
The Post-Translational Modification Networking in WNK-Centric Hypertension Regulation and Electrolyte Homeostasis

 , and
, and
Abstract
:1. Introduction
2. Familial Hypertension Stemming from Genetic Defects in WNK Signaling Network
2.1. WNKs in Pseudohypoaldosteronism Type II (PHAII)
2.2. The SGK1-WNK-SPAK/OSR1 Phosphorylation Pathway
2.3. The WNK Degradation Pathway: KLHL3 and CUL3
3. WNK Interactive Channels in the Proximal Tubule (PT)
3.1. Na+-H+ Exchanger 3 (NHE3)
3.2. Na+/3HCO3− Co-Transporter (NBC1)
4. WNK Interactive Channels in the Thick Ascending Limb (TAL)
4.1. Na/K/2Cl Co-Transporter 2 (NKCC2)
4.2. Renal Outer Medullary Potassium (ROMK) Channel
4.3. Uromodulin (UMOD)
5. WNK Interactive Channels in the Distal Convoluted Tubule (DCT)
Na+-Cl− Co-Transporter (NCC)
6. WNK Interacting Channels in the Cortical Collecting Duct (CCD)
6.1. SGK1-NEDD4L Degradation Mechanism
6.2. Epithelial Na+ Channel (ENaC)
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AKT | Protein kinase B |
| AME | Apparent mineralocorticoid excess |
| AT1R | Angiotensin receptor 1 |
| AVP | Arginine vasopressin |
| Ang II | Angiotensin 2 |
| cAMP | Cyclic adenosine monophosphate |
| CCD | Cortical collecting duct |
| CDK | Cyclin-dependent kinases |
| CHX | Cycloheximide |
| CNT | Connecting tubule |
| CUL3 | Cullin-ring ubiquitin 3 |
| CUL3Δex9 | Cullin 3 exon 9 deletion mutant |
| DCT | Distal convoluted tubule |
| DSS rat | Dahl salt sensitive rat |
| DUBs | Deubiquitinating enzymes |
| eGFR | Estimated glomerular filter rate |
| END1 | Endothelin-1 |
| ENaC | Epithelial sodium chloride co-transporter |
| ERAD | Endoplasmic reticulum (ER)-associated degradation |
| ERK | Extracellular signal-regulated kinases |
| FHHt | Familial hyperkalemic hypertension |
| GR | Glucocorticoid receptor |
| HECT domain | Homologous to the E6-AP carboxyl terminus |
| HTN | Hypertension |
| IR | Insulin receptor |
| KLHL3 | Kelch-like protein |
| KS-WNK1 | Kidney-specific WNK1 |
| L-wnk1 | Long-form WNK1 |
| MAGE-D2 | Melanoma-associated antigen D2 |
| MK pathway | MAPK-activated protein kinase pathway |
| MR | Mineralocorticoid receptor |
| mTORC2 | Mechanistic target of rapamycin complex 2 |
| NBC1 | Sodium bicarbonate co-transporter 1Na+/3HCO3− transporter |
| NCC | Sodium chloride co-transporter |
| NEDD4.2 | Neural precursor cell expressed developmentally down-regulated 4 ligase |
| NHE3 | Na+/H+ exchanger 3 |
| NHERF2 | Na+/H+ exchange regulatory factor 2 |
| NKCC2 | Na+/K+/2Cl− co-transporters |
| NMD | Nonsense mediated decay |
| OS9 | Osteosarcoma amplified 9 protein |
| OSR1 | Oxidative stress responsive kinase 1 (OXSR1) |
| PHAII | Pseudohypoaldosteronism type II |
| PI3K | Phosphoinositide 3-kinases |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PT | Proximal tubule |
| PTM | Post-translational modification |
| pRTA | Proximal renal tubular acidosis |
| PY motif | PPxY motif |
| RAAS | Renin aldosterone angiotensin system |
| ROMK | Renal outer medullary potassium channel |
| RTA | Renal tubular acidosis |
| SGK1 | Serum/glucocorticoid regulated kinase 1 |
| SPAK | Ste20-related proline alanine rich kinase (STK39) |
| TAL | Thick ascending loop |
| UCH-L3 | Ubiquitin carboxyl-terminal hydrolase isozyme L3 |
| UMOD | Uromodulin = Tamm horsfall protein (THP) |
| V2R | Vasopressin receptor 2 |
| VAMP | Vesicle-associated membrane protein |
| WNK | With-no-lysine (K) kinase |
| [K+]i | Intracellular potassium concentration |
| ΔIami | Amiloride-sensitive whole-cell currents |
References
- Baker, E.H. Ion channels and the control of blood pressure. Br. J. Clin. Pharmacol. 2000, 49, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y.; Ehrlich, B.E. Ion channels in renal disease. Chem. Rev. 2012, 112, 6353–6372. [Google Scholar] [CrossRef] [PubMed]
- Cunha, T.D.S.; Heilberg, I.P. Bartter syndrome: Causes, diagnosis, and treatment. Int. J. Nephrol. Renovasc. Dis. 2018, 11, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Healy, J.K. Pseudohypoaldosteronism type II: History, arguments, answers, and still some questions. Hypertension 2014, 63, 648–654. [Google Scholar] [CrossRef]
- Melander, O.; Orho-Melander, M.; Bengtsson, K.; Lindblad, U.; Rastam, L.; Groop, L.; Hulthen, U.L. Genetic variants of thiazide-sensitive NaCl-cotransporter in Gitelman’s syndrome and primary hypertension. Hypertension 2000, 36, 389–394. [Google Scholar] [CrossRef]
- Hansson, J.H.; Nelson-Williams, C.; Suzuki, H.; Schild, L.; Shimkets, R.; Lu, Y.; Canessa, C.; Iwasaki, T.; Rossier, B.; Lifton, R.P. Hypertension caused by a truncated epithelial sodium channel gamma subunit: Genetic heterogeneity of Liddle syndrome. Nat. Genet. 1995, 11, 76–82. [Google Scholar] [CrossRef]
- Brown, A.; Meor Azlan, N.F.; Wu, Z.; Zhang, J. WNK-SPAK/OSR1-NCC kinase signaling pathway as a novel target for the treatment of salt-sensitive hypertension. Acta Pharmacol. Sin. 2021, 42, 508–517. [Google Scholar] [CrossRef]
- Friedel, P.; Kahle, K.T.; Zhang, J.; Hertz, N.; Pisella, L.I.; Buhler, E.; Schaller, F.; Duan, J.; Khanna, A.R.; Bishop, P.N.; et al. WNK1-regulated inhibitory phosphorylation of the KCC2 cotransporter maintains the depolarizing action of GABA in immature neurons. Sci. Signal. 2015, 8, ra65. [Google Scholar] [CrossRef]
- Hadchouel, J.; Ellison, D.H.; Gamba, G. Regulation of Renal Electrolyte Transport by WNK and SPAK-OSR1 Kinases. Annu. Rev. Physiol. 2016, 78, 367–389. [Google Scholar] [CrossRef]
- Valinsky, W.C.; Touyz, R.M.; Shrier, A. Aldosterone, SGK1, and ion channels in the kidney. Clin. Sci. 2018, 132, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.; Rafiqi, F.H.; Karlsson, H.K.; Moleleki, N.; Vandewalle, A.; Campbell, D.G.; Morrice, N.A.; Alessi, D.R. Activation of the thiazide-sensitive Na+-Cl- cotransporter by the WNK-regulated kinases SPAK and OSR1. J. Cell Sci. 2008, 121, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Furusho, T.; Uchida, S.; Sohara, E. The WNK signaling pathway and salt-sensitive hypertension. Hypertens Res. 2020, 43, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.H.; Disse-Nicodeme, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.M.; et al. Human hypertension caused by mutations in WNK kinases. Science 2001, 293, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Boyden, L.M.; Choi, M.; Choate, K.A.; Nelson-Williams, C.J.; Farhi, A.; Toka, H.R.; Tikhonova, I.R.; Bjornson, R.; Mane, S.M.; Colussi, G.; et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature 2012, 482, 98–102. [Google Scholar] [CrossRef]
- Louis-Dit-Picard, H.; Barc, J.; Trujillano, D.; Miserey-Lenkei, S.; Bouatia-Naji, N.; Pylypenko, O.; Beaurain, G.; Bonnefond, A.; Sand, O.; Simian, C.; et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012, 44, 456–460. [Google Scholar] [CrossRef]
- Mansfield, T.A.; Simon, D.B.; Farfel, Z.; Bia, M.; Tucci, J.R.; Lebel, M.; Gutkin, M.; Vialettes, B.; Christofilis, M.A.; Kauppinen-Makelin, R.; et al. Multilocus linkage of familial hyperkalaemia and hypertension, pseudohypoaldosteronism type II, to chromosomes 1q31-42 and 17p11-q21. Nat. Genet. 1997, 16, 202–205. [Google Scholar] [CrossRef]
- Chavez-Canales, M.; Zhang, C.; Soukaseum, C.; Moreno, E.; Pacheco-Alvarez, D.; Vidal-Petiot, E.; Castaneda-Bueno, M.; Vazquez, N.; Rojas-Vega, L.; Meermeier, N.P.; et al. WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the Na-Cl cotransporter via SPAK, an effect antagonized by WNK4. Hypertension 2014, 64, 1047–1053. [Google Scholar] [CrossRef]
- Vitari, A.C.; Deak, M.; Morrice, N.A.; Alessi, D.R. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. BioChem. J. 2005, 391, 17–24. [Google Scholar] [CrossRef]
- Ohta, A.; Schumacher, F.R.; Mehellou, Y.; Johnson, C.; Knebel, A.; Macartney, T.J.; Wood, N.T.; Alessi, D.R.; Kurz, T. The CUL3-KLHL3 E3 ligase complex mutated in Gordon’s hypertension syndrome interacts with and ubiquitylates WNK isoforms: Disease-causing mutations in KLHL3 and WNK4 disrupt interaction. BioChem. J. 2013, 451, 111–122. [Google Scholar] [CrossRef]
- Na, T.; Wu, G.; Peng, J.B. Disease-causing mutations in the acidic motif of WNK4 impair the sensitivity of WNK4 kinase to calcium ions. BioChem. Biophys. Res. Commun. 2012, 419, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Ring, A.M.; Leng, Q.; Rinehart, J.; Wilson, F.H.; Kahle, K.T.; Hebert, S.C.; Lifton, R.P. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc. Natl. Acad. Sci. USA 2007, 104, 4025–4029. [Google Scholar] [CrossRef] [PubMed]
- Na, T.; Wu, G.; Zhang, W.; Dong, W.J.; Peng, J.B. Disease-causing R1185C mutation of WNK4 disrupts a regulatory mechanism involving calmodulin binding and SGK1 phosphorylation sites. Am. J. Physiol. Renal. Physiol. 2013, 304, F8–F18. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Zhang, C.; Lin, D.H.; Sun, P.; Wang, W.H. WNK4 inhibits Ca(2+)-activated big-conductance potassium channels (BK) via mitogen-activated protein kinase-dependent pathway. Biochim. Biophys. Acta 2013, 1833, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Rozansky, D.J.; Cornwall, T.; Subramanya, A.R.; Rogers, S.; Yang, Y.F.; David, L.L.; Zhu, X.; Yang, C.L.; Ellison, D.H. Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J. Clin. Investig. 2009, 119, 2601–2612. [Google Scholar] [CrossRef] [PubMed]
- Heise, C.J.; Xu, B.E.; Deaton, S.L.; Cha, S.K.; Cheng, C.J.; Earnest, S.; Sengupta, S.; Juang, Y.C.; Stippec, S.; Xu, Y.; et al. Serum and glucocorticoid-induced kinase (SGK) 1 and the epithelial sodium channel are regulated by multiple with no lysine (WNK) family members. J. Biol. Chem. 2010, 285, 25161–25167. [Google Scholar] [CrossRef]
- Castaneda-Bueno, M.; Arroyo, J.P.; Zhang, J.; Puthumana, J.; Yarborough, O., 3rd; Shibata, S.; Rojas-Vega, L.; Gamba, G.; Rinehart, J.; Lifton, R.P. Phosphorylation by PKC and PKA regulate the kinase activity and downstream signaling of WNK4. Proc. Natl. Acad. Sci. USA 2017, 114, E879–E886. [Google Scholar] [CrossRef]
- Argaiz, E.R.; Chavez-Canales, M.; Ostrosky-Frid, M.; Rodriguez-Gama, A.; Vazquez, N.; Gonzalez-Rodriguez, X.; Garcia-Valdes, J.; Hadchouel, J.; Ellison, D.; Gamba, G. Kidney-specific WNK1 isoform (KS-WNK1) is a potent activator of WNK4 and NCC. Am. J. Physiol. Renal. Physiol. 2018, 315, F734–F745. [Google Scholar] [CrossRef]
- Thastrup, J.O.; Rafiqi, F.H.; Vitari, A.C.; Pozo-Guisado, E.; Deak, M.; Mehellou, Y.; Alessi, D.R. SPAK/OSR1 regulate NKCC1 and WNK activity: Analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. BioChem. J. 2012, 441, 325–337. [Google Scholar] [CrossRef]
- Cai, H.; Cebotaru, V.; Wang, Y.H.; Zhang, X.M.; Cebotaru, L.; Guggino, S.E.; Guggino, W.B. WNK4 kinase regulates surface expression of the human sodium chloride cotransporter in mammalian cells. Kidney Int. 2006, 69, 2162–2170. [Google Scholar] [CrossRef]
- Mayan, H.; Munter, G.; Shaharabany, M.; Mouallem, M.; Pauzner, R.; Holtzman, E.J.; Farfel, Z. Hypercalciuria in familial hyperkalemia and hypertension accompanies hyperkalemia and precedes hypertension: Description of a large family with the Q565E WNK4 mutation. J. Clin. Endocrinol. Metab. 2004, 89, 4025–4030. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.S.; Morimoto, T.; Rai, T.; Chiga, M.; Sohara, E.; Ohno, M.; Uchida, K.; Lin, S.H.; Moriguchi, T.; Shibuya, H.; et al. Molecular pathogenesis of pseudohypoaldosteronism type II: Generation and analysis of a Wnk4(D561A/+) knockin mouse model. Cell Metab. 2007, 5, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Bazua-Valenti, S.; Chavez-Canales, M.; Rojas-Vega, L.; Gonzalez-Rodriguez, X.; Vazquez, N.; Rodriguez-Gama, A.; Argaiz, E.R.; Melo, Z.; Plata, C.; Ellison, D.H.; et al. The Effect of WNK4 on the Na+-Cl− Cotransporter Is Modulated by Intracellular Chloride. J. Am. Soc. Nephrol. 2015, 26, 1781–1786. [Google Scholar] [CrossRef]
- Maruyama, J.; Kobayashi, Y.; Umeda, T.; Vandewalle, A.; Takeda, K.; Ichijo, H.; Naguro, I. Osmotic stress induces the phosphorylation of WNK4 Ser575 via the p38MAPK-MK pathway. Sci. Rep. 2016, 6, 18710. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.J.; Huang, C.L. Activation of PI3-kinase stimulates endocytosis of ROMK via Akt1/SGK1-dependent phosphorylation of WNK1. J. Am. Soc. Nephrol. 2011, 22, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.R.; Liu, Z.; Huang, C.L. Domains of WNK1 kinase in the regulation of ROMK1. Am. J. Physiol. Renal. Physiol. 2008, 295, F438–F445. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; English, J.M.; Wilsbacher, J.L.; Stippec, S.; Goldsmith, E.J.; Cobb, M.H. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J. Biol. Chem. 2000, 275, 16795–16801. [Google Scholar] [CrossRef]
- Min, X.; Lee, B.H.; Cobb, M.H.; Goldsmith, E.J. Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure 2004, 12, 1303–1311. [Google Scholar] [CrossRef]
- McCormick, J.A.; Ellison, D.H. The WNKs: Atypical protein kinases with pleiotropic actions. Physiol. Rev. 2011, 91, 177–219. [Google Scholar] [CrossRef]
- Vidal-Petiot, E.; Elvira-Matelot, E.; Mutig, K.; Soukaseum, C.; Baudrie, V.; Wu, S.; Cheval, L.; Huc, E.; Cambillau, M.; Bachmann, S.; et al. WNK1-related Familial Hyperkalemic Hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc. Natl. Acad. Sci. USA 2013, 110, 14366–14371. [Google Scholar] [CrossRef]
- Akella, R.; Drozdz, M.A.; Humphreys, J.M.; Jiou, J.; Durbacz, M.Z.; Mohammed, Z.J.; He, H.; Liwocha, J.; Sekulski, K.; Goldsmith, E.J. A Phosphorylated Intermediate in the Activation of WNK Kinases. Biochemistry 2020, 59, 1747–1755. [Google Scholar] [CrossRef]
- AlAmri, M.A.; Kadri, H.; Alderwick, L.J.; Simpkins, N.S.; Mehellou, Y. Rafoxanide and Closantel Inhibit SPAK and OSR1 Kinases by Binding to a Highly Conserved Allosteric Site on Their C-terminal Domains. ChemMedChem 2017, 12, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, K.B.; England, R.; Delpire, E. Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol. Cell Biol. 2006, 26, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Lorente-Rodriguez, A.; Earnest, S.; Stippec, S.; Guo, X.; Trudgian, D.C.; Mirzaei, H.; Cobb, M.H. Regulation of OSR1 and the sodium, potassium, two chloride cotransporter by convergent signals. Proc. Natl. Acad. Sci. USA 2013, 110, 18826–18831. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Charnock-Jones, D.S.; Burton, G.J. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS ONE 2011, 6, e17894. [Google Scholar] [CrossRef]
- Yan, L.; Mieulet, V.; Lamb, R.F. mTORC2 is the hydrophobic motif kinase for SGK1. BioChem. J. 2008, 416, e19–e21. [Google Scholar] [CrossRef]
- Debonneville, C.; Flores, S.Y.; Kamynina, E.; Plant, P.J.; Tauxe, C.; Thomas, M.A.; Munster, C.; Chraibi, A.; Pratt, J.H.; Horisberger, J.D.; et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 2001, 20, 7052–7059. [Google Scholar] [CrossRef]
- Lee, I.H.; Dinudom, A.; Sanchez-Perez, A.; Kumar, S.; Cook, D.I. Akt mediates the effect of insulin on epithelial sodium channels by inhibiting Nedd4-2. J. Biol. Chem. 2007, 282, 29866–29873. [Google Scholar] [CrossRef]
- Pohl, P.; Joshi, R.; Petrvalska, O.; Obsil, T.; Obsilova, V. 14-3-3-protein regulates Nedd4-2 by modulating interactions between HECT and WW domains. Commun. Biol. 2021, 4, 899. [Google Scholar] [CrossRef]
- Manning, J.A.; Kumar, S. Physiological Functions of Nedd4-2: Lessons from Knockout Mouse Models. Trends BioChem. Sci. 2018, 43, 635–647. [Google Scholar] [CrossRef]
- Snyder, P.M.; Olson, D.R.; Kabra, R.; Zhou, R.; Steines, J.C. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na(+) channel through convergent phosphorylation of Nedd4-2. J. Biol. Chem. 2004, 279, 45753–45758. [Google Scholar] [CrossRef] [Green Version]
- Bruce, M.C.; Kanelis, V.; Fouladkou, F.; Debonneville, A.; Staub, O.; Rotin, D. Regulation of Nedd4-2 self-ubiquitination and stability by a PY motif located within its HECT-domain. BioChem. J. 2008, 415, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Cui, L.; Lu, J.; Lang, Y.; Bottillo, I.; Zhao, X. A novel mutation in exon 9 of Cullin 3 gene contributes to aberrant splicing in pseudohypoaldosteronism type II. FEBS Open Bio. 2018, 8, 461–469. [Google Scholar] [CrossRef]
- Yoshizaki, Y.; Mori, Y.; Tsuzaki, Y.; Mori, T.; Nomura, N.; Wakabayashi, M.; Takahashi, D.; Zeniya, M.; Kikuchi, E.; Araki, Y.; et al. Impaired degradation of WNK by Akt and PKA phosphorylation of KLHL3. BioChem. Biophys. Res. Commun. 2015, 467, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Arroyo, J.P.; Castaneda-Bueno, M.; Puthumana, J.; Zhang, J.; Uchida, S.; Stone, K.L.; Lam, T.T.; Lifton, R.P. Angiotensin II signaling via protein kinase C phosphorylates Kelch-like 3, preventing WNK4 degradation. Proc. Natl. Acad. Sci. USA 2014, 111, 15556–15561. [Google Scholar] [CrossRef]
- Murillo-de-Ozores, A.R.; Rodriguez-Gama, A.; Carbajal-Contreras, H.; Gamba, G.; Castaneda-Bueno, M. WNK4 Kinase: From structure to physiology. Am. J. Physiol. Renal. Physiol. 2021, 320, F378–F403. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sun, H.; Lang, F.; Yun, C.C. Activation of NHE3 by dexamethasone requires phosphorylation of NHE3 at Ser663 by SGK1. Am. J. Physiol. Cell Physiol. 2005, 289, C802–C810. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wiederkehr, M.R.; Fan, L.; Collazo, R.L.; Crowder, L.A.; Moe, O.W. Acute inhibition of Na/H exchanger NHE-3 by cAMP. Role of protein kinase a and NHE-3 phosphoserines 552 and 605. J. Biol. Chem. 1999, 274, 3978–3987. [Google Scholar] [CrossRef]
- Gross, E.; Fedotoff, O.; Pushkin, A.; Abuladze, N.; Newman, D.; Kurtz, I. Phosphorylation-induced modulation of pNBC1 function: Distinct roles for the amino- and carboxy-termini. J. Physiol. 2003, 549, 673–682. [Google Scholar] [CrossRef]
- Farfel, Z.; Mayan, H.; Karlish, S.J.D. Familial hyperkalemia and hypertension and a hypothesis to explain proximal renal tubular acidosis. Proc. Natl. Acad. Sci. USA 2019, 116, 16173–16174. [Google Scholar] [CrossRef]
- Pedrosa, R.; Goncalves, N.; Hopfer, U.; Jose, P.A.; Soares-da-Silva, P. Activity and regulation of Na+-HCO3− cotransporter in immortalized spontaneously hypertensive rat and Wistar-Kyoto rat proximal tubular epithelial cells. Hypertension 2007, 49, 1186–1193. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.; Fraser, S.A.; Galic, S.; Choy, S.W.; Katerelos, M.; Gleich, K.; Kemp, B.E.; Mount, P.F.; Power, D.A. Novel mechanisms of Na+ retention in obesity: Phosphorylation of NKCC2 and regulation of SPAK/OSR1 by AMPK. Am. J. Physiol. Renal. Physiol. 2014, 307, F96–F106. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, A.; Flatman, P.W. Phosphorylation and transport in the Na-K-2Cl cotransporters, NKCC1 and NKCC2A, compared in HEK-293 cells. PLoS ONE 2011, 6, e17992. [Google Scholar] [CrossRef]
- Gonzales, P.A.; Pisitkun, T.; Hoffert, J.D.; Tchapyjnikov, D.; Star, R.A.; Kleta, R.; Wang, N.S.; Knepper, M.A. Large-scale proteomics and phosphoproteomics of urinary exosomes. J. Am. Soc. Nephrol. 2009, 20, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Flemmer, A.W.; Gimenez, I.; Dowd, B.F.; Darman, R.B.; Forbush, B. Activation of the Na-K-Cl cotransporter NKCC1 detected with a phospho-specific antibody. J. Biol. Chem. 2002, 277, 37551–37558. [Google Scholar] [CrossRef] [PubMed]
- Darman, R.B.; Forbush, B. A regulatory locus of phosphorylation in the N terminus of the Na-K-Cl cotransporter, NKCC1. J. Biol. Chem. 2002, 277, 37542–37550. [Google Scholar] [CrossRef] [PubMed]
- Ponce-Coria, J.; Markadieu, N.; Austin, T.M.; Flammang, L.; Rios, K.; Welling, P.A.; Delpire, E. A novel Ste20-related proline/alanine-rich kinase (SPAK)-independent pathway involving calcium-binding protein 39 (Cab39) and serine threonine kinase with no lysine member 4 (WNK4) in the activation of Na-K-Cl cotransporters. J. Biol. Chem. 2014, 289, 17680–17688. [Google Scholar] [CrossRef]
- Yoo, D.; Kim, B.Y.; Campo, C.; Nance, L.; King, A.; Maouyo, D.; Welling, P.A. Cell surface expression of the ROMK (Kir 1.1) channel is regulated by the aldosterone-induced kinase, SGK-1, and protein kinase A. J. Biol. Chem. 2003, 278, 23066–23075. [Google Scholar] [CrossRef]
- Pearce, D.; Soundararajan, R.; Trimpert, C.; Kashlan, O.B.; Deen, P.M.; Kohan, D.E. Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 135–146. [Google Scholar] [CrossRef]
- O’Connell, A.D.; Leng, Q.; Dong, K.; MacGregor, G.G.; Giebisch, G.; Hebert, S.C. Phosphorylation-regulated endoplasmic reticulum retention signal in the renal outer-medullary K+ channel (ROMK). Proc. Natl. Acad. Sci. USA 2005, 102, 9954–9959. [Google Scholar] [CrossRef]
- Welling, P.A.; Ho, K. A comprehensive guide to the ROMK potassium channel: Form and function in health and disease. Am. J. Physiol. Renal Physiol. 2009, 297, F849–F863. [Google Scholar] [CrossRef] [Green Version]
- Staub, O.; Dho, S.; Henry, P.; Correa, J.; Ishikawa, T.; McGlade, J.; Rotin, D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle’s syndrome. EMBO J. 1996, 15, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Patel, S.V.; Snyder, P.M. Nedd4-2 catalyzes ubiquitination and degradation of cell surface ENaC. J. Biol. Chem. 2007, 282, 20207–20212. [Google Scholar] [CrossRef]
- Shimkets, R.A.; Warnock, D.G.; Bositis, C.M.; Nelson-Williams, C.; Hansson, J.H.; Schambelan, M.; Gill, J.R., Jr.; Ulick, S.; Milora, R.V.; Findling, J.W.; et al. Liddle’s syndrome: Heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 1994, 79, 407–414. [Google Scholar] [CrossRef]
- Harris, M.; Garcia-Caballero, A.; Stutts, M.J.; Firsov, D.; Rossier, B.C. Preferential assembly of epithelial sodium channel (ENaC) subunits in Xenopus oocytes: Role of furin-mediated endogenous proteolysis. J. Biol. Chem. 2008, 283, 7455–7463. [Google Scholar] [CrossRef] [PubMed]
- Hughey, R.P.; Bruns, J.B.; Kinlough, C.L.; Harkleroad, K.L.; Tong, Q.; Carattino, M.D.; Johnson, J.P.; Stockand, J.D.; Kleyman, T.R. Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 2004, 279, 18111–18114. [Google Scholar] [CrossRef] [PubMed]
- Hughey, R.P.; Bruns, J.B.; Kinlough, C.L.; Kleyman, T.R. Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J. Biol. Chem. 2004, 279, 48491–48494. [Google Scholar] [CrossRef]
- Kleyman, T.R.; Kashlan, O.B.; Hughey, R.P. Epithelial Na(+) Channel Regulation by Extracellular and Intracellular Factors. Annu. Rev. Physiol. 2018, 80, 263–281. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, L.; Su, X.T.; Zhang, J.; Lin, D.H.; Wang, W.H. ENaC and ROMK activity are inhibited in the DCT2/CNT of TgWnk4(PHAII) mice. Am. J. Physiol. Renal. Physiol. 2017, 312, F682–F688. [Google Scholar] [CrossRef]
- Kahle, K.T.; Wilson, F.H.; Leng, Q.; Lalioti, M.D.; O’Connell, A.D.; Dong, K.; Rapson, A.K.; MacGregor, G.G.; Giebisch, G.; Hebert, S.C.; et al. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat. Genet. 2003, 35, 372–376. [Google Scholar] [CrossRef]
- Gordon, R.D.; Hodsman, G.P. The syndrome of hypertension and hyperkalaemia without renal failure: Long term correction by thiazide diuretic. Scott Med. J. 1986, 31, 43–44. [Google Scholar] [CrossRef]
- Morsing, P.; Velazquez, H.; Wright, F.S.; Ellison, D.H. Adaptation of distal convoluted tubule of rats. II. Effects of chronic thiazide infusion. Am. J. Physiol. 1991, 261, F137–F143. [Google Scholar] [CrossRef]
- Sica, D.A. Diuretic-related side effects: Development and treatment. J. Clin. Hypertens 2004, 6, 532–540. [Google Scholar] [CrossRef]
- Mori, T.; Kikuchi, E.; Watanabe, Y.; Fujii, S.; Ishigami-Yuasa, M.; Kagechika, H.; Sohara, E.; Rai, T.; Sasaki, S.; Uchida, S. Chemical library screening for WNK signalling inhibitors using fluorescence correlation spectroscopy. BioChem. J. 2013, 455, 339–345. [Google Scholar] [CrossRef]
- Kikuchi, E.; Mori, T.; Zeniya, M.; Isobe, K.; Ishigami-Yuasa, M.; Fujii, S.; Kagechika, H.; Ishihara, T.; Mizushima, T.; Sasaki, S.; et al. Discovery of Novel SPAK Inhibitors That Block WNK Kinase Signaling to Cation Chloride Transporters. J. Am. Soc. Nephrol. 2015, 26, 1525–1536. [Google Scholar] [CrossRef]
- Zhang, J.; Bhuiyan, M.I.H.; Zhang, T.; Karimy, J.K.; Wu, Z.; Fiesler, V.M.; Zhang, J.; Huang, H.; Hasan, M.N.; Skrzypiec, A.E.; et al. Modulation of brain cation-Cl(-) cotransport via the SPAK kinase inhibitor ZT-1a. Nat. Commun. 2020, 11, 78. [Google Scholar] [CrossRef]
- Jonniya, N.A.; Zhang, J.; Kar, P. Molecular Mechanism of Inhibiting WNK Binding to OSR1 by Targeting the Allosteric Pocket of the OSR1-CCT Domain with Potential Antihypertensive Inhibitors: An In Silico Study. J. Phys Chem. B 2021, 125, 9115–9129. [Google Scholar] [CrossRef]
- Xu, Q.; Modrek, B.; Lee, C. Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res. 2002, 30, 3754–3766. [Google Scholar] [CrossRef]
- O’Reilly, M.; Marshall, E.; Speirs, H.J.; Brown, R.W. WNK1, a gene within a novel blood pressure control pathway, tissue-specifically generates radically different isoforms with and without a kinase domain. J. Am. Soc. Nephrol. 2003, 14, 2447–2456. [Google Scholar] [CrossRef]
- Delaloy, C.; Lu, J.; Houot, A.M.; Disse-Nicodeme, S.; Gasc, J.M.; Corvol, P.; Jeunemaitre, X. Multiple promoters in the WNK1 gene: One controls expression of a kidney-specific kinase-defective isoform. Mol. Cell Biol. 2003, 23, 9208–9221. [Google Scholar] [CrossRef]
- Abboud, H.E.; Luke, R.G.; Galla, J.H.; Kotchen, T.A. Stimulation of renin by acute selective chloride depletion in the rat. Circ. Res. 1979, 44, 815–821. [Google Scholar] [CrossRef] [Green Version]
- Kotchen, T.A.; Luke, R.G.; Ott, C.E.; Galla, J.H.; Whitescarver, S. Effect of chloride on renin and blood pressure responses to sodium chloride. Ann. Intern. Med. 1983, 98, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.N.; Forman, J.P.; Farwell, W.R. Serum anion gap and blood pressure in the national health and nutrition examination survey. Hypertension 2007, 50, 320–324. [Google Scholar] [CrossRef] [PubMed]
- McCallum, L.; Jeemon, P.; Hastie, C.E.; Patel, R.K.; Williamson, C.; Redzuan, A.M.; Dawson, J.; Sloan, W.; Muir, S.; Morrison, D.; et al. Serum chloride is an independent predictor of mortality in hypertensive patients. Hypertension 2013, 62, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Lo, Y.F.; Lin, Y.W.; Lin, S.H.; Huang, C.L.; Cheng, C.J. WNK4 kinase is a physiological intracellular chloride sensor. Proc. Natl. Acad. Sci. USA 2019, 116, 4502–4507. [Google Scholar] [CrossRef]
- Piala, A.T.; Moon, T.M.; Akella, R.; He, H.; Cobb, M.H.; Goldsmith, E.J. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci. Signal. 2014, 7, ra41. [Google Scholar] [CrossRef]
- Pleinis, J.M.; Norrell, L.; Akella, R.; Humphreys, J.M.; He, H.; Sun, Q.; Zhang, F.; Sosa-Pagan, J.; Morrison, D.E.; Schellinger, J.N.; et al. WNKs are potassium-sensitive kinases. Am. J. Physiol. Cell Physiol. 2021, 320, C703–C721. [Google Scholar] [CrossRef]
- Nishida, H.; Sohara, E.; Nomura, N.; Chiga, M.; Alessi, D.R.; Rai, T.; Sasaki, S.; Uchida, S. Phosphatidylinositol 3-kinase/Akt signaling pathway activates the WNK-OSR1/SPAK-NCC phosphorylation cascade in hyperinsulinemic db/db mice. Hypertension 2012, 60, 981–990. [Google Scholar] [CrossRef]
- Shiojima, I.; Walsh, K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 2002, 90, 1243–1250. [Google Scholar] [CrossRef]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef] [Green Version]
- Nanamatsu, A.; Mori, T.; Ando, F.; Furusho, T.; Mandai, S.; Susa, K.; Sohara, E.; Rai, T.; Uchida, S. Vasopressin Induces Urinary Uromodulin Secretion By Activating PKA (Protein Kinase A). Hypertension 2021, 77, 1953–1963. [Google Scholar] [CrossRef]
- Aoi, W.; Niisato, N.; Sawabe, Y.; Miyazaki, H.; Marunaka, Y. Aldosterone-induced abnormal regulation of ENaC and SGK1 in Dahl salt-sensitive rat. BioChem. Biophys. Res. Commun. 2006, 341, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Masilamani, S.; Turner, R.; Mitchell, C.; Wade, J.B.; Knepper, M.A. The thiazide-sensitive Na-Cl cotransporter is an aldosterone-induced protein. Proc. Natl. Acad. Sci. USA 1998, 95, 14552–14557. [Google Scholar] [CrossRef]
- Wang, J.; Barbry, P.; Maiyar, A.C.; Rozansky, D.J.; Bhargava, A.; Leong, M.; Firestone, G.L.; Pearce, D. SGK integrates insulin and mineralocorticoid regulation of epithelial sodium transport. Am. J. Physiol. Renal Physiol. 2001, 280, F303–F313. [Google Scholar] [CrossRef]
- Tong, Q.; Booth, R.E.; Worrell, R.T.; Stockand, J.D. Regulation of Na+ transport by aldosterone: Signaling convergence and cross talk between the PI3-K and MAPK1/2 cascades. Am. J. Physiol. Renal Physiol. 2004, 286, F1232–F1238. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.E.; Stippec, S.; Lazrak, A.; Huang, C.L.; Cobb, M.H. WNK1 activates SGK1 by a phosphatidylinositol 3-kinase-dependent and non-catalytic mechanism. J. Biol. Chem. 2005, 280, 34218–34223. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.E.; Stippec, S.; Chu, P.Y.; Lazrak, A.; Li, X.J.; Lee, B.H.; English, J.M.; Ortega, B.; Huang, C.L.; Cobb, M.H. WNK1 activates SGK1 to regulate the epithelial sodium channel. Proc. Natl. Acad. Sci. USA 2005, 102, 10315–10320. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Park, H.M.; Rigel, D.F.; DiPetrillo, K.; Whalen, E.J.; Anisowicz, A.; Beil, M.; Berstler, J.; Brocklehurst, C.E.; Burdick, D.A.; et al. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat. Chem. Biol. 2016, 12, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Disse-Nicodeme, S.; Achard, J.M.; Desitter, I.; Houot, A.M.; Fournier, A.; Corvol, P.; Jeunemaitre, X. A new locus on chromosome 12p13.3 for pseudohypoaldosteronism type II, an autosomal dominant form of hypertension. Am. J. Hum Genet. 2000, 67, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Armando, I.; Upadhyay, K.; Pascua, A.; Jose, P.A. The regulation of proximal tubular salt transport in hypertension: An update. Curr. Opin. Nephrol. Hypertens. 2009, 18, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Fenton, R.A.; Poulsen, S.B.; de la Mora Chavez, S.; Soleimani, M.; Dominguez Rieg, J.A.; Rieg, T. Renal tubular NHE3 is required in the maintenance of water and sodium chloride homeostasis. Kidney Int. 2017, 92, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Shull, G.E.; Miguel-Qin, E.; Zhuo, J.L. Role of the Na+/H+ exchanger 3 in angiotensin II-induced hypertension. Physiol. Genom. 2015, 47, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Zhu, D.; Chen, X.; Zheng, X.; Zhao, C.; Zhang, J.; Soleimani, M.; Rubera, I.; Tauc, M.; Zhou, X.; et al. Proximal Tubule-Specific Deletion of the NHE3 (Na(+)/H(+) Exchanger 3) in the Kidney Attenuates Ang II (Angiotensin II)-Induced Hypertension in Mice. Hypertension 2019, 74, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Zheng, X.; Chen, X.; Zhao, C.; Zhu, D.; Zhang, J.; Zhuo, J.L. Genetic and genomic evidence for an important role of the Na(+)/H(+) exchanger 3 in blood pressure regulation and angiotensin II-induced hypertension. Physiol. Genom. 2019, 51, 97–108. [Google Scholar] [CrossRef]
- Yin, J.; Tse, C.M.; Cha, B.; Sarker, R.; Zhu, X.C.; Walentinsson, A.; Greasley, P.J.; Donowitz, M. A common NHE3 single-nucleotide polymorphism has normal function and sensitivity to regulatory ligands. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G129–G137. [Google Scholar] [CrossRef] [PubMed]
- Baum, M.; Moe, O.W.; Gentry, D.L.; Alpern, R.J. Effect of glucocorticoids on renal cortical NHE-3 and NHE-1 mRNA. Am. J. Physiol. 1994, 267, F437–F442. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, H.; Lang, F.; Yun, C.C. Acute activation of NHE3 by dexamethasone correlates with activation of SGK1 and requires a functional glucocorticoid receptor. Am. J. Physiol. Cell Physiol. 2007, 292, C396–C404. [Google Scholar] [CrossRef]
- Baum, M.; Cano, A.; Alpern, R.J. Glucocorticoids stimulate Na+/H+ antiporter in OKP cells. Am. J. Physiol. 1993, 264, F1027–F1031. [Google Scholar] [CrossRef]
- Chen, S.Y.; Bhargava, A.; Mastroberardino, L.; Meijer, O.C.; Wang, J.; Buse, P.; Firestone, G.L.; Verrey, F.; Pearce, D. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc. Natl. Acad. Sci. USA 1999, 96, 2514–2519. [Google Scholar] [CrossRef]
- Fuster, D.G.; Bobulescu, I.A.; Zhang, J.; Wade, J.; Moe, O.W. Characterization of the regulation of renal Na+/H+ exchanger NHE3 by insulin. Am. J. Physiol. Renal. Physiol. 2007, 292, F577–F585. [Google Scholar] [CrossRef] [Green Version]
- Klisic, J.; Hu, M.C.; Nief, V.; Reyes, L.; Fuster, D.; Moe, O.W.; Ambuhl, P.M. Insulin activates Na(+)/H(+) exchanger 3: Biphasic response and glucocorticoid dependence. Am. J. Physiol. Renal. Physiol. 2002, 283, F532–F539. [Google Scholar] [CrossRef]
- Bobulescu, I.A.; Dwarakanath, V.; Zou, L.; Zhang, J.; Baum, M.; Moe, O.W. Glucocorticoids acutely increase cell surface Na+/H+ exchanger-3 (NHE3) by activation of NHE3 exocytosis. Am. J. Physiol. Renal. Physiol. 2005, 289, F685–F691. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.S.; Wu, K.D.; Wu, M.S.; Hsieh, B.S. Endothelin-1 chronically inhibits Na/H exchanger-3 in ET(B)-overexpressing OKP cells. BioChem. Biophys. Res. Commun. 2000, 271, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Kocinsky, H.S.; Dynia, D.W.; Wang, T.; Aronson, P.S. NHE3 phosphorylation at serines 552 and 605 does not directly affect NHE3 activity. Am. J. Physiol. Renal. Physiol. 2007, 293, F212–F218. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, R.; Braucht, D.W.; Rinschen, M.M.; Chou, C.L.; Hoffert, J.D.; Pisitkun, T.; Knepper, M.A. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc. Natl. Acad. Sci. USA 2010, 107, 15653–15658. [Google Scholar] [CrossRef]
- Chen, T.; Kocinsky, H.S.; Cha, B.; Murtazina, R.; Yang, J.; Tse, C.M.; Singh, V.; Cole, R.; Aronson, P.S.; de Jonge, H.; et al. Cyclic GMP kinase II (cGKII) inhibits NHE3 by altering its trafficking and phosphorylating NHE3 at three required sites: Identification of a multifunctional phosphorylation site. J. Biol. Chem. 2015, 290, 1952–1965. [Google Scholar] [CrossRef]
- Lo, Y.F.; Yang, S.S.; Seki, G.; Yamada, H.; Horita, S.; Yamazaki, O.; Fujita, T.; Usui, T.; Tsai, J.D.; Yu, I.S.; et al. Severe metabolic acidosis causes early lethality in NBC1 W516X knock-in mice as a model of human isolated proximal renal tubular acidosis. Kidney Int. 2011, 79, 730–741. [Google Scholar] [CrossRef]
- Li, Y.; Yamada, H.; Kita, Y.; Kunimi, M.; Horita, S.; Suzuki, M.; Endo, Y.; Shimizu, T.; Seki, G.; Fujita, T. Roles of ERK and cPLA2 in the angiotensin II-mediated biphasic regulation of Na+-HCO3(-) transport. J. Am. Soc. Nephrol. 2008, 19, 252–259. [Google Scholar] [CrossRef]
- Zheng, Y.; Yamada, H.; Sakamoto, K.; Horita, S.; Kunimi, M.; Endo, Y.; Li, Y.; Tobe, K.; Terauchi, Y.; Kadowaki, T.; et al. Roles of insulin receptor substrates in insulin-induced stimulation of renal proximal bicarbonate absorption. J. Am. Soc. Nephrol. 2005, 16, 2288–2295. [Google Scholar] [CrossRef]
- Sonalker, P.A.; Tofovic, S.P.; Bastacky, S.I.; Jackson, E.K. Chronic noradrenaline increases renal expression of NHE-3, NBC-1, BSC-1 and aquaporin-2. Clin. Exp Pharmacol. Physiol. 2008, 35, 594–600. [Google Scholar] [CrossRef]
- Nakamura, M.; Shirai, A.; Yamazaki, O.; Satoh, N.; Suzuki, M.; Horita, S.; Yamada, H.; Seki, G. Roles of renal proximal tubule transport in acid/base balance and blood pressure regulation. BioMed. Res. Int. 2014, 2014, 504808. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Capasso, G.; Schwab, M.; Waldegger, S. Renal tubular transport and the genetic basis of hypertensive disease. Clin. Exp Nephrol. 2005, 9, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Hebert, S.C.; Andreoli, T.E. Ionic conductance pathways in the mouse medullary thick ascending limb of Henle. The paracellular pathway and electrogenic Cl- absorption. J. Gen Physiol. 1986, 87, 567–590. [Google Scholar] [CrossRef] [PubMed]
- Castrop, H.; Schnermann, J. Isoforms of renal Na-K-2Cl cotransporter NKCC2: Expression and functional significance. Am. J. Physiol. Renal. Physiol. 2008, 295, F859–F866. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.A.; Xu, J.C.; Haas, M.; Lytle, C.Y.; Ward, D.; Forbush, B., 3rd. Primary structure, functional expression, and chromosomal localization of the bumetanide-sensitive Na-K-Cl cotransporter in human colon. J. Biol. Chem. 1995, 270, 17977–17985. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, M.; Mizel, D.; Kim, S.M.; Chen, L.; Faulhaber-Walter, R.; Huang, Y.; Li, C.; Deng, C.; Briggs, J.; Schnermann, J.; et al. Renal function in mice with targeted disruption of the A isoform of the Na-K-2Cl co-transporter. J. Am. Soc. Nephrol. 2007, 18, 440–448. [Google Scholar] [CrossRef]
- Ji, W.; Foo, J.N.; O’Roak, B.J.; Zhao, H.; Larson, M.G.; Simon, D.B.; Newton-Cheh, C.; State, M.W.; Levy, D.; Lifton, R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008, 40, 592–599. [Google Scholar] [CrossRef]
- Adachi, M.; Asakura, Y.; Sato, Y.; Tajima, T.; Nakajima, T.; Yamamoto, T.; Fujieda, K. Novel SLC12A1 (NKCC2) mutations in two families with Bartter syndrome type 1. Endocr J. 2007, 54, 1003–1007. [Google Scholar] [CrossRef]
- Mutig, K.; Kahl, T.; Saritas, T.; Godes, M.; Persson, P.; Bates, J.; Raffi, H.; Rampoldi, L.; Uchida, S.; Hille, C.; et al. Activation of the bumetanide-sensitive Na+,K+,2Cl- cotransporter (NKCC2) is facilitated by Tamm-Horsfall protein in a chloride-sensitive manner. J. Biol. Chem. 2011, 286, 30200–30210. [Google Scholar] [CrossRef]
- Ares, G.R.; Haque, M.Z.; Delpire, E.; Ortiz, P.A. Hyperphosphorylation of Na-K-2Cl cotransporter in thick ascending limbs of Dahl salt-sensitive rats. Hypertension 2012, 60, 1464–1470. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Basile, D.P.; Pratt, J.H. Sodium reabsorption in the thick ascending limb in relation to blood pressure: A clinical perspective. Hypertension 2011, 57, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Glorioso, N.; Filigheddu, F.; Troffa, C.; Soro, A.; Parpaglia, P.P.; Tsikoudakis, A.; Myers, R.H.; Herrera, V.L.; Ruiz-Opazo, N. Interaction of alpha(1)-Na,K-ATPase and Na,K,2Cl-cotransporter genes in human essential hypertension. Hypertension 2001, 38, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Seaayfan, E.; Defontaine, N.; Demaretz, S.; Zaarour, N.; Laghmani, K. OS9 Protein Interacts with Na-K-2Cl Co-transporter (NKCC2) and Targets Its Immature Form for the Endoplasmic Reticulum-associated Degradation Pathway. J. Biol. Chem. 2016, 291, 4487–4502. [Google Scholar] [CrossRef] [PubMed]
- Caceres, P.S.; Ares, G.R.; Ortiz, P.A. cAMP stimulates apical exocytosis of the renal Na(+)-K(+)-2Cl(-) cotransporter NKCC2 in the thick ascending limb: Role of protein kinase A. J. Biol. Chem. 2009, 284, 24965–24971. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Urushiyama, S.; Hisamoto, N.; Iemura, S.; Uchida, S.; Natsume, T.; Matsumoto, K.; Shibuya, H. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J. Biol. Chem. 2005, 280, 42685–42693. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Yu, I.S.; Jiang, S.T.; Lin, S.W.; Chu, P.; Chen, A.; Sytwu, H.K.; Sohara, E.; Uchida, S.; Sasaki, S.; et al. Impaired phosphorylation of Na(+)-K(+)-2Cl(-) cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 17538–17543. [Google Scholar] [CrossRef]
- McCormick, J.A.; Mutig, K.; Nelson, J.H.; Saritas, T.; Hoorn, E.J.; Yang, C.L.; Rogers, S.; Curry, J.; Delpire, E.; Bachmann, S.; et al. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 2011, 14, 352–364. [Google Scholar] [CrossRef]
- Yang, S.S.; Lo, Y.F.; Wu, C.C.; Lin, S.W.; Yeh, C.J.; Chu, P.; Sytwu, H.K.; Uchida, S.; Sasaki, S.; Lin, S.H. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J. Am. Soc. Nephrol. 2010, 21, 1868–1877. [Google Scholar] [CrossRef]
- Ponce-Coria, J.; San-Cristobal, P.; Kahle, K.T.; Vazquez, N.; Pacheco-Alvarez, D.; de Los Heros, P.; Juarez, P.; Munoz, E.; Michel, G.; Bobadilla, N.A.; et al. Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc. Natl. Acad. Sci. USA 2008, 105, 8458–8463. [Google Scholar] [CrossRef]
- Filippi, B.M.; de los Heros, P.; Mehellou, Y.; Navratilova, I.; Gourlay, R.; Deak, M.; Plater, L.; Toth, R.; Zeqiraj, E.; Alessi, D.R. MO25 is a master regulator of SPAK/OSR1 and MST3/MST4/YSK1 protein kinases. EMBO J. 2011, 30, 1730–1741. [Google Scholar] [CrossRef] [Green Version]
- Gimenez, I.; Forbush, B. Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J. Biol. Chem. 2003, 278, 26946–26951. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, I.; Forbush, B. Regulatory phosphorylation sites in the NH2 terminus of the renal Na-K-Cl cotransporter (NKCC2). Am. J. Physiol. Renal. Physiol. 2005, 289, F1341–F1345. [Google Scholar] [CrossRef]
- Piechotta, K.; Lu, J.; Delpire, E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J. Biol. Chem. 2002, 277, 50812–50819. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Sakamoto, K.; de los Heros, P.; Deak, M.; Campbell, D.G.; Prescott, A.R.; Alessi, D.R. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J. Cell Sci. 2011, 124, 789–800. [Google Scholar] [CrossRef]
- Mutig, K.; Paliege, A.; Kahl, T.; Jons, T.; Muller-Esterl, W.; Bachmann, S. Vasopressin V2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. Am. J. Physiol. Renal. Physiol. 2007, 293, F1166–F1177. [Google Scholar] [CrossRef]
- Feric, M.; Zhao, B.; Hoffert, J.D.; Pisitkun, T.; Knepper, M.A. Large-scale phosphoproteomic analysis of membrane proteins in renal proximal and distal tubule. Am. J. Physiol. Cell Physiol. 2011, 300, C755–C770. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, P.A. cAMP increases surface expression of NKCC2 in rat thick ascending limbs: Role of VAMP. Am. J. Physiol. Renal. Physiol. 2006, 290, F608–F616. [Google Scholar] [CrossRef]
- Ares, G.R.; Caceres, P.S.; Ortiz, P.A. Molecular regulation of NKCC2 in the thick ascending limb. Am. J. Physiol. Renal. Physiol. 2011, 301, F1143–F1159. [Google Scholar] [CrossRef] [PubMed]
- Bhalla, V.; Oyster, N.M.; Fitch, A.C.; Wijngaarden, M.A.; Neumann, D.; Schlattner, U.; Pearce, D.; Hallows, K.R. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4-2. J. Biol. Chem. 2006, 281, 26159–26169. [Google Scholar] [CrossRef]
- Carattino, M.D.; Edinger, R.S.; Grieser, H.J.; Wise, R.; Neumann, D.; Schlattner, U.; Johnson, J.P.; Kleyman, T.R.; Hallows, K.R. Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J. Biol. Chem. 2005, 280, 17608–17616. [Google Scholar] [CrossRef] [Green Version]
- Reiche, J.; Theilig, F.; Rafiqi, F.H.; Carlo, A.S.; Militz, D.; Mutig, K.; Todiras, M.; Christensen, E.I.; Ellison, D.H.; Bader, M.; et al. SORLA/SORL1 functionally interacts with SPAK to control renal activation of Na(+)-K(+)-Cl(-) cotransporter 2. Mol. Cell Biol. 2010, 30, 3027–3037. [Google Scholar] [CrossRef] [PubMed]
- Borschewski, A.; Himmerkus, N.; Boldt, C.; Blankenstein, K.I.; McCormick, J.A.; Lazelle, R.; Willnow, T.E.; Jankowski, V.; Plain, A.; Bleich, M.; et al. Calcineurin and Sorting-Related Receptor with A-Type Repeats Interact to Regulate the Renal Na(+)-K(+)-2Cl(-) Cotransporter. J. Am. Soc. Nephrol. 2016, 27, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Paredes, A.; Plata, C.; Rivera, M.; Moreno, E.; Vazquez, N.; Munoz-Clares, R.; Hebert, S.C.; Gamba, G. Activity of the renal Na+-K+-2Cl− cotransporter is reduced by mutagenesis of N-glycosylation sites: Role for protein surface charge in Cl- transport. Am. J. Physiol. Renal. Physiol. 2006, 290, F1094–F1102. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Alzamora, R.; Smolak, C.; Li, H.; Naveed, S.; Neumann, D.; Hallows, K.R.; Pastor-Soler, N.M. Vacuolar H+-ATPase apical accumulation in kidney intercalated cells is regulated by PKA and AMP-activated protein kinase. Am. J. Physiol. Renal. Physiol. 2010, 298, F1162–F1169. [Google Scholar] [CrossRef]
- Greger, R.; Bleich, M.; Schlatter, E. Ion channels in the thick ascending limb of Henle’s loop. Ren. Physiol. BioChem. 1990, 13, 37–50. [Google Scholar] [CrossRef]
- Graham, L.A.; Dominiczak, A.F.; Ferreri, N.R. Role of renal transporters and novel regulatory interactions in the TAL that control blood pressure. Physiol. Genomics 2017, 49, 261–276. [Google Scholar] [CrossRef]
- MacGregor, G.A.; Fenton, S.; Alaghband-Zadeh, J.; Markandu, N.D.; Roulston, J.E.; de Wardener, H.E. An increase in a circulating inhibitor of Na+,K+-dependent ATPase: A possible link between salt intake and the development of essential hypertension. Clin. Sci. 1981, 61 (Suppl. 7), 17s–20s. [Google Scholar] [CrossRef]
- O’Brien, W.J.; Lingrel, J.B.; Wallick, E.T. Ouabain binding kinetics of the rat alpha two and alpha three isoforms of the sodium-potassium adenosine triphosphate. Arch BioChem. Biophys. 1994, 310, 32–39. [Google Scholar] [CrossRef]
- Haque, M.Z.; Ares, G.R.; Caceres, P.S.; Ortiz, P.A. High salt differentially regulates surface NKCC2 expression in thick ascending limbs of Dahl salt-sensitive and salt-resistant rats. Am. J. Physiol. Renal Physiol. 2011, 300, F1096–F1104. [Google Scholar] [CrossRef]
- Fang, L.; Li, D.; Welling, P.A. Hypertension resistance polymorphisms in ROMK (Kir1.1) alter channel function by different mechanisms. Am. J. Physiol. Renal Physiol. 2010, 299, F1359–F1364. [Google Scholar] [CrossRef] [Green Version]
- Battula, S.; Hao, S.; Pedraza, P.L.; Stier, C.T.; Ferreri, N.R. Tumor necrosis factor-alpha is an endogenous inhibitor of Na+-K+-2Cl− cotransporter (NKCC2) isoform A in the thick ascending limb. Am. J. Physiol. Renal Physiol. 2011, 301, F94–F100. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.; Nichols, C.G.; Lederer, W.J.; Lytton, J.; Vassilev, P.M.; Kanazirska, M.V.; Hebert, S.C. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 1993, 362, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Mennitt, P.A.; Wade, J.B.; Ecelbarger, C.A.; Palmer, L.G.; Frindt, G. Localization of ROMK channels in the rat kidney. J. Am. Soc. Nephrol. 1997, 8, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wang, T.; Yan, Q.; Yang, X.; Dong, K.; Knepper, M.A.; Wang, W.; Giebisch, G.; Shull, G.E.; Hebert, S.C. Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter’s) knockout mice. J. Biol. Chem. 2002, 277, 37881–37887. [Google Scholar] [CrossRef]
- Muto, S.; Asano, Y.; Okazaki, H.; Kano, S. Renal potassium wasting in distal renal tubular acidosis: Role of aldosterone. Intern. Med. 1992, 31, 1047–1051. [Google Scholar] [CrossRef]
- Valles, P.G.; Batlle, D. Hypokalemic Distal Renal Tubular Acidosis. Adv Chronic Kidney Dis. 2018, 25, 303–320. [Google Scholar] [CrossRef]
- Simon, D.B.; Karet, F.E.; Rodriguez-Soriano, J.; Hamdan, J.H.; DiPietro, A.; Trachtman, H.; Sanjad, S.A.; Lifton, R.P. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat. Genet. 1996, 14, 152–156. [Google Scholar] [CrossRef]
- Leung, Y.M.; Zeng, W.Z.; Liou, H.H.; Solaro, C.R.; Huang, C.L. Phosphatidylinositol 4,5-bisphosphate and intracellular pH regulate the ROMK1 potassium channel via separate but interrelated mechanisms. J. Biol. Chem. 2000, 275, 10182–10189. [Google Scholar] [CrossRef]
- Leng, Q.; Kahle, K.T.; Rinehart, J.; MacGregor, G.G.; Wilson, F.H.; Canessa, C.M.; Lifton, R.P.; Hebert, S.C. WNK3, a kinase related to genes mutated in hereditary hypertension with hyperkalaemia, regulates the K+ channel ROMK1 (Kir1.1). J. Physiol. 2006, 571, 275–286. [Google Scholar] [CrossRef]
- Yang, L.; Xu, Y.; Gravotta, D.; Frindt, G.; Weinstein, A.M.; Palmer, L.G. ENaC and ROMK channels in the connecting tubule regulate renal K+ secretion. J. Gen Physiol. 2021, 153, e202112902. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, Z.; Shin, M.K.; Horwitz, S.B.; Levorse, J.M.; Zhu, L.; Sharif-Rodriguez, W.; Streltsov, D.Y.; Dajee, M.; Hernandez, M.; et al. Heterozygous disruption of renal outer medullary potassium channel in rats is associated with reduced blood pressure. Hypertension 2013, 62, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Forrest, M.J.; Sharif-Rodriguez, W.; Forrest, G.; Szeto, D.; Urosevic-Price, O.; Zhu, Y.; Stevenson, A.S.; Zhou, Y.; Stribling, S.; et al. Chronic Inhibition of Renal Outer Medullary Potassium Channel Not Only Prevented but Also Reversed Development of Hypertension and End-Organ Damage in Dahl Salt-Sensitive Rats. Hypertension 2017, 69, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhu, Y.; Teumelsan, N.; Walsh, S.P.; Shahripour, A.; Priest, B.T.; Swensen, A.M.; Felix, J.P.; Brochu, R.M.; Bailey, T.; et al. Discovery of MK-7145, an Oral Small Molecule ROMK Inhibitor for the Treatment of Hypertension and Heart Failure. ACS Med. Chem. Lett. 2016, 7, 697–701. [Google Scholar] [CrossRef]
- Wolf, M.T.F.; Zhang, J.; Nie, M. Uromodulin in mineral metabolism. Curr. Opin. Nephrol. Hypertens. 2019, 28, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Pennica, D.; Kohr, W.J.; Kuang, W.J.; Glaister, D.; Aggarwal, B.B.; Chen, E.Y.; Goeddel, D.V. Identification of human uromodulin as the Tamm-Horsfall urinary glycoprotein. Science 1987, 236, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Tokonami, N.; Takata, T.; Beyeler, J.; Ehrbar, I.; Yoshifuji, A.; Christensen, E.I.; Loffing, J.; Devuyst, O.; Olinger, E.G. Uromodulin is expressed in the distal convoluted tubule, where it is critical for regulation of the sodium chloride cotransporter NCC. Kidney Int. 2018, 94, 701–715. [Google Scholar] [CrossRef]
- Devuyst, O.; Olinger, E.; Rampoldi, L. Uromodulin: From physiology to rare and complex kidney disorders. Nat. Rev. Nephrol. 2017, 13, 525–544. [Google Scholar] [CrossRef]
- Devuyst, O.; Dahan, K.; Pirson, Y. Tamm-Horsfall protein or uromodulin: New ideas about an old molecule. Nephrol. Dial Transpl. 2005, 20, 1290–1294. [Google Scholar] [CrossRef]
- Olinger, E.; Lake, J.; Sheehan, S.; Schiano, G.; Takata, T.; Tokonami, N.; Debaix, H.; Consolato, F.; Rampoldi, L.; Korstanje, R.; et al. Hepsin-mediated Processing of Uromodulin is Crucial for Salt-sensitivity and Thick Ascending Limb Homeostasis. Sci. Rep. 2019, 9, 12287. [Google Scholar] [CrossRef]
- Paladino, S.; Lebreton, S.; Tivodar, S.; Campana, V.; Tempre, R.; Zurzolo, C. Different GPI-attachment signals affect the oligomerisation of GPI-anchored proteins and their apical sorting. J. Cell Sci. 2008, 121, 4001–4007. [Google Scholar] [CrossRef] [Green Version]
- Welker, P.; Bohlick, A.; Mutig, K.; Salanova, M.; Kahl, T.; Schluter, H.; Blottner, D.; Ponce-Coria, J.; Gamba, G.; Bachmann, S. Renal Na+-K+-Cl− cotransporter activity and vasopressin-induced trafficking are lipid raft-dependent. Am. J. Physiol. Renal. Physiol. 2008, 295, F789–F802. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.S.; Steinberg, S.L.; Liederman, K.; Pfeiffer, J.R.; Surviladze, Z.; Zhang, J.; Samelson, L.E.; Yang, L.H.; Kotula, P.G.; Oliver, J.M. Markers for detergent-resistant lipid rafts occupy distinct and dynamic domains in native membranes. Mol. Biol. Cell 2004, 15, 2580–2592. [Google Scholar] [CrossRef] [PubMed]
- Trudu, M.; Janas, S.; Lanzani, C.; Debaix, H.; Schaeffer, C.; Ikehata, M.; Citterio, L.; Demaretz, S.; Trevisani, F.; Ristagno, G.; et al. Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat. Med. 2013, 19, 1655–1660. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, S.; Mutig, K.; Bates, J.; Welker, P.; Geist, B.; Gross, V.; Luft, F.C.; Alenina, N.; Bader, M.; Thiele, B.J.; et al. Renal effects of Tamm-Horsfall protein (uromodulin) deficiency in mice. Am. J. Physiol. Renal. Physiol. 2005, 288, F559–F567. [Google Scholar] [CrossRef]
- Renigunta, A.; Renigunta, V.; Saritas, T.; Decher, N.; Mutig, K.; Waldegger, S. Tamm-Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function. J. Biol. Chem. 2011, 286, 2224–2235. [Google Scholar] [CrossRef]
- Rampoldi, L.; Caridi, G.; Santon, D.; Boaretto, F.; Bernascone, I.; Lamorte, G.; Tardanico, R.; Dagnino, M.; Colussi, G.; Scolari, F.; et al. Allelism of MCKD, FJHN and GCKD caused by impairment of uromodulin export dynamics. Hum Mol. Genet. 2003, 12, 3369–3384. [Google Scholar] [CrossRef] [PubMed]
- Gudbjartsson, D.F.; Holm, H.; Indridason, O.S.; Thorleifsson, G.; Edvardsson, V.; Sulem, P.; de Vegt, F.; d’Ancona, F.C.; den Heijer, M.; Wetzels, J.F.; et al. Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet. 2010, 6, e1001039. [Google Scholar] [CrossRef]
- Troyanov, S.; Delmas-Frenette, C.; Bollee, G.; Youhanna, S.; Bruat, V.; Awadalla, P.; Devuyst, O.; Madore, F. Clinical, Genetic, and Urinary Factors Associated with Uromodulin Excretion. Clin. J. Am. Soc. Nephrol. 2016, 11, 62–69. [Google Scholar] [CrossRef]
- Kottgen, A.; Hwang, S.J.; Larson, M.G.; Van Eyk, J.E.; Fu, Q.; Benjamin, E.J.; Dehghan, A.; Glazer, N.L.; Kao, W.H.; Harris, T.B.; et al. Uromodulin levels associate with a common UMOD variant and risk for incident CKD. J. Am. Soc. Nephrol. 2010, 21, 337–344. [Google Scholar] [CrossRef]
- Kottgen, A.; Glazer, N.L.; Dehghan, A.; Hwang, S.J.; Katz, R.; Li, M.; Yang, Q.; Gudnason, V.; Launer, L.J.; Harris, T.B.; et al. Multiple loci associated with indices of renal function and chronic kidney disease. Nat. Genet. 2009, 41, 712–717. [Google Scholar] [CrossRef] [Green Version]
- Nqebelele, N.U.; Dickens, C.; Dix-Peek, T.; Duarte, R.; Naicker, S. Urinary Uromodulin Levels and UMOD Variants in Black South Africans with Hypertension-Attributed Chronic Kidney Disease. Int. J. Nephrol. 2019, 2019, 8094049. [Google Scholar] [CrossRef]
- Chun, J.; Wang, M.; Wilkins, M.S.; Knob, A.U.; Benjamin, A.; Bu, L.; Pollak, M.R. Autosomal Dominant Tubulointerstitial Kidney Disease-Uromodulin Misclassified as Focal Segmental Glomerulosclerosis or Hereditary Glomerular Disease. Kidney Int. Rep. 2020, 5, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wolley, M.; Stowasser, M. The interplay of renal potassium and sodium handling in blood pressure regulation: Critical role of the WNK-SPAK-NCC pathway. J. Hum. Hypertens. 2019, 33, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Wardak, H.; Tutakhel, O.A.Z.; Van Der Wijst, J. Role of the alternative splice variant of NCC in blood pressure control. Channels 2018, 12, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H.; Velazquez, H.; Wright, F.S. Thiazide-sensitive sodium chloride cotransport in early distal tubule. Am. J. Physiol. 1987, 253, F546–F554. [Google Scholar] [CrossRef]
- Moreno, E.; Plata, C.; Rodriguez-Gama, A.; Argaiz, E.R.; Vazquez, N.; Leyva-Rios, K.; Islas, L.; Cutler, C.; Pacheco-Alvarez, D.; Mercado, A.; et al. The European Eel NCCbeta Gene Encodes a Thiazide-resistant Na-Cl Cotransporter. J. Biol. Chem. 2016, 291, 22472–22481. [Google Scholar] [CrossRef]
- Castaneda-Bueno, M.; Vazquez, N.; Bustos-Jaimes, I.; Hernandez, D.; Rodriguez-Lobato, E.; Pacheco-Alvarez, D.; Carino-Cortes, R.; Moreno, E.; Bobadilla, N.A.; Gamba, G. A single residue in transmembrane domain 11 defines the different affinity for thiazides between the mammalian and flounder NaCl transporters. Am. J. Physiol. Renal Physiol. 2010, 299, F1111–F1119. [Google Scholar] [CrossRef]
- Valdez-Flores, M.A.; Vargas-Poussou, R.; Verkaart, S.; Tutakhel, O.A.; Valdez-Ortiz, A.; Blanchard, A.; Treard, C.; Hoenderop, J.G.; Bindels, R.J.; Jelen, S. Functionomics of NCC mutations in Gitelman syndrome using a novel mammalian cell-based activity assay. Am. J. Physiol. Renal Physiol. 2016, 311, F1159–F1167. [Google Scholar] [CrossRef]
- Simon, D.B.; Nelson-Williams, C.; Bia, M.J.; Ellison, D.; Karet, F.E.; Molina, A.M.; Vaara, I.; Iwata, F.; Cushner, H.M.; Koolen, M.; et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 1996, 12, 24–30. [Google Scholar] [CrossRef]
- Pacheco-Alvarez, D.; Cristobal, P.S.; Meade, P.; Moreno, E.; Vazquez, N.; Munoz, E.; Diaz, A.; Juarez, M.E.; Gimenez, I.; Gamba, G. The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J. Biol. Chem. 2006, 281, 28755–28763. [Google Scholar] [CrossRef] [PubMed]
- Saritas, T.; Borschewski, A.; McCormick, J.A.; Paliege, A.; Dathe, C.; Uchida, S.; Terker, A.; Himmerkus, N.; Bleich, M.; Demaretz, S.; et al. SPAK differentially mediates vasopressin effects on sodium cotransporters. J. Am. Soc. Nephrol. 2013, 24, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Lang, Y.; Wang, Y.; Gao, Y.; Zhang, W.; Niu, H.; Liu, S.; Chen, N. High-frequency variant p.T60M in NaCl cotransporter and blood pressure variability in Han Chinese. Am. J. Nephrol. 2012, 35, 515–519. [Google Scholar] [CrossRef]
- Yang, S.S.; Fang, Y.W.; Tseng, M.H.; Chu, P.Y.; Yu, I.S.; Wu, H.C.; Lin, S.W.; Chau, T.; Uchida, S.; Sasaki, S.; et al. Phosphorylation regulates NCC stability and transporter activity in vivo. J. Am. Soc. Nephrol. 2013, 24, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaek, L.L.; Assentoft, M.; Pedersen, N.B.; MacAulay, N.; Fenton, R.A. Characterization of a novel phosphorylation site in the sodium-chloride cotransporter, NCC. J. Physiol. 2012, 590, 6121–6139. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Gamba, G. Regulation of the renal Na+-Cl− cotransporter by phosphorylation and ubiquitylation. Am. J. Physiol. Renal. Physiol. 2012, 303, F1573–F1583. [Google Scholar] [CrossRef]
- Nesterov, V.; Krueger, B.; Bertog, M.; Dahlmann, A.; Palmisano, R.; Korbmacher, C. In Liddle Syndrome, Epithelial Sodium Channel Is Hyperactive Mainly in the Early Part of the Aldosterone-Sensitive Distal Nephron. Hypertension 2016, 67, 1256–1262. [Google Scholar] [CrossRef]
- Fotia, A.B.; Dinudom, A.; Shearwin, K.E.; Koch, J.P.; Korbmacher, C.; Cook, D.I.; Kumar, S. The role of individual Nedd4-2 (KIAA0439) WW domains in binding and regulating epithelial sodium channels. FASEB J. 2003, 17, 70–72. [Google Scholar] [CrossRef]
- Oberfeld, B.; Ruffieux-Daidie, D.; Vitagliano, J.J.; Pos, K.M.; Verrey, F.; Staub, O. Ubiquitin-specific protease 2-45 (Usp2-45) binds to epithelial Na+ channel (ENaC)-ubiquitylating enzyme Nedd4-2. Am. J. Physiol. Renal Physiol. 2011, 301, F189–F196. [Google Scholar] [CrossRef]
- Staub, O.; Gautschi, I.; Ishikawa, T.; Breitschopf, K.; Ciechanover, A.; Schild, L.; Rotin, D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J. 1997, 16, 6325–6336. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.P.; Lagnaz, D.; Ronzaud, C.; Vazquez, N.; Ko, B.S.; Moddes, L.; Ruffieux-Daidie, D.; Hausel, P.; Koesters, R.; Yang, B.; et al. Nedd4-2 modulates renal Na+-Cl− cotransporter via the aldosterone-SGK1-Nedd4-2 pathway. J. Am. Soc. Nephrol. 2011, 22, 1707–1719. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Kawabe, H.; Rotin, D. The Ubiquitin Ligase Nedd4L Regulates the Na/K/2Cl Co-transporter NKCC1/SLC12A2 in the Colon. J. Biol. Chem. 2017, 292, 3137–3145. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, X.; Lai, G.; Yang, X.; Wang, L.; Zhao, Y. Synergistical effect of 20-HETE and high salt on NKCC2 protein and blood pressure via ubiquitin-proteasome pathway. Hum. Genet. 2013, 132, 179–187. [Google Scholar] [CrossRef]
- Meyer-Schwesinger, C. The ubiquitin-proteasome system in kidney physiology and disease. Nat. Rev. Nephrol. 2019, 15, 393–411. [Google Scholar] [CrossRef]
- Rosenbaek, L.L.; Kortenoeven, M.L.; Aroankins, T.S.; Fenton, R.A. Phosphorylation decreases ubiquitylation of the thiazide-sensitive cotransporter NCC and subsequent clathrin-mediated endocytosis. J. Biol. Chem. 2014, 289, 13347–13361. [Google Scholar] [CrossRef]
- Canessa, C.M.; Schild, L.; Buell, G.; Thorens, B.; Gautschi, I.; Horisberger, J.D.; Rossier, B.C. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 1994, 367, 463–467. [Google Scholar] [CrossRef]
- Snyder, P.M.; McDonald, F.J.; Stokes, J.B.; Welsh, M.J. Membrane topology of the amiloride-sensitive epithelial sodium channel. J. Biol. Chem. 1994, 269, 24379–24383. [Google Scholar] [CrossRef]
- Shimkets, R.A.; Lifton, R.; Canessa, C.M. In vivo phosphorylation of the epithelial sodium channel. Proc. Natl. Acad. Sci. USA 1998, 95, 3301–3305. [Google Scholar] [CrossRef]
- May, A.; Puoti, A.; Gaeggeler, H.P.; Horisberger, J.D.; Rossier, B.C. Early effect of aldosterone on the rate of synthesis of the epithelial sodium channel alpha subunit in A6 renal cells. J. Am. Soc. Nephrol. 1997, 8, 1813–1822. [Google Scholar] [CrossRef]
- Thomas, S.V.; Kathpalia, P.P.; Rajagopal, M.; Charlton, C.; Zhang, J.; Eaton, D.C.; Helms, M.N.; Pao, A.C. Epithelial sodium channel regulation by cell surface-associated serum- and glucocorticoid-regulated kinase 1. J. Biol. Chem. 2011, 286, 32074–32085. [Google Scholar] [CrossRef] [PubMed]
- Levanovich, P.E.; Diaczok, A.; Rossi, N.F. Clinical and Molecular Perspectives of Monogenic Hypertension. Curr. Hypertens. Rev. 2020, 16, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Corvol, P. Liddle’s syndrome: Heritable human hypertension caused by mutations in the Beta subunit of the epithelial sodium channel. J. Endocrinol. Investig. 1995, 18, 592–594. [Google Scholar] [CrossRef]
- Enslow, B.T.; Stockand, J.D.; Berman, J.M. Liddle’s syndrome mechanisms, diagnosis and management. Integr. Blood Press. Control 2019, 12, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Pitzer, A.L.; Van Beusecum, J.P.; Kleyman, T.R.; Kirabo, A. ENaC in Salt-Sensitive Hypertension: Kidney and Beyond. Curr. Hypertens. Rep. 2020, 22, 69. [Google Scholar] [CrossRef]
- Tetti, M.; Monticone, S.; Burrello, J.; Matarazzo, P.; Veglio, F.; Pasini, B.; Jeunemaitre, X.; Mulatero, P. Liddle Syndrome: Review of the Literature and Description of a New Case. Int. J. Mol. Sci. 2018, 19, 812. [Google Scholar] [CrossRef]
- Diakov, A.; Korbmacher, C. A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the C terminus of the channel’s alpha-subunit. J. Biol. Chem. 2004, 279, 38134–38142. [Google Scholar] [CrossRef]
- Rauh, R.; Diakov, A.; Tzschoppe, A.; Korbmacher, J.; Azad, A.K.; Cuppens, H.; Cassiman, J.J.; Dotsch, J.; Sticht, H.; Korbmacher, C. A mutation of the epithelial sodium channel associated with atypical cystic fibrosis increases channel open probability and reduces Na+ self inhibition. J. Physiol. 2010, 588, 1211–1225. [Google Scholar] [CrossRef]
- Rauh, R.; Dinudom, A.; Fotia, A.B.; Paulides, M.; Kumar, S.; Korbmacher, C.; Cook, D.I. Stimulation of the epithelial sodium channel (ENaC) by the serum- and glucocorticoid-inducible kinase (Sgk) involves the PY motifs of the channel but is independent of sodium feedback inhibition. Pflugers Arch. 2006, 452, 290–299. [Google Scholar] [CrossRef]
- Christensen, B.M.; Perrier, R.; Wang, Q.; Zuber, A.M.; Maillard, M.; Mordasini, D.; Malsure, S.; Ronzaud, C.; Stehle, J.C.; Rossier, B.C.; et al. Sodium and potassium balance depends on alphaENaC expression in connecting tubule. J. Am. Soc. Nephrol. 2010, 21, 1942–1951. [Google Scholar] [CrossRef] [Green Version]
- Bhogaraju, S.; Kalayil, S.; Liu, Y.; Bonn, F.; Colby, T.; Matic, I.; Dikic, I. Phosphoribosylation of Ubiquitin Promotes Serine Ubiquitination and Impairs Conventional Ubiquitination. Cell 2016, 167, 1636–1649.e1613. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski, J.; Beal, R.; Hoffman, L.; Wilkinson, K.D.; Cohen, R.E.; Pickart, C.M. Inhibition of the 26 S proteasome by polyubiquitin chains synthesized to have defined lengths. J. Biol. Chem. 1997, 272, 23712–23721. [Google Scholar] [CrossRef] [PubMed]
- Alli, A.A.; Bao, H.F.; Alli, A.A.; Aldrugh, Y.; Song, J.Z.; Ma, H.P.; Yu, L.; Al-Khalili, O.; Eaton, D.C. Phosphatidylinositol phosphate-dependent regulation of Xenopus ENaC by MARCKS protein. Am. J. Physiol. Renal. Physiol. 2012, 303, F800–F811. [Google Scholar] [CrossRef] [PubMed]
- Krueger, B.; Yang, L.; Korbmacher, C.; Rauh, R. The phosphorylation site T613 in the beta-subunit of rat epithelial Na(+) channel (ENaC) modulates channel inhibition by Nedd4-2. Pflugers Arch. 2018, 470, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.M.; Rinke, R.; Korbmacher, C. Stimulation of the epithelial sodium channel (ENaC) by cAMP involves putative ERK phosphorylation sites in the C termini of the channel’s beta- and gamma-subunit. J. Biol. Chem. 2006, 281, 9859–9868. [Google Scholar] [CrossRef]
- Zachar, R.; Mikkelsen, M.K.; Skjodt, K.; Marcussen, N.; Zamani, R.; Jensen, B.L.; Svenningsen, P. The epithelial Na(+) channel alpha- and gamma-subunits are cleaved at predicted furin-cleavage sites, glycosylated and membrane associated in human kidney. Pflugers Arch. 2019, 471, 1383–1396. [Google Scholar] [CrossRef]
- Kota, P.; Gentzsch, M.; Dang, Y.L.; Boucher, R.C.; Stutts, M.J. The N terminus of alpha-ENaC mediates ENaC cleavage and activation by furin. J. Gen Physiol. 2018, 150, 1179–1187. [Google Scholar] [CrossRef]
- Carattino, M.D.; Mueller, G.M.; Palmer, L.G.; Frindt, G.; Rued, A.C.; Hughey, R.P.; Kleyman, T.R. Prostasin interacts with the epithelial Na+ channel and facilitates cleavage of the gamma-subunit by a second protease. Am. J. Physiol. Renal. Physiol. 2014, 307, F1080–F1087. [Google Scholar] [CrossRef]
- Kashlan, O.B.; Kinlough, C.L.; Myerburg, M.M.; Shi, S.; Chen, J.; Blobner, B.M.; Buck, T.M.; Brodsky, J.L.; Hughey, R.P.; Kleyman, T.R. N-linked glycans are required on epithelial Na(+) channel subunits for maturation and surface expression. Am. J. Physiol. Renal. Physiol. 2018, 314, F483–F492. [Google Scholar] [CrossRef]
- Kleyman, T.R.; Eaton, D.C. Regulating ENaC’s gate. Am. J. Physiol. Cell Physiol. 2020, 318, C150–C162. [Google Scholar] [CrossRef]
- Mueller, G.M.; Maarouf, A.B.; Kinlough, C.L.; Sheng, N.; Kashlan, O.B.; Okumura, S.; Luthy, S.; Kleyman, T.R.; Hughey, R.P. Cys palmitoylation of the beta subunit modulates gating of the epithelial sodium channel. J. Biol. Chem. 2010, 285, 30453–30462. [Google Scholar] [CrossRef]
- Mukherjee, A.; Mueller, G.M.; Kinlough, C.L.; Sheng, N.; Wang, Z.; Mustafa, S.A.; Kashlan, O.B.; Kleyman, T.R.; Hughey, R.P. Cysteine palmitoylation of the gamma subunit has a dominant role in modulating activity of the epithelial sodium channel. J. Biol. Chem. 2014, 289, 14351–14359. [Google Scholar] [CrossRef] [PubMed]
- Kashlan, O.B.; Blobner, B.M.; Zuzek, Z.; Tolino, M.; Kleyman, T.R. Na+ inhibits the epithelial Na+ channel by binding to a site in an extracellular acidic cleft. J. Biol. Chem. 2015, 290, 568–576. [Google Scholar] [CrossRef]
- Bize, V.; Horisberger, J.D. Sodium self-inhibition of human epithelial sodium channel: Selectivity and affinity of the extracellular sodium sensing site. Am. J. Physiol. Renal Physiol. 2007, 293, F1137–F1146. [Google Scholar] [CrossRef] [PubMed]
- Noreng, S.; Posert, R.; Bharadwaj, A.; Houser, A.; Baconguis, I. Molecular principles of assembly, activation, and inhibition in epithelial sodium channel. eLife 2020, 9, e59038. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.A.; Boucher, R.C.; Stutts, M.J. Serine protease activation of near-silent epithelial Na+ channels. Am. J. Physiol. Cell Physiol. 2004, 286, C190–C194. [Google Scholar] [CrossRef] [PubMed]
- Collier, D.M.; Snyder, P.M. Extracellular protons regulate human ENaC by modulating Na+ self-inhibition. J. Biol. Chem. 2009, 284, 792–798. [Google Scholar] [CrossRef]
- Subramanya, A.R.; Ellison, D.H. Distal convoluted tubule. Clin. J. Am. Soc. Nephrol. 2014, 9, 2147–2163. [Google Scholar] [CrossRef] [Green Version]


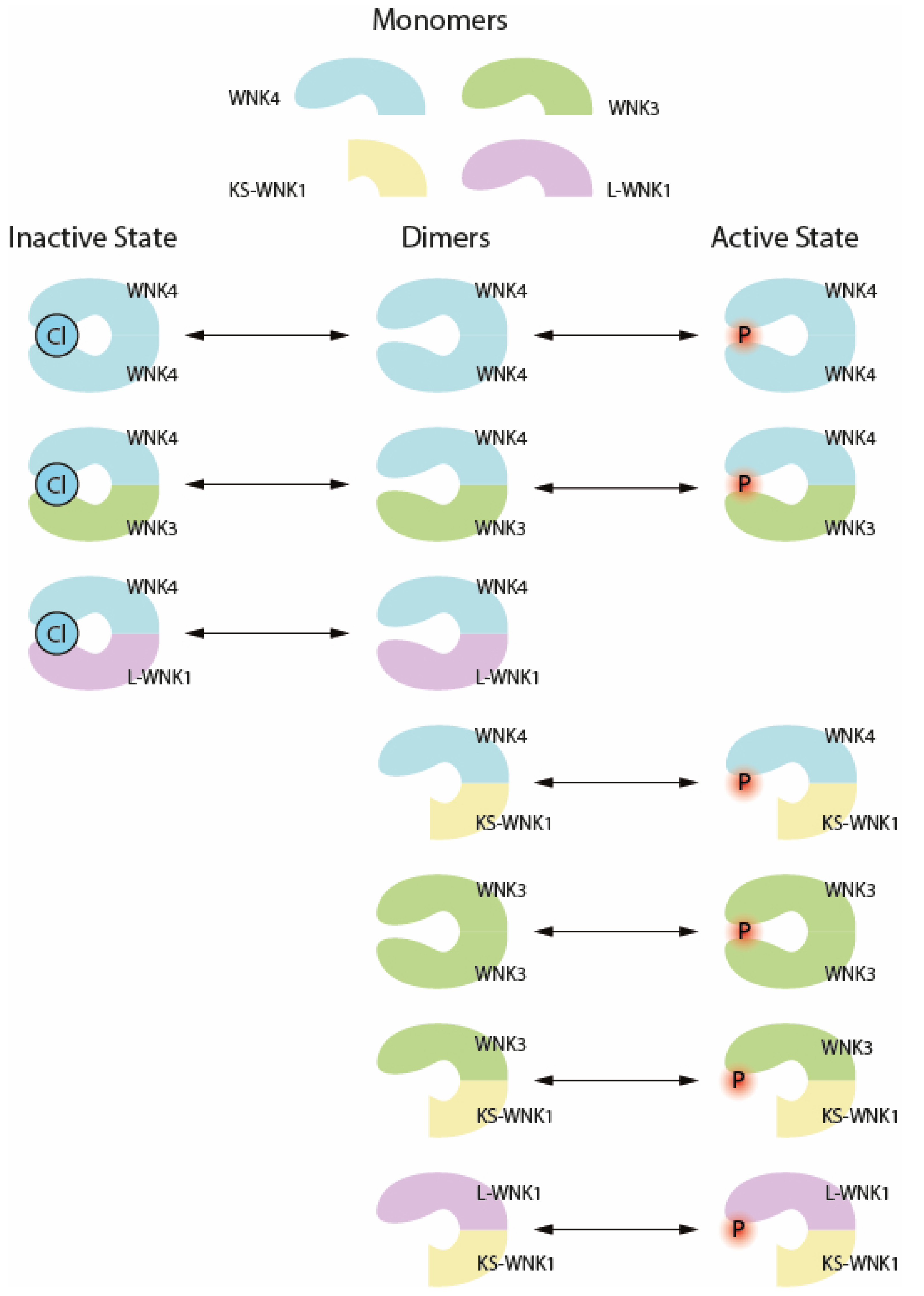
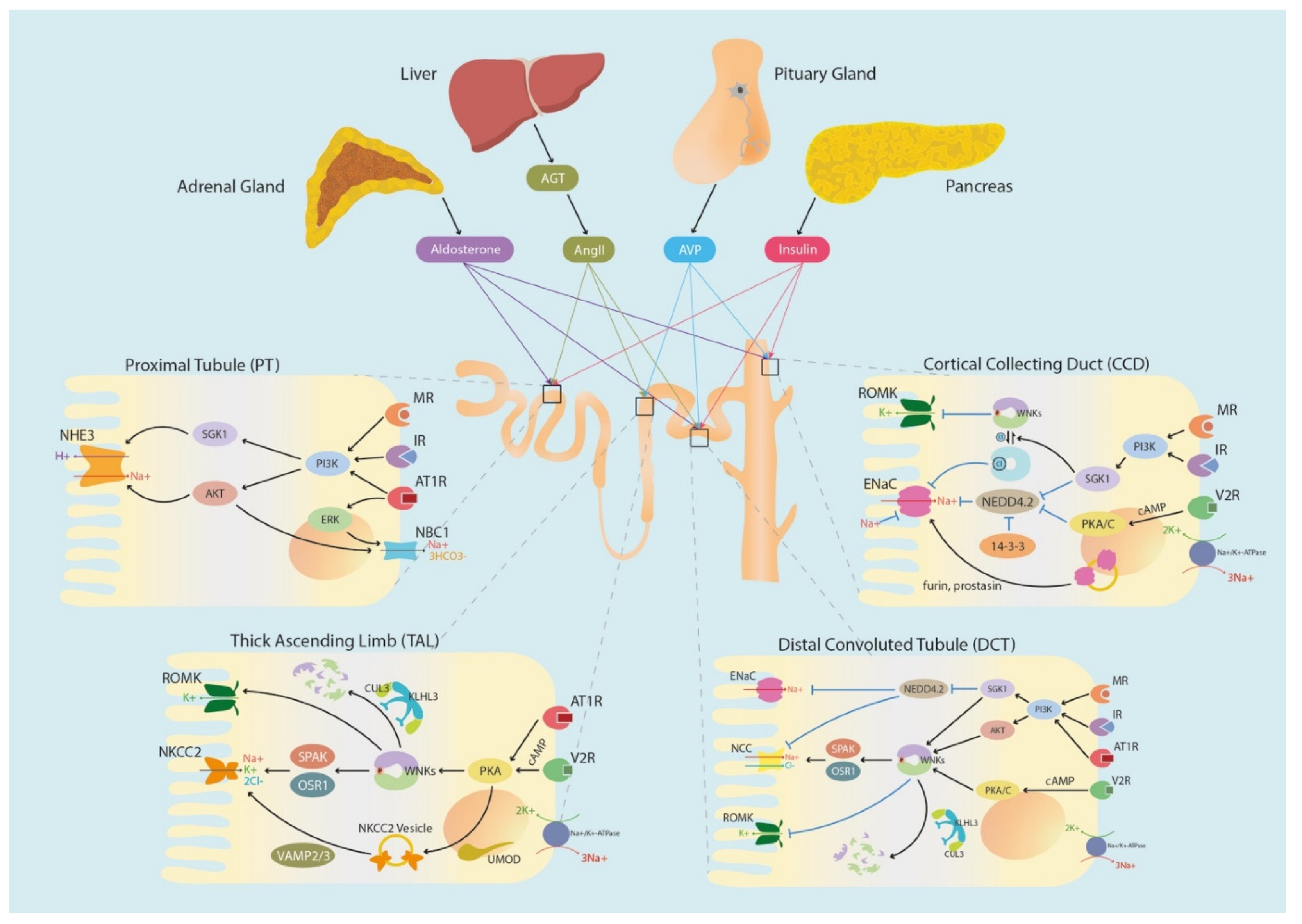

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | Upstream Regulator (Direct) | PTM Sites | PTM Modified Motif & Domain | PTM Effect | Effect on BP1 | Ref | Mutation Sites | Clinical Significance * | Motif & Domain | Mutation Effect | Effect on BP | Ref |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| The WNK Signaling | ||||||||||||
| WNK4 | SGK1 & PKA & PKC | p-S1169(m) = p-S1190(h) | COOH termini, RRXS motifs | inhibit ENaC | ↓ | [21,22] | S1169D | COOH termini | WNK4 gain of function; PHAII | ↑ | [23] | |
| S1169A | COOH termini | inhibit ENaC | ↓ | [21] | ||||||||
| p-S1196(m) = p-S1217(h) | COOH termini, RRXS motifs | WNK4 S332 autophosphorylation, reduces NCC inhibition | ↑ | [22,24] | S1196D | COOH termini | WNK4 gain of function | ↑ | [23] | |||
| AKT & SGK1 | p-S47(h) | NH2 termini, RRXS motifs | WNK4 S332 autophosphorylation, reduces NCC inhibition | ↑ | [25] | |||||||
| PKA&PKC | p-S64(h) | NH2 termini, RRXS motifs | WNK4 S332 autophosphorylation | ↑ | [26] | |||||||
| SGK1 | p-S1800(m) = p-S1201(h) | COOH termini, RRXS motifs | activate WNK4 | ↑ | [22] | |||||||
| autophosphorylation | p-S332(m) = p-S335(h) | activation T-loop, kinase domain | activate WNK4 | ↑ | [27] | S335A | activation T-loop, kinase domain | WNK4 loss of function | ↓ | [28] | ||
| L322F | T-loop kinase domain | WNK4 gain of function | ↑ | |||||||||
| R1185C | CaM binding domain | WNK4 partially loss of function | ↓ | [29] | ||||||||
| D561A | acidic motif | prevent KLHL3 bind to WNK4 | ↑ | [29,30,31] | ||||||||
| Q565E | Pathogenic | |||||||||||
| E562K | Pathogenic | |||||||||||
| D321A or D321K, K186D | T-loop kinase domain | WNK4 kinase dead | ↓ | [32] | ||||||||
| p38MAPK- MK pathway | p-S575(h) | near the acidic motif | activate WNK4 | ↑ | [33] | |||||||
| WNK1 | SGK1 & AKT | p-T58(m) = p-Thr60(h) | proline-rich domain (PRD) | inhibit ROMK | ↑ | [34,35] | T58A(m) | proline-rich domain (PRD) | WNK1 loss of function | ↓ | [34] | |
| K233M | subdomain I, kinase domain | WNK1 kinase dead | ↓ | [36] | ||||||||
| auto | p-S382(h) | activation T-loop, kinase domain | activate WNK4 | ↑ | [17] | S382A | activation T-loop, kinase domain | WNK1 kinase dead | ↓ | [37] | ||
| D368A | activation T-loop, kinase domain | WNK1 kinase dead | ↓ | [38] | ||||||||
| F316A | kinase domain | inhibit NCC | ↓ | [27] | ||||||||
| HQ/AA | coiled-coil domain, HQ motif | inhibit NCC | ↓ | [27] | ||||||||
| intron 1 479–667 deletion | WNK1 overexpression, PHAII | ↑ | [19,39] | |||||||||
| WNK3 | auto | p-S308, p-S304 | activation T-loop, kinase domain | activate WNK3 | ↑ | [33,40] | ||||||
| SPAK | WNK1, WNK3 & WNK4 | T233 | activation T-loop, kinase domain | activate SPAK | ↑ | [7] | T233E | activation T-loop, kinase domain | SPAK gain of function | ↑ | [41] | |
| S373, S387 | COOH termini, S-motif | activate SPAK | ↑ | [7] | ||||||||
| T243A | activation T-loop, kinase domain | SPAK loss of function, Gitelman’s Syndrome | ↓ | [28,42] | ||||||||
| OSR1 | WNK1, WNK3 & WNK4 | T185 | activation T-loop, kinase domain | activate OSR1 | ↑ | [7] | T185E | activation T-loop, kinase domain | OSR1 gain of function | ↑ | [41] | |
| S325, S339 | COOH termini, S-motif | activate OSR1 | ↑ | [7] | kidney-specific inactivation: KSP-OSR1-/- | OSR1 loss of function, Bartter’s Syndrome | ↓ | [43] | ||||
| AKT | mTORC2 | p-S473 | hydrophobic motif | activate WNK1 | ↑ | [44] | ||||||
| p-T308 | activation T-loop, kinase domain | activate WNK1 | ↑ | [44] | ||||||||
| SGK1 | mTORC2 | p-S422 | hydrophobic motif | inhibit ROMK | ↑ | [17,45] | S422D | hydrophobic motif | SGK1 gain of function | ↑ | [24] | |
| p-T256 | activation T-loop, kinase domain | activate WNK1 | ↑ | [17,45] | ||||||||
| K127M | ATP-binding site in kinase domain | SGK1 kinase dead | ↓ | [24] | ||||||||
| NEDD4.2 | SGK1 & 14-3-3 | p-S342, p-T367, p-S448 | 14-3-3 binding motifs | increase ENaC expression | ↑ | [46,47,48] | ||||||
| AKT | p-S342, p-S428 | between the WW domains | increase ENaC expression | ↑ | [47,49] | |||||||
| PKA/PKC | p-S327, p-S221, p-T246 | between the WW domains | inhibit Nedd4-2 | ↑ | [50] | |||||||
| NEDD4.2 WW domain | n/a | HECT domain, LPxY motif | increase ENaC expression | ↑ | [51] | |||||||
| CUL3 | exon 9 deletion | NH2 termini | CUL3 loss of function, PHAIIE | ↑ | [52] | |||||||
| KLHL3 | AKT, PKC & PKA | p-S433 | kelch repeat region | increase WNK4 expression | ↑ | [53] | S433E | Kelch repeat region | KLHL3 loss of function | ↑ | [54] | |
| R528H | Pathogenic | Kelch repeat region | KLHL3 loss of function | ↑ | [55] | |||||||
| Proximal Tubule (PT) | ||||||||||||
| NHE3 | SGK1 | p-S663 | RRxS motifs | increase NHE3 expression | ↑ | [56] | S663A | RRxS motifs | NHE3 loss of function | ↓ | [56] | |
| PKA | p-S555, p-S607 | RRxS motifs | inhibit NHE3 expression | ↓ | [56] | S607A | RRxS motif | NHE3 gain of function | ↑ | [57] | ||
| NBC1 | PKA | p-S1026 | COOH termini | increase NBC1 expression | ↑ | [58] | ||||||
| p-T49 | NH2 termini | increase NBC1 expression | ↑ | [58] | ||||||||
| T485S or A799V or R881C | NBC1 gain of function | pRTA | [59,60] | |||||||||
| Thick Ascending Tubule (TAL) | ||||||||||||
| NKCC2 | SPAK & OSR1 | p-S91, p-T95, p-T100, p-T105, p-T118, p-S120, p-S130 | amino acid permease N-termini | NKCC2 activated | ↑ | [61,62,63] | ||||||
| NKCC1 | SPAK & OSR1 | p-T203, p-T207, p-T212, p-T230 | amino acid permease N-termini | NKCC1 activated | ↑ | [64,65,66] | ||||||
| Cortical Collecting Duct (CCD) | ||||||||||||
| ROMK | WNK4 | p-S44 | inward rectifier potassium region | decrease ROMK expression | ↓ | [67,68] | S44D | inward rectifier potassium region | ROMK gain of function | ↑ | [69] | |
| SGK1 & PKA & PKC | p-S44 | inward rectifier potassium region | increase ROMK expression | ↑ | [70] | S44A | inward rectifier potassium region | ROMK loss of function | ↓ | [69] | ||
| ENaC | NEDD4.2 | u-βT613, u-γT623 | PY motif | inhibit ENaC | ↓ | [71,72] | Truncation -SCNN1B: COOH termini SCNN1C: the PY motif, the Nedd4.2 binding site | ROMK gain of function Liddle’s Syndrome | ↑ | [6,73] | ||
| Furin | cleaves α-subunit twice (after residue 181 and 204) | extracellular domain | activate ENaC | ↑ | [74,75,76] | |||||||
| cleaves γ-subunit once (after residue 138) | extracellular domain | activate ENaC | ↑ | [74,75,76] | ||||||||
| prostasin | cleaves γ subunit at a defined polybasic (RKRK) tract | finger domain | activate ENaC | ↑ | [77] | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, S.-C.; Ma, C.; Chang, K.-J.; Cheong, H.-P.; Lee, M.-C.; Lan, Y.-T.; Wang, C.-Y.; Chiou, S.-H.; Huo, T.-I.; Hsu, T.-K.; et al. The Post-Translational Modification Networking in WNK-Centric Hypertension Regulation and Electrolyte Homeostasis. Biomedicines 2022, 10, 2169. https://doi.org/10.3390/biomedicines10092169
Lin S-C, Ma C, Chang K-J, Cheong H-P, Lee M-C, Lan Y-T, Wang C-Y, Chiou S-H, Huo T-I, Hsu T-K, et al. The Post-Translational Modification Networking in WNK-Centric Hypertension Regulation and Electrolyte Homeostasis. Biomedicines. 2022; 10(9):2169. https://doi.org/10.3390/biomedicines10092169
Chicago/Turabian StyleLin, Shiuan-Chen, Chun Ma, Kao-Jung Chang, Han-Ping Cheong, Ming-Cheng Lee, Yuan-Tzu Lan, Chien-Ying Wang, Shih-Hwa Chiou, Teh-Ia Huo, Tsui-Kang Hsu, and et al. 2022. "The Post-Translational Modification Networking in WNK-Centric Hypertension Regulation and Electrolyte Homeostasis" Biomedicines 10, no. 9: 2169. https://doi.org/10.3390/biomedicines10092169