
Navigating Rarity: Pathological Challenges and Diagnostic Ambiguity in Rare Gliomas—A Case Series with a Focus on Personalized Treatment and Quality of Life

 , and
, and
Simple Summary
Abstract
1. Introduction
2. Methodology
3. Case Series
3.1. Case 1: Pleomorphic Xanthoastrocytoma to High-Grade Glioma with Pleomorphic and Pseudopapillary Features (HPAP) to Neuroepithelial Tumor with PATZ1 Fusion
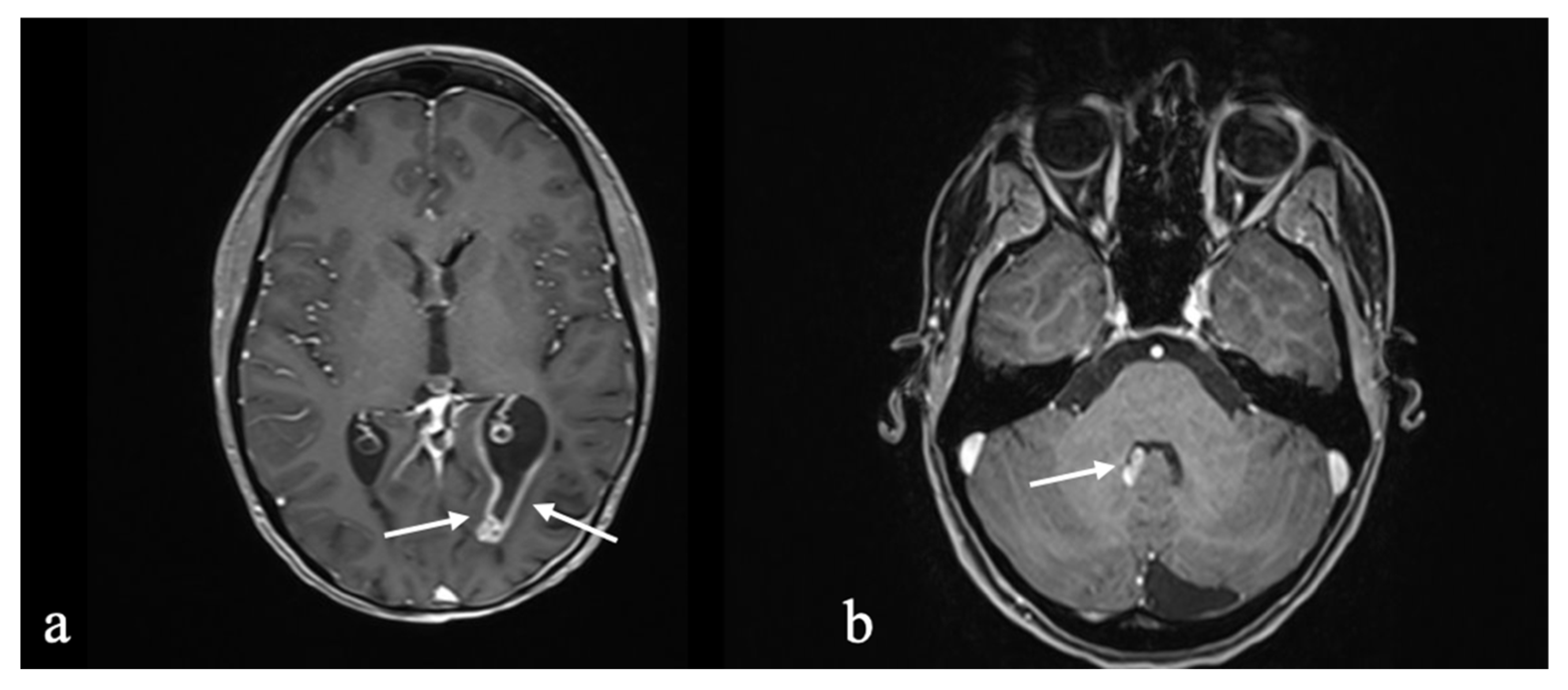
3.1.1. Initial Diagnosis
3.1.2. Molecular Profiling and Treatment Timeline
3.1.3. Long-Term Outcome and Life Quality
3.2. Case 2: Astroblastoma
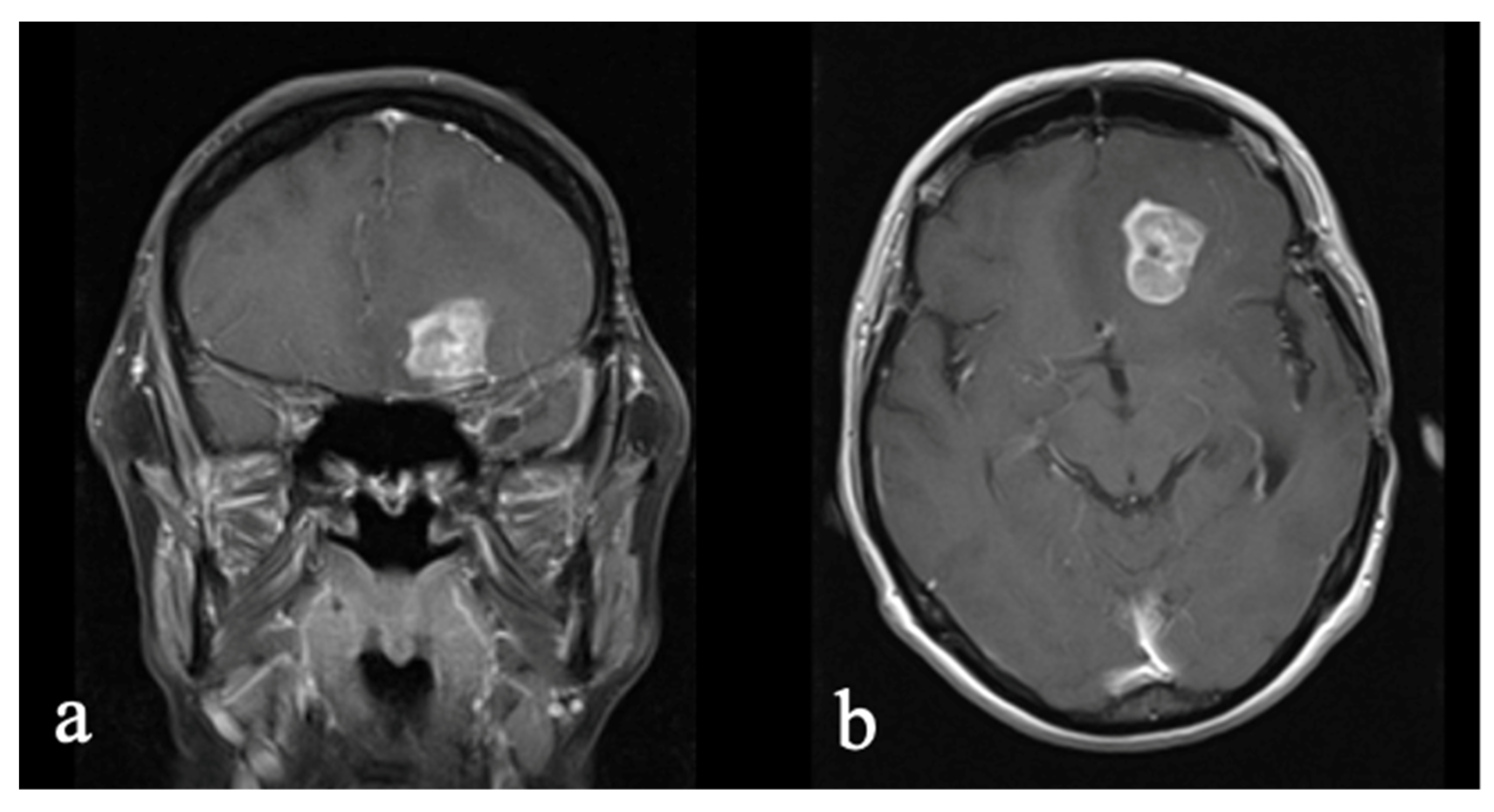
3.2.1. Initial Diagnosis
3.2.2. Molecular Profiling and Treatment Timeline
3.2.3. Long-Term Outcome and Life Quality
3.3. Case 3: Oligodendroglioma to Glioblastoma
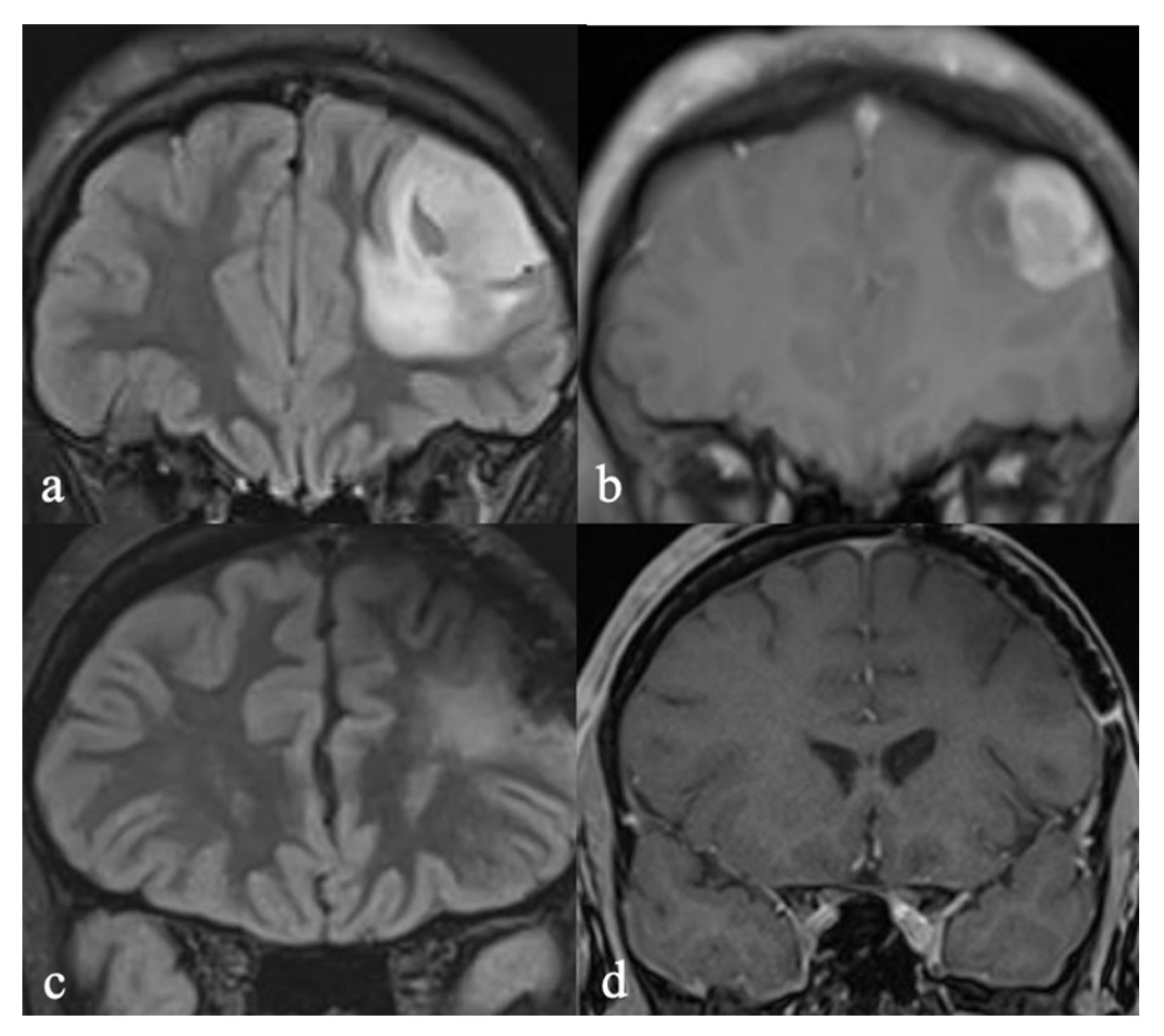
3.3.1. Initial Diagnosis
3.3.2. Molecular Profiling and Treatment Timeline
3.3.3. Long-Term Outcome and Life Quality
3.4. Case 4: Glioblastoma to Low-Grade, SEGA-like Astrocytoma
3.4.1. Initial Diagnosis
3.4.2. Molecular Profiling and Treatment Timeline
3.4.3. Long-Term Outcome and Life Quality
4. Discussion
4.1. Diagnostic Ambiguity
4.2. Treatment Options
4.3. Quality of Life Considerations
4.4. Mental Health
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AB | Astroblastoma |
| APXA | Anaplastic pleomorphic xanthoastrocytoma |
| CNS | Central nervous system |
| CT | Computer tomography |
| CR | Complete tumor response |
| DNA | Deoxyribonucleic acid |
| EQ-5D-5L | European Quality of Life 5 Dimensions 5-Level Version |
| EQ VAS | European Quality Visual Analog Scale |
| GFAP | Glial fibrillary acidic protein |
| GTR | Gross total resection |
| GBM | Glioblastoma multiforme |
| GY | Gray |
| IDH | Isocitrate dehydrogenase |
| KPS | Karnofsky performance score |
| MGMT | O (6)-methylguanine-DNA methyltransferase |
| MRI | Magnetic resonance imaging |
| MoCA | Montreal Cognitive Assessment |
| MRC | Medical Research Council |
| NGS | Next-generation sequencing |
| NF 1 | Neurofibromatosis 1 |
| HPAP | High-grade glioma with pleomorphic and pseudopapillary features |
| KPS | Karnofsky performance score |
| PXA | Pleomorphic xanthoastrocytoma |
| QoL | Quality of life |
| SEGA | Subependymal giant cell astrocytoma |
| TTF | Tumor-treating field |
| WHO | World Health Organization |
References
- Duffau, H. A Personalized Longitudinal Strategy in Low-Grade Glioma Patients: Predicting Oncological and Neural Interindividual Variability and Its Changes over Years to Think One Step Ahead. J. Pers. Med. 2022, 12, 1621. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.; Pan, P.C. Management of Low-Grade Gliomas. Cancer J. 2025, 31, e0760. [Google Scholar] [CrossRef]
- Morshed, R.A.; Young, J.S.; Hervey-Jumper, S.L.; Berger, M.S. The Management of Low-Grade Gliomas in Adults. J. Neurosurg. Sci. 2019, 63, 450–457. [Google Scholar] [CrossRef]
- Franceschi, E.; Frappaz, D.; Rudà, R.; Hau, P.; Preusser, M.; Houillier, C.; Lombardi, G.; Asioli, S.; Dehais, C.; Bielle, F.; et al. Rare Primary Central Nervous System Tumors in Adults: An Overview. Front. Oncol. 2020, 10, 996. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Figarella-Branger, D.; Appay, R.; Metais, A.; Tauziède-Espariat, A.; Colin, C.; Rousseau, A.; Varlet, P. The 2021 WHO Classification of Tumours of the Central Nervous System. Ann. Pathol. 2022, 42, 367–382. [Google Scholar] [CrossRef]
- Pratt, D.; Penas-Prado, M.; Gilbert, M.R. Clinical Impact of Molecular Profiling in Rare Brain Tumors. Curr. Opin. Neurol. 2023, 36, 579–586. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA Methylation-Based Classification of Central Nervous System Tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Saaid, A.; Monticelli, M.; Ricci, A.A.; Orlando, G.; Botta, C.; Zeppa, P.; Bianconi, A.; Osella-Abate, S.; Bruno, F.; Pellerino, A.; et al. Prognostic Analysis of the IDH1 G105G (Rs11554137) SNP in IDH-Wildtype Glioblastoma. Genes 2022, 13, 1439. [Google Scholar] [CrossRef]
- Duffau, H.; Mandonnet, E. The “Onco-Functional Balance” in Surgery for Diffuse Low-Grade Glioma: Integrating the Extent of Resection with Quality of Life. Acta Neurochir. 2013, 155, 951–957. [Google Scholar] [CrossRef]
- Dirven, L.; Aaronson, N.K.; Heimans, J.J.; Taphoorn, M.J.B. Health-Related Quality of Life in High-Grade Glioma Patients. Chin. J. Cancer 2014, 33, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Coomans, M.; Dirven, L.; Aaronson, N.K.; Baumert, B.G.; van den Bent, M.; Bottomley, A.; Brandes, A.A.; Chinot, O.; Coens, C.; Gorlia, T.; et al. The Added Value of Health-Related Quality of Life as a Prognostic Indicator of Overall Survival and Progression-Free Survival in Glioma Patients: A Meta-Analysis Based on Individual Patient Data from Randomised Controlled Trials. Eur. J. Cancer 2019, 116, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Aaronson, N.K.; Taphoorn, M.J.B.; Heimans, J.J.; Postma, T.J.; Gundy, C.M.; Beute, G.N.; Slotman, B.J.; Klein, M. Compromised Health-Related Quality of Life in Patients with Low-Grade Glioma. J. Clin. Oncol. 2011, 29, 4430–4435. [Google Scholar] [CrossRef]
- Duffau, H. Diffuse Low-Grade Glioma, Oncological Outcome and Quality of Life: A Surgical Perspective. Curr. Opin. Oncol. 2018, 30, 383–389. [Google Scholar] [CrossRef]
- Boele, F.W.; Klein, M.; Reijneveld, J.C.; Verdonck-de Leeuw, I.M.; Heimans, J.J. Symptom Management and Quality of Life in Glioma Patients. CNS Oncol. 2014, 3, 37–47. [Google Scholar] [CrossRef]
- Alhalabi, K.T.; Stichel, D.; Sievers, P.; Peterziel, H.; Sommerkamp, A.C.; Sturm, D.; Wittmann, A.; Sill, M.; Jäger, N.; Beck, P.; et al. PATZ1 Fusions Define a Novel Molecularly Distinct Neuroepithelial Tumor Entity with a Broad Histological Spectrum. Acta Neuropathol. 2021, 142, 841–857. [Google Scholar] [CrossRef]
- Smith, H.L.; Wadhwani, N.; Horbinski, C. Major Features of the 2021 WHO Classification of CNS Tumors. Neurotherapeutics 2022, 19, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Borchert, K.; Jacob, C.; Wetzel, N.; Jänicke, M.; Eggers, E.; Sauer, A.; Marschner, N.; Altevers, J.; Mittendorf, T.; Greiner, W. Application Study of the EQ-5D-5L in Oncology: Linking Self-Reported Quality of Life of Patients with Advanced or Metastatic Colorectal Cancer to Clinical Data from a German Tumor Registry. Health Econ. Rev. 2020, 10, 40. [Google Scholar] [CrossRef]
- Ownby, K.K. Use of the Distress Thermometer in Clinical Practice. J. Adv. Pract. Oncol. 2019, 10, 175. [Google Scholar] [CrossRef]
- Jammula, V.; Rogers, J.L.; Vera, E.; Christ, A.; Leeper, H.E.; Acquaye, A.; Briceno, N.; Choi, A.; Grajkowska, E.; Levine, J.E.; et al. The Montreal Cognitive Assessment (MoCA) in Neuro-Oncology: A Pilot Study of Feasibility and Utility in Telehealth and in-Person Clinical Assessments. Neurooncol. Pract. 2022, 9, 429–440. [Google Scholar] [CrossRef]
- Wick, W.; Hartmann, C.; Engel, C.; Stoffels, M.; Felsberg, J.; Stockhammer, F.; Sabel, M.C.; Koeppen, S.; Ketter, R.; Meyermann, R.; et al. NOA-04 Randomized Phase III Trial of Sequential Radiochemotherapy of Anaplastic Glioma with Procarbazine, Lomustine, and Vincristine or Temozolomide. J. Clin. Oncol. 2009, 27, 5874–5880. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Jóźwiak, S.; Mandera, M.; Młynarski, W. Natural History and Current Treatment Options for Subependymal Giant Cell Astrocytoma in Tuberous Sclerosis Complex. Semin. Pediatr. Neurol. 2015, 22, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Pucko, E.; Sulejczak, D.; Ostrowski, R.P. Subependymal Giant Cell Astrocytoma: The Molecular Landscape and Treatment Advances. Cancers 2024, 16, 3406. [Google Scholar] [CrossRef]
- Gao, C.; Zabielska, B.; Jiao, F.; Mei, D.; Wang, X.; Kotulska, K.; Jozwiak, S. Subependymal Giant Cell Astrocytomas in Tuberous Sclerosis Complex-Current Views on Their Pathogenesis and Management. J. Clin. Med. 2023, 12, 956. [Google Scholar] [CrossRef]
- Yamada, S.; Tanikawa, M.; Matsushita, Y.; Fujinami, R.; Yamada, H.; Sakomi, K.; Sakata, T.; Inagaki, H.; Yokoo, H.; Ichimura, K.; et al. SEGA-like Circumscribed Astrocytoma in a Non-NF1 Patient, Harboring Molecular Profile of GBM. A Case Report. Neuropathology 2024, 44, 190–199. [Google Scholar] [CrossRef]
- Movahed-Ezazi, M.; Schwartz, P.J.; Zimmerman, D.L.; Song, X. Subependymal Giant Cell Astrocytoma in a 79-Year-Old Woman without Clinical Features of Tuberous Sclerosis: A Case Report. J. Neuropathol. Exp. Neurol. 2021, 80, 199–201. [Google Scholar] [CrossRef]
- Soffietti, R.; Rudà, R.; Reardon, D. Rare Glial Tumors. In Handbook of Clinical Neurology; Springer: Berlin/Heidelberg, Germany, 2016; Volume 134, pp. 399–415. [Google Scholar] [CrossRef]
- Mallick, S.; Giridhar, P.; Benson, R.; Melgandi, W.; Kishor Rath, G. Demography, Pattern of Care, and Survival in Patients with Xanthoastrocytoma: A Systematic Review and Individual Patient Data Analysis of 325 Cases. Neurosci. Rural. Pr. 2019, 10, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Ishikawa, E.; Kohzuki, H.; Sakamoto, N.; Zaboronok, A.; Matsuda, M.; Shibuya, M.; Matsumura, A. Malignant Transformation of Pleomorphic Xanthoastrocytoma and Differential Diagnosis: Case Report. BMC Neurol. 2020, 20, 21. [Google Scholar] [CrossRef]
- Pratt, D.; Abdullaev, Z.; Papanicolau-Sengos, A.; Ketchum, C.; Panneer Selvam, P.; Chung, H.J.; Lee, I.; Raffeld, M.; Gilbert, M.R.; Armstrong, T.S.; et al. High-Grade Glioma with Pleomorphic and Pseudopapillary Features (HPAP): A Proposed Type of Circumscribed Glioma in Adults Harboring Frequent TP53 Mutations and Recurrent Monosomy 13. Acta Neuropathol. 2022, 143, 403–414. [Google Scholar] [CrossRef]
- Joshi, P.R.; Pandey, S.B.; Manandhar, U.; GC, S.; Sedain, G. Cerebral Astroblastoma Radiologically Mimicking Pilocytic Astrocytoma: A Case Report. Clin. Case Rep. 2022, 10, e05781. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Hirose, Y.; Cohen, K.J.; Feuerstein, B.G.; Burger, P.C. Astroblastoma: Clinicopathologic Features and Chromosomal Abnormalities Defined by Comparative Genomic Hybridization. Brain Pathol. 2000, 10, 342. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Nobusawa, S.; Sugiyama, K.; Amatya, V.J.; Fujimoto, N.; Sasaki, A.; Mikami, Y.; Kakita, A.; Tanaka, S.; Yokoo, H. Astroblastoma: A Distinct Tumor Entity Characterized by Alterations of the X Chromosome and MN1 Rearrangement. Brain Pathol. 2018, 28, 684–694. [Google Scholar] [CrossRef]
- Mallick, S.; Benson, R.; Venkatesulu, B.; Melgandi, W.; Rath, G.K. Patterns of Care and Survival Outcomes in Patients with Astroblastoma: An Individual Patient Data Analysis of 152 Cases. Childs Nerv. Syst. 2017, 33, 1295–1302. [Google Scholar] [CrossRef]
- Merfeld, E.C.; Dahiya, S.; Perkins, S.M. Patterns of Care and Treatment Outcomes of Patients with Astroblastoma: A National Cancer Database Analysis. CNS Oncol. 2018, 7, CNS13. [Google Scholar] [CrossRef]
- Wen, P.Y.; Kesari, S. Malignant Gliomas in Adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Witthayanuwat, S.; Pesee, M.; Supaadirek, C.; Supakalin, N.; Thamronganantasakul, K.; Krusun, S. Survival Analysis of Glioblastoma Multiforme. Asian Pac. J. Cancer Prev. 2018, 19, 2613–2617. [Google Scholar] [CrossRef]
- Häni, L.; Kopcic, M.; Branca, M.; Schütz, A.; Murek, M.; Söll, N.; Vassella, E.; Raabe, A.; Hewer, E.; Schucht, P. Quantitative Analysis of the MGMT Methylation Status of Glioblastomas in Light of the 2021 WHO Classification. Cancers 2022, 14, 3149. [Google Scholar] [CrossRef]
- Romo, C.G.; Piotrowski, A.F.; Campian, J.L.; Diarte, J.; Rodriguez, F.J.; Bale, T.A.; Dahiya, S.; Gutmann, D.H.; Lucas, C.H.G.; Prichett, L.; et al. Clinical, Histological, and Molecular Features of Gliomas in Adults with Neurofibromatosis Type 1. Neuro Oncol. 2023, 25, 1474–1486. [Google Scholar] [CrossRef]
- Klein, M.; Taphoorn, M.J.B.; Heimans, J.J.; Van der Ploeg, H.M.; Vandertop, W.P.; Smit, E.F.; Leenstra, S.; Tulleken, C.A.F.; Boogerd, W.; Belderbos, J.S.A.; et al. Neurobehavioral Status and Health-Related Quality of Life in Newly Diagnosed High-Grade Glioma Patients. J. Clin. Oncol. 2001, 19, 4037–4047. [Google Scholar] [CrossRef]
- Lucas, M.R. Psychosocial Implications for the Patient with a High-Grade Glioma. J. Neurosci. Nurs. 2010, 42, 104–108. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample I, Dura | Methylation grade neuroepithelial tumor with PATZ1 fusion. |
| Sample II: Frontotemporal | No allocation for an established reference class. |
| Sample III: Frontobasal | No allocation for an established reference class. |
| Sample IV: Temporal | Methylation grade neuroepithelial tumor with PATZ1 fusion. |
| Histological examination | Glioma with pleomorphic and pseudopapillary features. |
| Molecular genetic analysis | neuroepithelial tumor with PATZ1 fusion, IDH1 R132H negative, nuclear ATRX expression present, BRAF V600E negative, H3 G34R negative, and an unmethylated MGMT promoter. |
| Integrated Diagnosis | Glioma, not classified elsewhere. |
| Case | Diagnosis (Initial → Final) | Age */Sex | WHO | Molecular Findings | Survival | Current Neurological Status | QoL Scores (Latest) | KPS |
| 1 | PXA → APXA → HPAP → Neuroepithelial tumor with PATZ1 fusion | 51/M | NEC | PATZ1 fusion, IDH1 wildtype, MGMT unmethylated | >12 years | No deficits; returned to work | EQ-VAS: 90%; Distress: 1/10; EQ-5D-5L: mild issues in two domains | 90 |
| 2 | Astroblastoma → MN1-altered astroblastoma | 4/F | Not defined | MN1:BEND2 fusion, MGMT unmethylated | >25 years | No deficits; mild memory decline (MoCA 24) | EQ-VAS: 100%; Distress: 0/10; EQ-5D-5L: no problems | 90 |
| 3 | Anaplastic oligodendroglioma → GBM with oligodendroglial component | 40/F | 4 | IDH mutant, 1p/19q codeletion, MGMT 11% methylated | >20 years (10 years therapy-free) | No deficits | EQ-VAS: 70%; Distress: 3/10; EQ-5D-5L: Pain: moderate problems | 90 |
| 4 | Glioblastoma → SEGA-like astrocytoma | 24/F | NEC | NF1; t-SNE proximity to pilocytic astrocytoma | ~3 years | Chronic pain, fatigue | EQ-VAS: 75%; Distress: 3/10; EQ-5D-5L: pain: moderate problems | 80 |
| WHO grade | Not defined |
| Histological examination | Higher-grade glioma with astroblastic rosettes. |
| Molecular genetic analysis | Astroblastoma, fusion of MN1:BEND2, unmethylated MGMT promoter, no relevant variations in NGS |
| Integrated Diagnosis | Astroblastoma, MN1-altered |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grübel, N.; Wickert, A.; Sahm, F.; Schmitz, B.; Osterloh, A.; Kassubek, R.; König, R.; Wirtz, C.R.; Engelke, J.; Pala, A.; et al. Navigating Rarity: Pathological Challenges and Diagnostic Ambiguity in Rare Gliomas—A Case Series with a Focus on Personalized Treatment and Quality of Life. Onco 2025, 5, 28. https://doi.org/10.3390/onco5020028
Grübel N, Wickert A, Sahm F, Schmitz B, Osterloh A, Kassubek R, König R, Wirtz CR, Engelke J, Pala A, et al. Navigating Rarity: Pathological Challenges and Diagnostic Ambiguity in Rare Gliomas—A Case Series with a Focus on Personalized Treatment and Quality of Life. Onco. 2025; 5(2):28. https://doi.org/10.3390/onco5020028
Chicago/Turabian StyleGrübel, Nadja, Anika Wickert, Felix Sahm, Bernd Schmitz, Anja Osterloh, Rebecca Kassubek, Ralph König, Christian Rainer Wirtz, Jens Engelke, Andrej Pala, and et al. 2025. "Navigating Rarity: Pathological Challenges and Diagnostic Ambiguity in Rare Gliomas—A Case Series with a Focus on Personalized Treatment and Quality of Life" Onco 5, no. 2: 28. https://doi.org/10.3390/onco5020028
APA StyleGrübel, N., Wickert, A., Sahm, F., Schmitz, B., Osterloh, A., Kassubek, R., König, R., Wirtz, C. R., Engelke, J., Pala, A., & Laible, M. (2025). Navigating Rarity: Pathological Challenges and Diagnostic Ambiguity in Rare Gliomas—A Case Series with a Focus on Personalized Treatment and Quality of Life. Onco, 5(2), 28. https://doi.org/10.3390/onco5020028