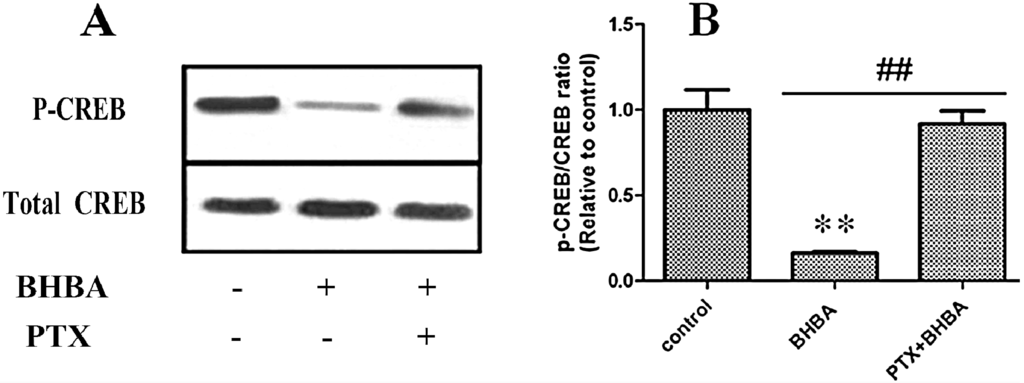
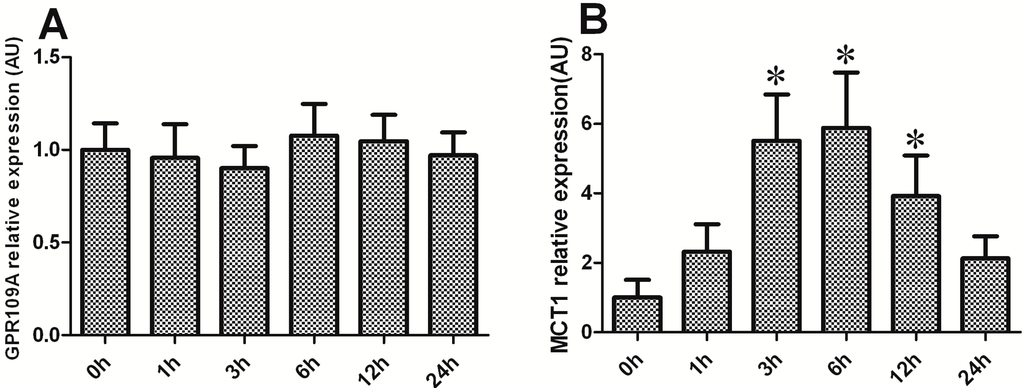
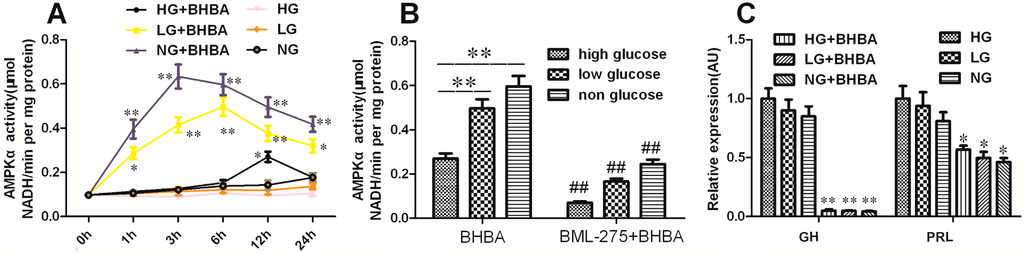
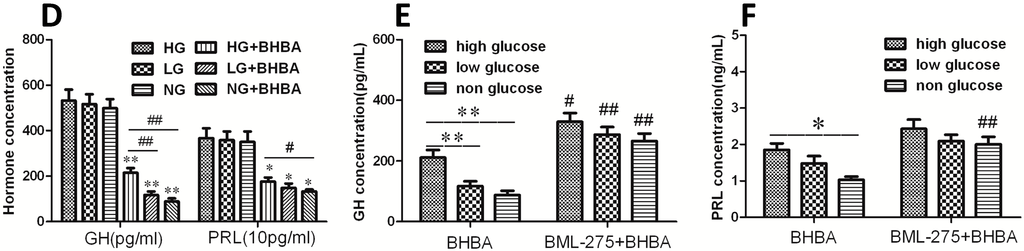
The 5-HT2A serotonin receptor (HTR2A) has been reported to be involved in the serotonin- or serotonin receptor 2A agonist-induced melanogenesis in human melanoma cells. However, the molecular mechanisms underlying HTR2A in regulating melanogenesis remain poorly understood. In this research, cultured mouse melanoma cell line B16F10, human skin, and zebrafish embryos were used to elucidate the downstream signaling of HTR2A in regulating melanogenesis and to verify the potential application of HTR2A in the treatment of pigment-associated cutaneous diseases. We demonstrated that HTR2A antagonists (AT1015 and ketanserin) attenuated the melanogenesis induction of serotonin in both mouse melanoma cells and zebrafish embryos. The agonists of HTR2A (DOI and TCB-2) increased melanin synthesis and transfer in B16F10 cells, human skin tissue, and zebrafish embryos. Furthermore, the HTR2A agonists increased the expression of proteins related to melanosome organization and melanocyte dendrites to facilitate the melanocyte migration and melanosome transport. HTR2A antagonists and genetic knockout of zebrafish htr2aa (the homologue of mammalian HTR2A gene) were also used to clarify that HTR2A mediates serotonin and DOI in regulating melanogenesis. Finally, through small scale screening of the candidate downstream pathway, we demonstrated that HTR2A mediates the melanogenesis induction of its ligands by activating the PKA/CREB signaling pathway. In this research, we further confirmed that HTR2A is a crucial protein to mediate melanocyte function. Meanwhile, this research supports that HTR2A could be designed as a drug target for the development of chemicals to treat cutaneous diseases with melanocytes or melanogenesis abnormality.
Full article
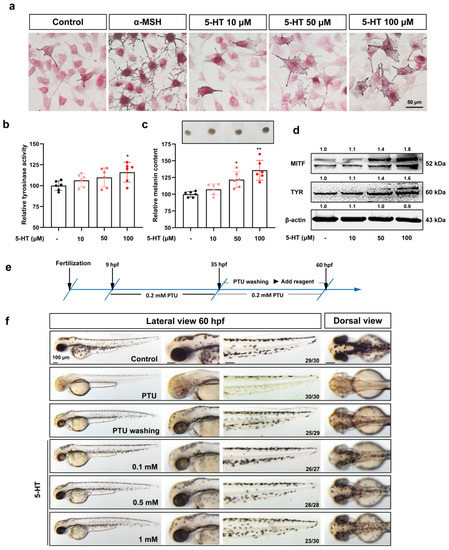
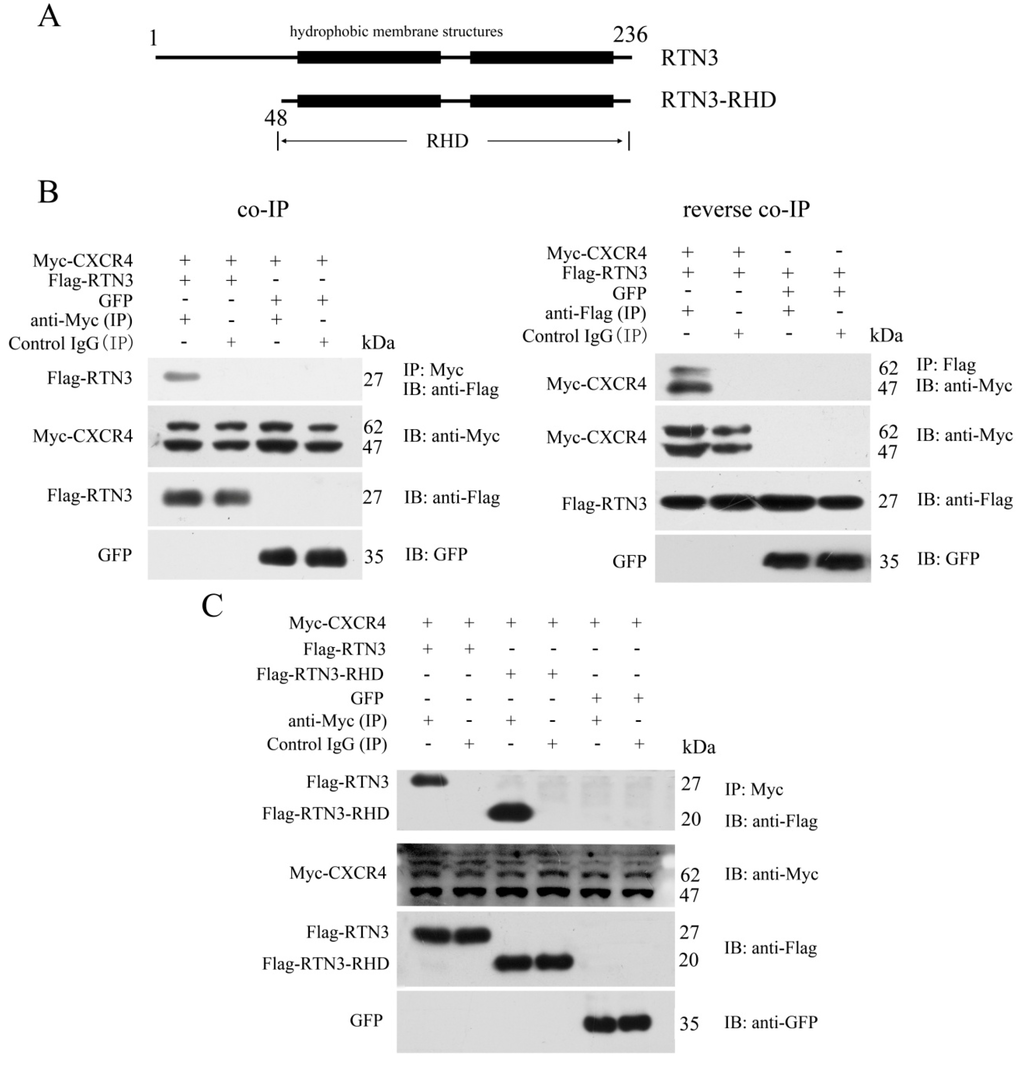
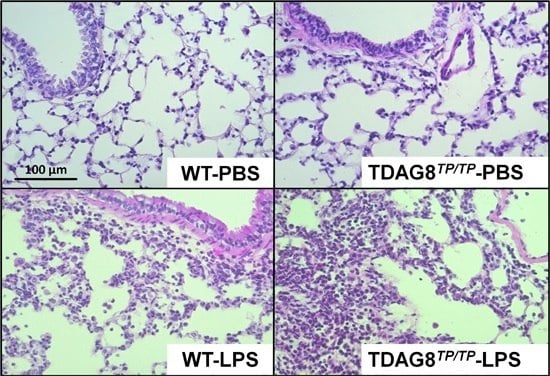
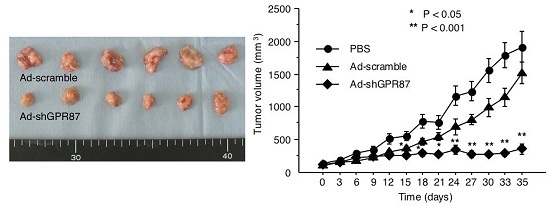
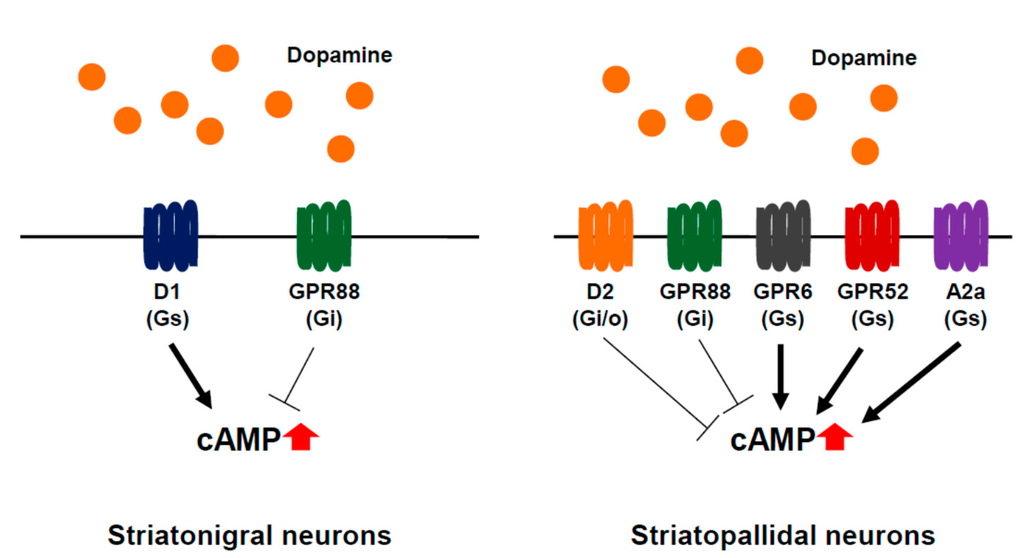
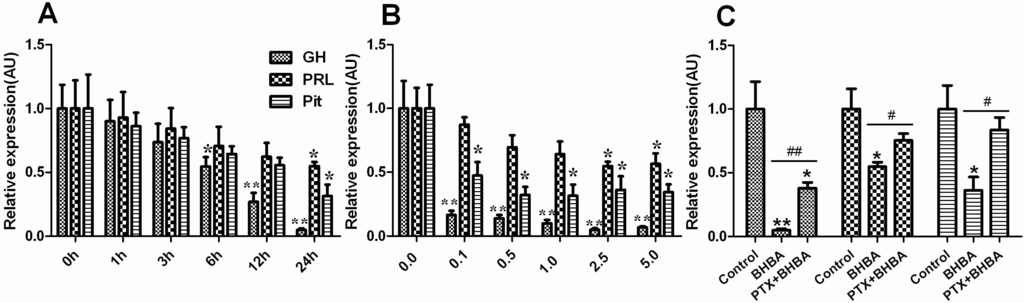
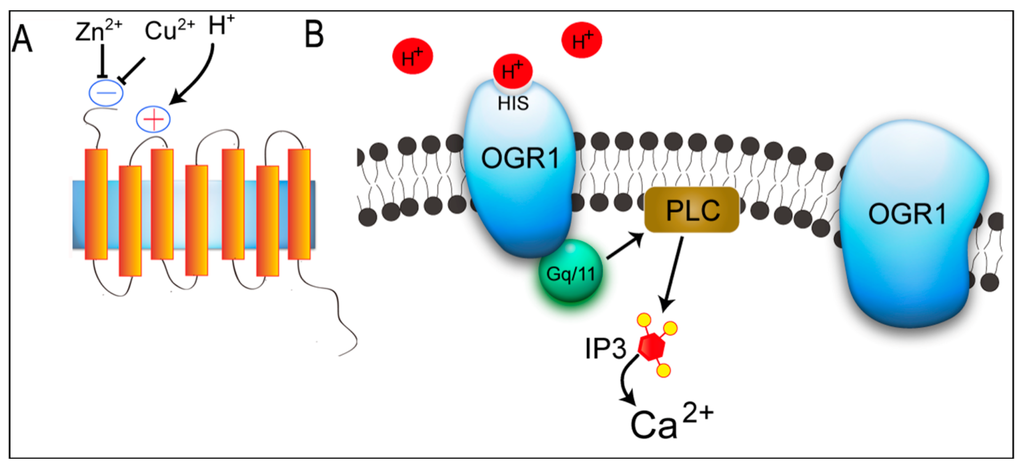
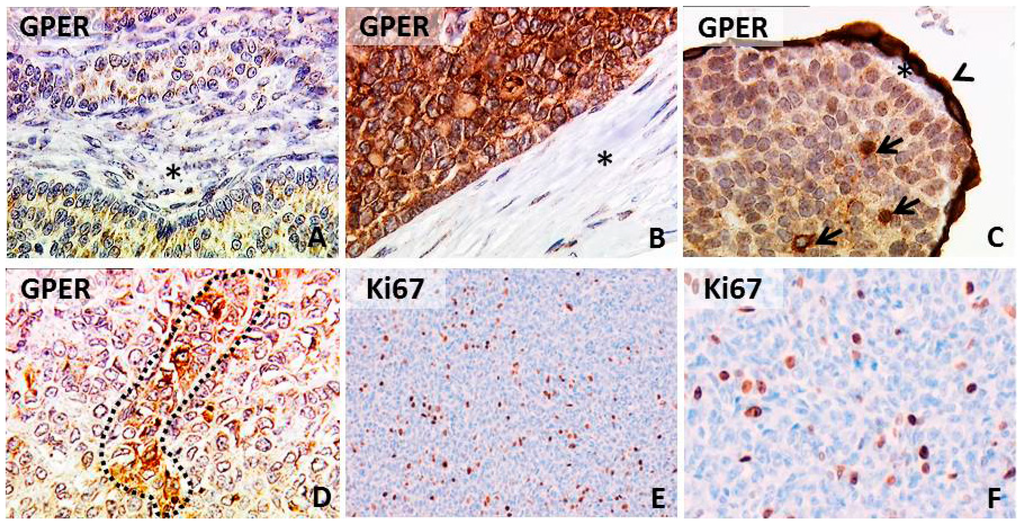
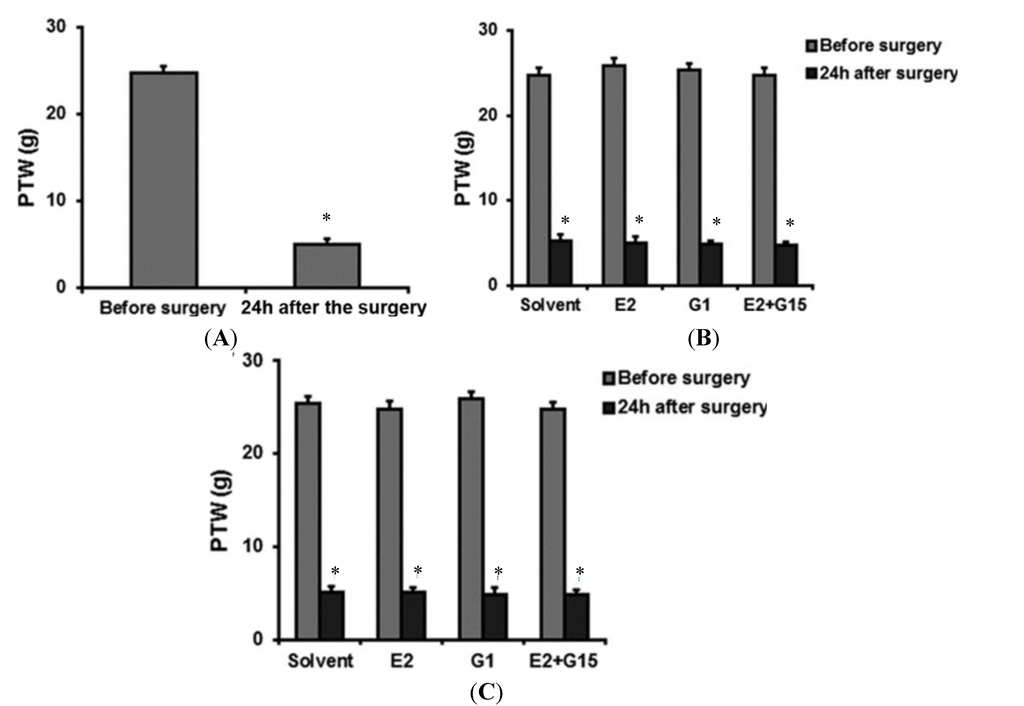
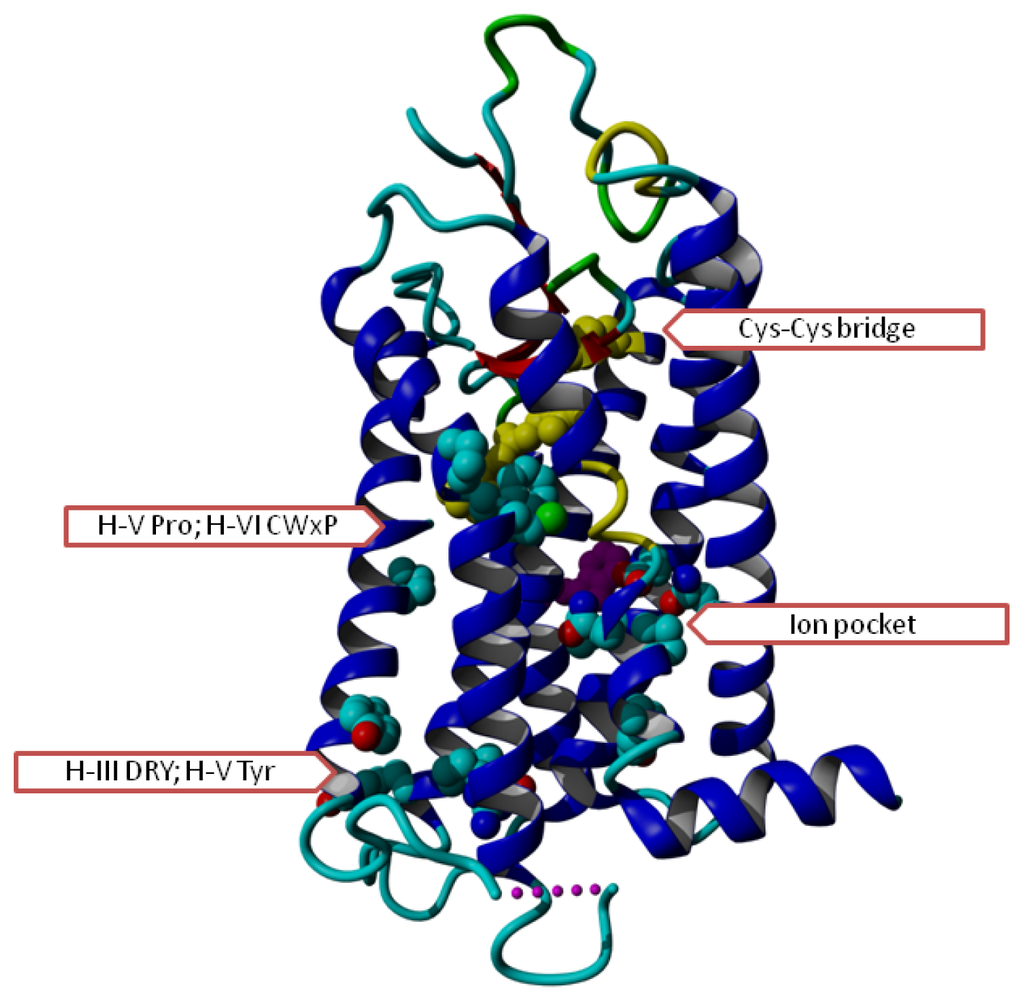
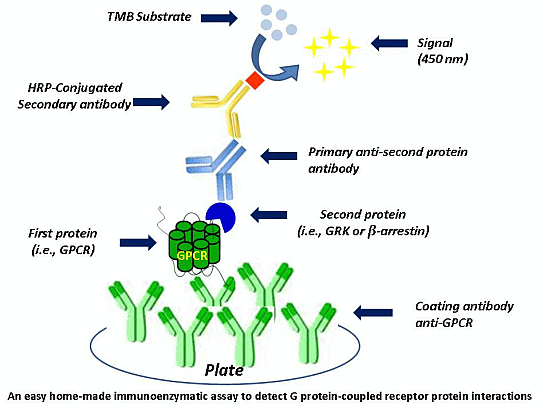
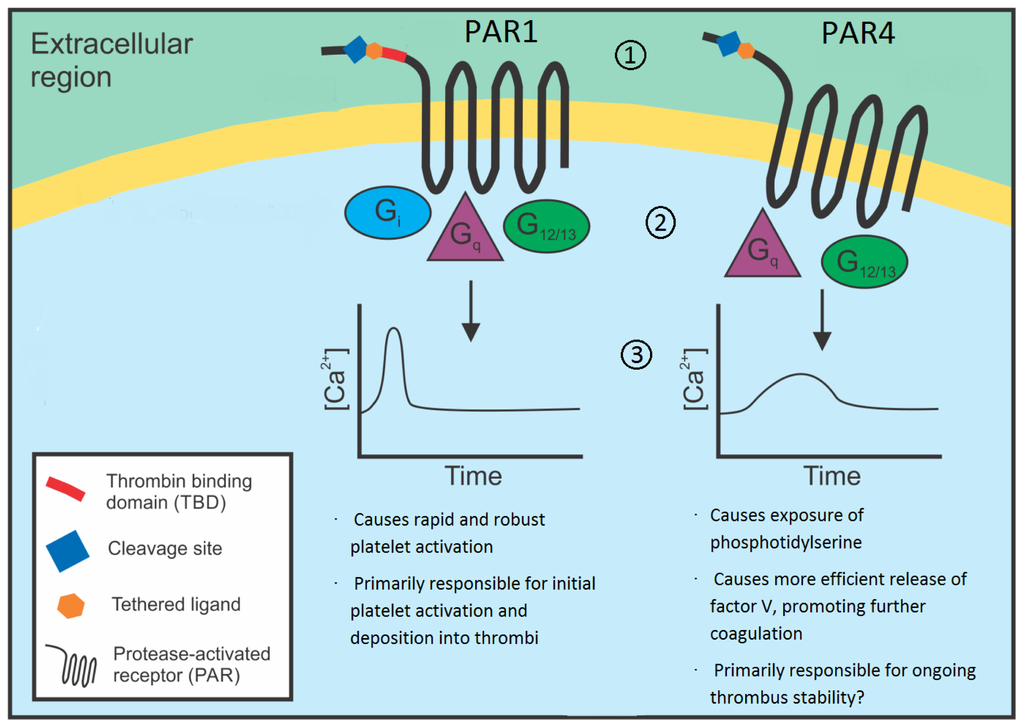
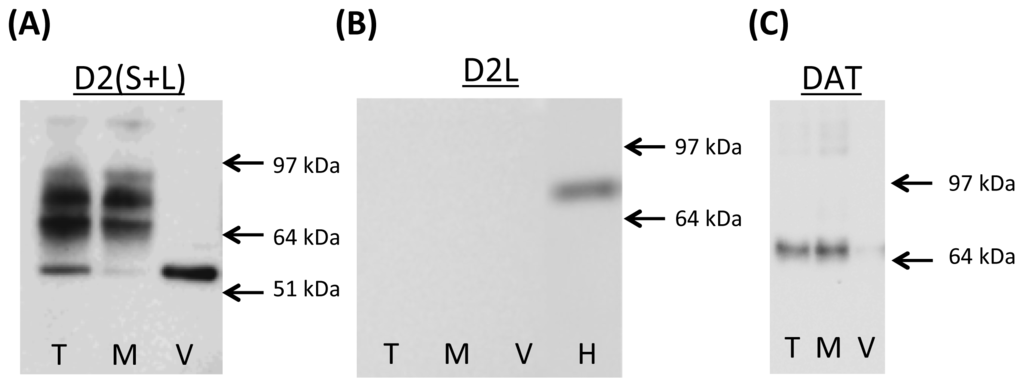
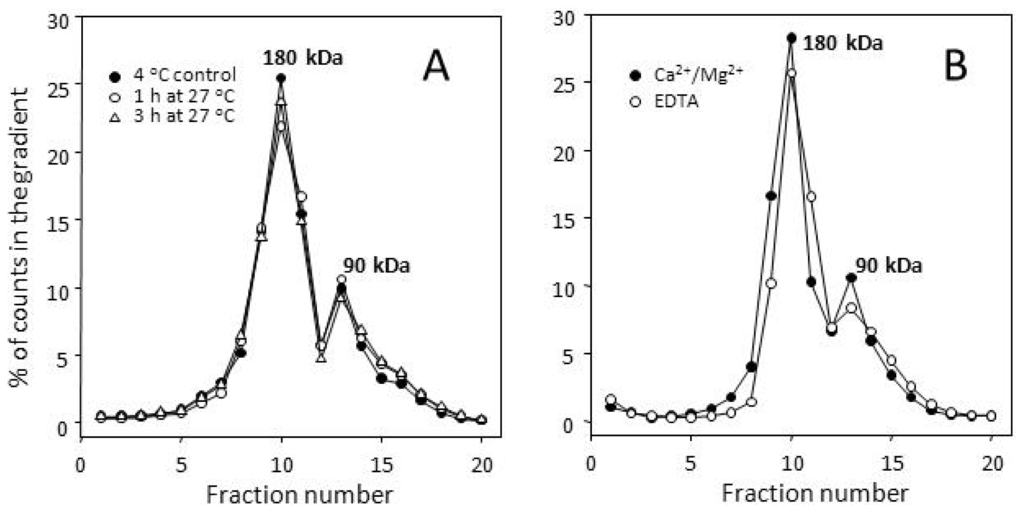
Figure 1















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}