
An In Silico Approach for Comparative Characterization of Imidazolonepropionase from Agrobacterium fabrum & Bacillus subtilis: An Imperative Enzyme for Histidine Degradation †



Abstract
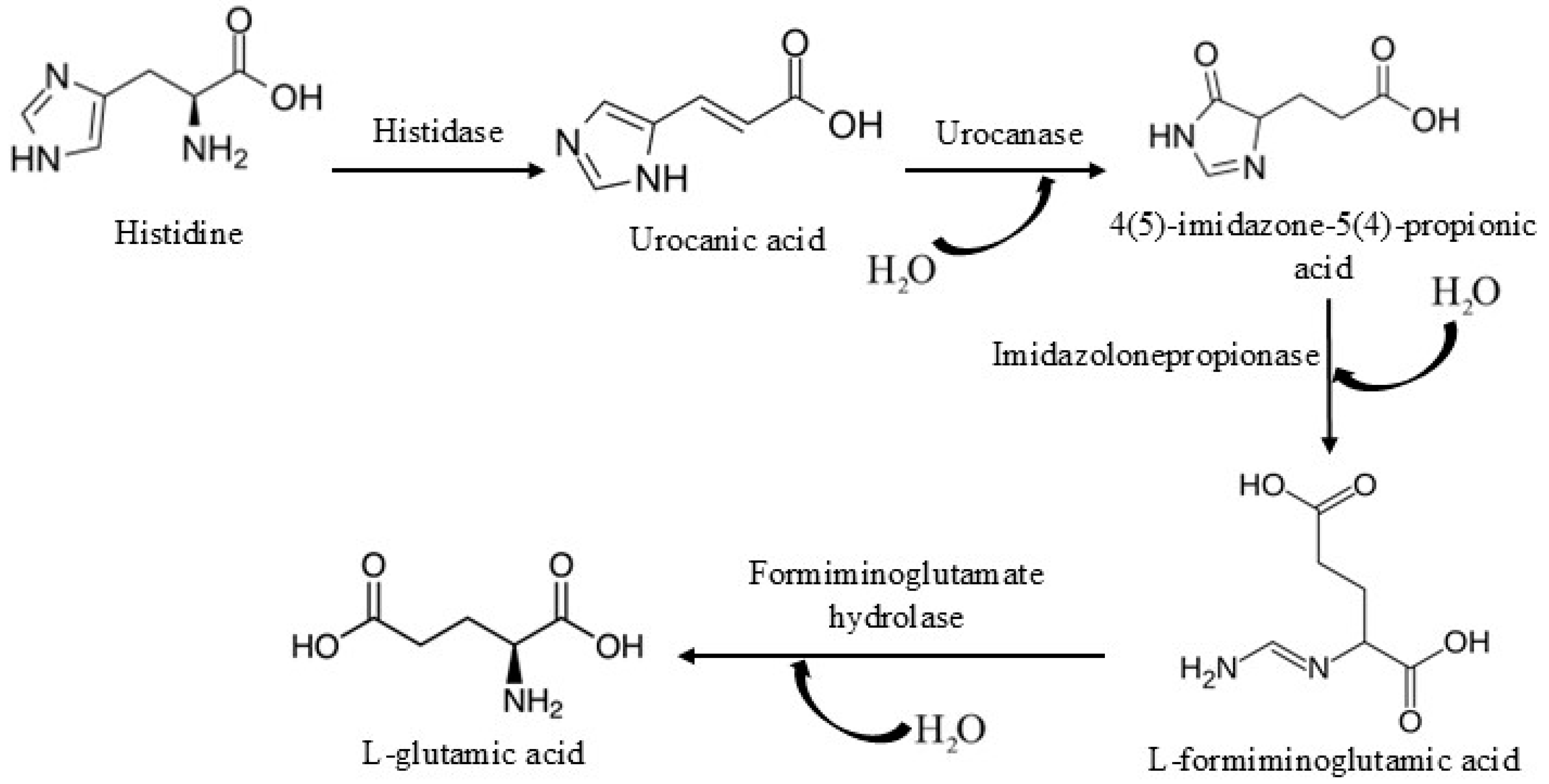
1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Analysis of Protein Sequences
2.3. Analysis of Protein Structures
2.4. Molecular Dynamic Simulations
3. Results and Discussions
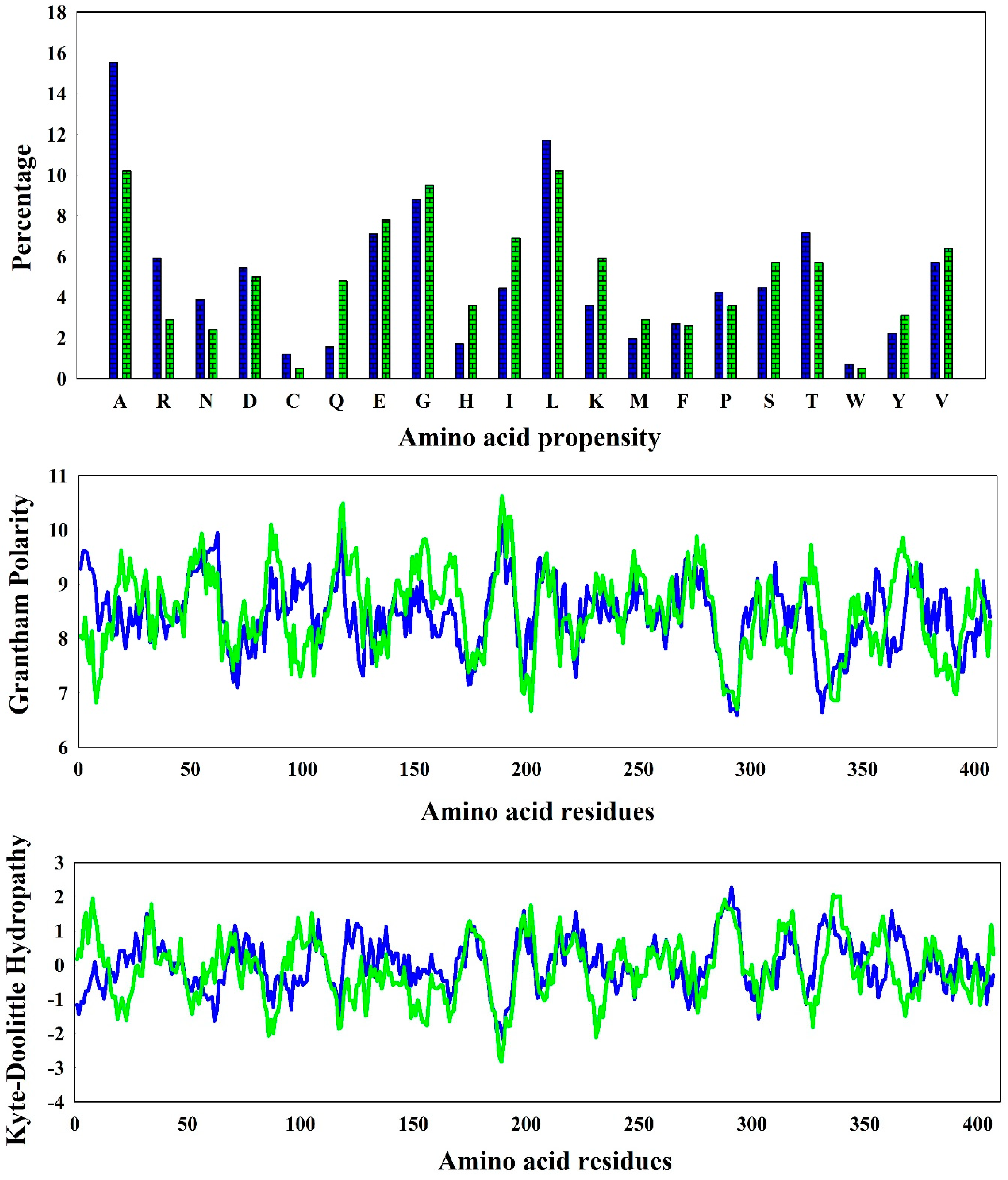
3.1. Amino Acid Diversity and Secondary Structure Formation
3.2. Formation of Intra-Protein Interactions
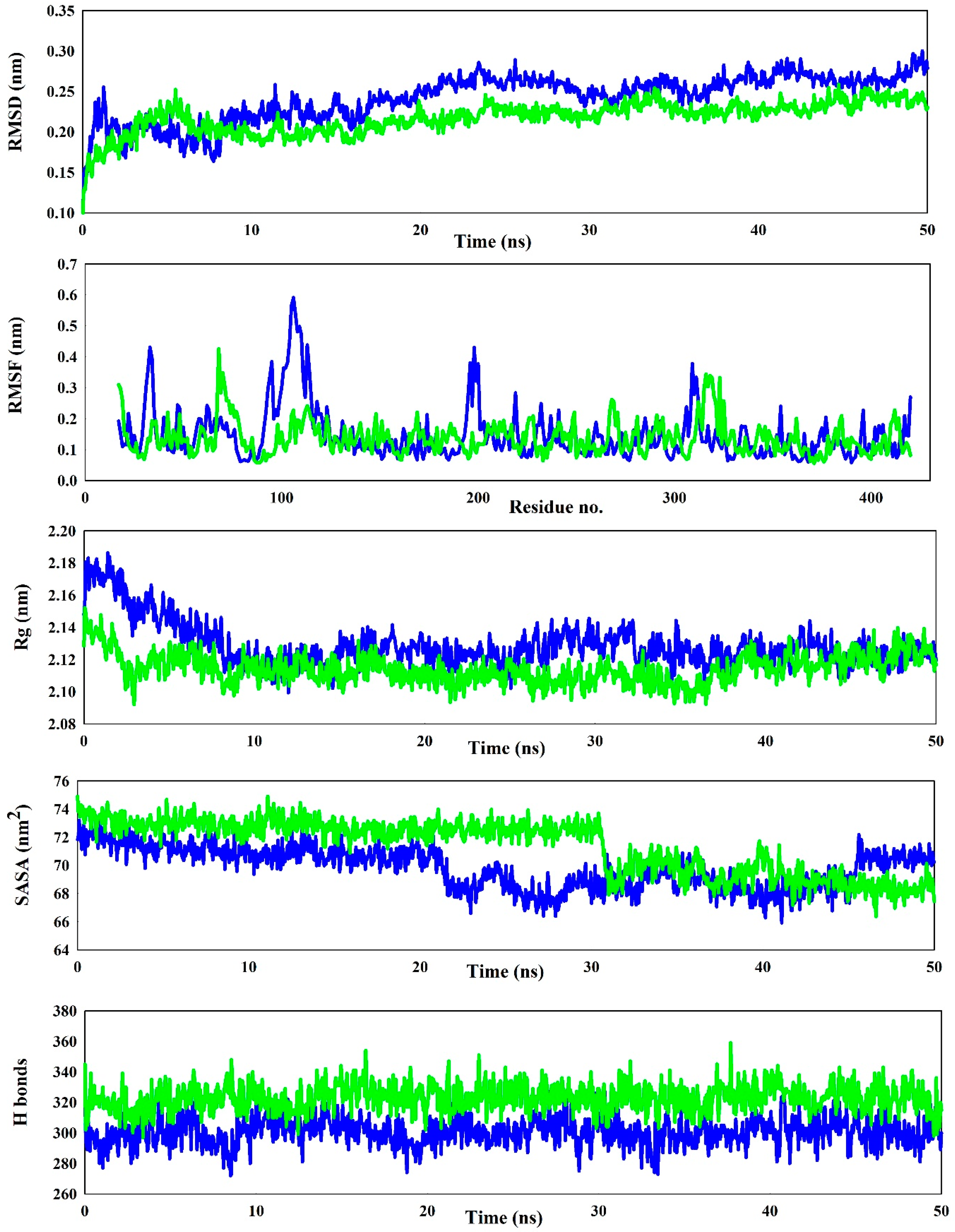
3.3. Stability Checks Through Simulation Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tyagi, R.; Kumaran, D.; Burley, S.; Swaminathan, S. X-ray structure of imidazolonepropionase from Agrobacterium tumefaciens at 1.87 angstrom resolution. Proteins Struct. Funct. Bioinform. 2007, 69, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liang, Y.H.; Brostromer, E.; Quan, J.M.; Panjikar, S.; Dong, Y.H.; Su, X.D. A catalytic mechanism revealed by the crystal structures of the imidazolonepropionase from Bacillus subtilis. J. Biol. Chem. 2006, 281, 36929–36936. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, R.; Eswaramoorthy, S.; Burley, S.K.; Raushel, F.M.; Swaminathan, S. A common catalytic mechanism for proteins of the HutI family. Biochemistry 2008, 47, 5608–5615. [Google Scholar] [CrossRef] [PubMed]
- Snyder, S.H.; Silva, O.L.; Kies, M.W. The mammalian metabolism of L-Histidine: IV. purification and properties of imidazolone propionic acid hydrolase. J. Biol. Chem. 1961, 236, 2996–2998. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, L.; Zheng, X.; Liang, Y.H.; Su, X.D. Protein preparation, crystallization and preliminary X-ray analysis of imidazolonepropionase from Bacillus subtilis. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2006, 1764, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chu, W.; Yu, M.; Wang, Y.; Ma, S.; Dong, Y.; Wu, Z. Local structure investigation of the active site of the imidazolonepropionase from Bacillus subtilis by XANES spectroscopy and ab initio calculations. J. Synchrotron Radiat. 2008, 15, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.E.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Mitra, D.; Pal, A.K.; Das Mohapatra, P.K. Intra-protein interactions of SARS-CoV-2 and SARS: A bioinformatic analysis for plausible explanation regarding stability, divergency, and severity. Syst. Microbiol. Biomanuf. 2022, 2, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Das Mohapatra, P.K. In silico comparative structural compositional analysis of glycoproteins of RSV to study the nature of stability transmissibility of RSV A. Syst. Microbiol. Biomanuf. 2023, 3, 312–327. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Díaz-Pascual, F.; Lempp, M.; Nosho, K.; Jeckel, H.; Jo, J.K.; Neuhaus, K.; Hartmann, R.; Jelli, E.; Hansen, M.F.; Price-Whelan, A.; et al. Spatial alanine metabolism determines local growth dynamics of Escherichia coli colonies. Elife 2021, 10, e70794. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Das Mohapatra, P.K. Discovery of novel cyclic salt bridge in thermophilic bacterial protease and study of its sequence and structure. Appl. Biochem. Biotechnol. 2021, 193, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Biswas, I.; Mitra, D. Insilico sequence-structure based analysis of bacterial chromate reductase to unravel enzymatic specificity towards chromium pollution. Biocatal. Agric. Biotechnol. 2024, 60, 103339. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [PubMed]
- Mitra, D.; Afreen, S.; Das Mohapatra, P.K.; Abdalla, M. Threat of respiratory syncytial virus infection knocking the door: A proposed potential drug candidate through molecular dynamics simulations, a future alternative. J. Mol. Model. 2023, 29, 91. [Google Scholar] [CrossRef] [PubMed]
- Biswas, I.; Mitra, D. November. Comparative Analysis of RuBisCO Evolution and Intrinsic Differences: Insights from In Silico Assessment in Cyanobacteria, Monocot, and Dicot Plants. Biol. Life Sci. Forum 2023, 27, 44. [Google Scholar]
- Mitra, D.; Afreen, S.; Das Mohapatra, P.K.; Abdalla, M. Inhibition of respiratory syncytial virus by Daclatasvir and its derivatives: Synthesis of computational derivatives as a new drug development. J. Biomol. Struct. Dyn. 2025, 43, 2440–2462. [Google Scholar] [CrossRef] [PubMed]
- Mitra, D.; Paul, M.; Thatoi, H.; Das Mohapatra, P.K. Potentiality of bioactive compounds as inhibitor of M protein and F protein function of human respiratory syncytial virus. Silico Pharmacol. 2023, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, Z.S.; Lee, L.; Derewenda, U. The occurence of C–H··· O hydrogen bonds in proteins. J. Mol. Biol. 1995, 252, 248–262. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Name | Sheets | Beta-Alpha-Beta | Beta Hairpins | Beta Bulges | Strands | Helices | Helix-Helix Interacs | Beta Turns | Gamma Turns |
|---|---|---|---|---|---|---|---|---|---|
| Agrobacterium fabrum | 2 | 7 | 4 | 7 | 16 | 19 | 21 | 21 | 6 |
| Bacillus subtilis | 2 | 7 | 4 | 8 | 17 | 18 | 21 | 22 | 6 |
| Name | Salt Bridge | Aromatic-Aromatic | Cation-Pi | |||
|---|---|---|---|---|---|---|
| Isolated | Network | Isolated | Network | Isolated | Network | |
| Agrobacterium fabrum | 19 | 0 | 3 | 2 | 4 | 0 |
| Bacillus subtilis | 13 | 3 | 2 | 5 | 4 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biswas, I.; Biswas, T.; Mitra, D. An In Silico Approach for Comparative Characterization of Imidazolonepropionase from Agrobacterium fabrum & Bacillus subtilis: An Imperative Enzyme for Histidine Degradation. Biol. Life Sci. Forum 2025, 41, 3. https://doi.org/10.3390/blsf2025041003
Biswas I, Biswas T, Mitra D. An In Silico Approach for Comparative Characterization of Imidazolonepropionase from Agrobacterium fabrum & Bacillus subtilis: An Imperative Enzyme for Histidine Degradation. Biology and Life Sciences Forum. 2025; 41(1):3. https://doi.org/10.3390/blsf2025041003
Chicago/Turabian StyleBiswas, Ishita, Trishanjan Biswas, and Debanjan Mitra. 2025. "An In Silico Approach for Comparative Characterization of Imidazolonepropionase from Agrobacterium fabrum & Bacillus subtilis: An Imperative Enzyme for Histidine Degradation" Biology and Life Sciences Forum 41, no. 1: 3. https://doi.org/10.3390/blsf2025041003
APA StyleBiswas, I., Biswas, T., & Mitra, D. (2025). An In Silico Approach for Comparative Characterization of Imidazolonepropionase from Agrobacterium fabrum & Bacillus subtilis: An Imperative Enzyme for Histidine Degradation. Biology and Life Sciences Forum, 41(1), 3. https://doi.org/10.3390/blsf2025041003