
Bioinformatics Tools for NGS-Based Identification of Single Nucleotide Variants and Large-Scale Rearrangements in Mitochondrial DNA

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation for NGS
2.2. Bioinformatic Data Analysis
perl MitoSAlt1.1.1.pl config_human.txt \
sample_name_L001_R1_001.fastq.gz \
sample_name_L001_R2_001.fastq.gz \
sample_name.
singularity run -B \
/path_to_main_fastq_folder:/mitopore_data \
/path/mitopore_workflow.sif python \
/home/ag-rossi/projects/mitopore_workflow/mitopore_local/mitopore_indel.py /mitopore_data/ illumina.
3. Results
3.1. SLSMDSs Analysis
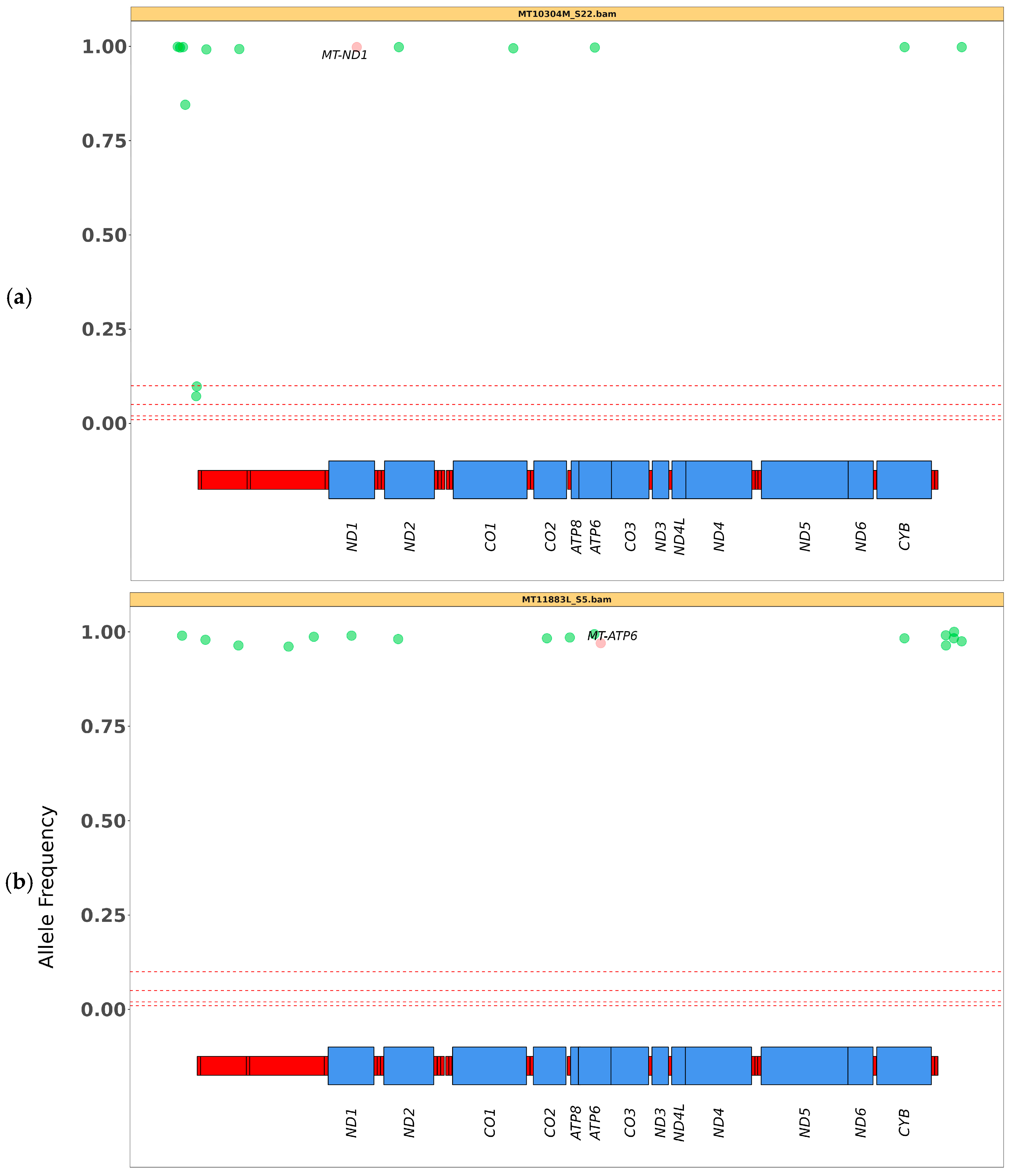
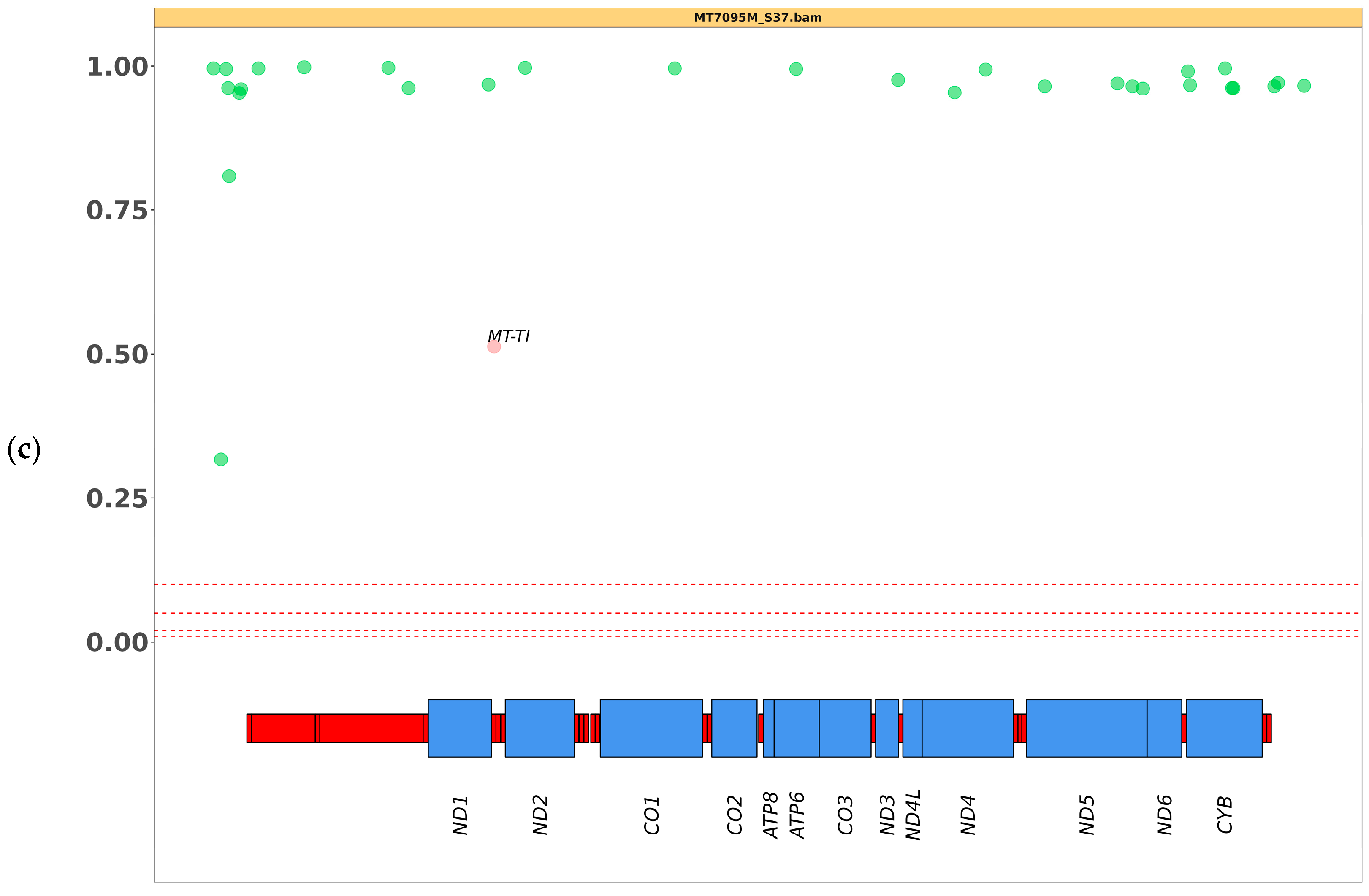
3.2. SNV Analysis
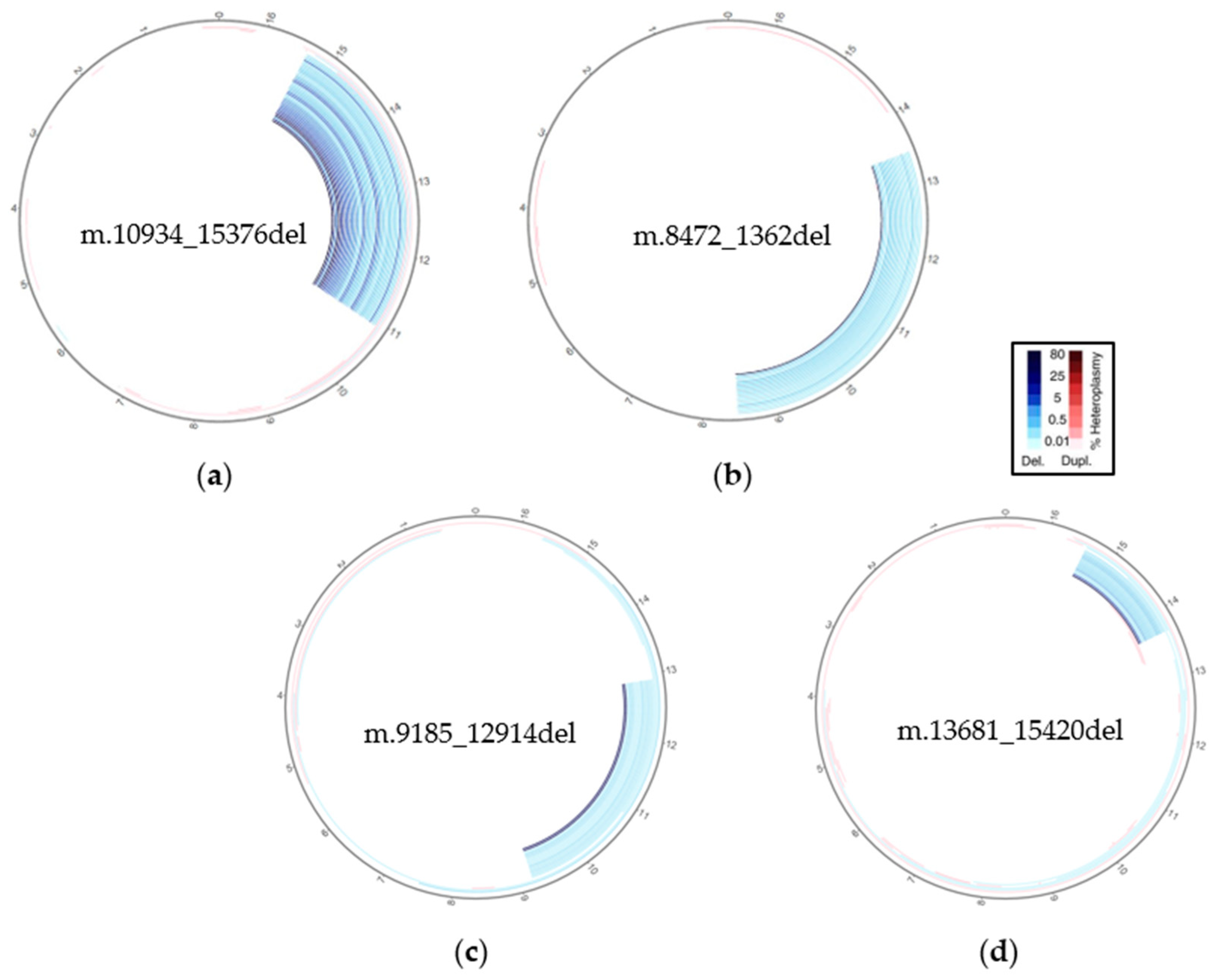
3.3. SVs Analysis by Mitopore
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Habbane, M.; Montoya, J.; Rhouda, T.; Sbaoui, Y.; Radallah, D.; Emperador, S. Human Mitochondrial DNA: Particularities and Diseases. Biomedicines 2021, 9, 1364. [Google Scholar] [CrossRef] [PubMed]
- Tuppen, H.A.L.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta BBA Bioenerg. 2010, 1797, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Parakatselaki, M.E.; Ladoukakis, E.D. mtDNA Heteroplasmy: Origin, Detection, Significance, and Evolutionary Consequences. Life 2021, 11, 633. [Google Scholar] [CrossRef]
- Lax, N.Z.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Mutations: Newly discovered players in neuronal degeneration. NeuroSci 2011, 17, 645–658. [Google Scholar] [CrossRef]
- Salazar, D.; Kloke, K.M.; Guerrero, R.B.; Ferreira, C.R.; Blau, N. Clinical and biochemical footprints of inherited metabolic disorders. XI. Gastrointestinal symptoms. Mol. Genet. Metab. 2023, 138, 107528. [Google Scholar] [CrossRef]
- Chinnery, P.F. Primary Mitochondrial Disorders Overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Eds.; University of Washington: Seattle, WA, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1224/ (accessed on 20 September 2024).
- Xue, L.; Moreira, J.D.; Smith, K.K.; Fetterman, J.L. The Mighty NUMT: Mitochondrial DNA Flexing Its Code in the Nuclear Genome. Biomolecules 2023, 13, 753. [Google Scholar] [CrossRef]
- Parr, R.L.; Maki, J.; Reguly, B.; Dakubo, G.D.; Aguirre, A.; Wittock, R.; Robinson, K.; Jakupciak, J.P.; Thayer, R.E. The pseudo-mitochondrial genome influences mistakes in heteroplasmy interpretation. BMC Genom. 2006, 7, 185. [Google Scholar] [CrossRef]
- Rahman, S.; Poulton, J.; Marchington, D.; Suomalainen, A. Decrease of 3243 A→G mtDNA Mutation from Blood in MELAS Syndrome: A Longitudinal Study. Am. J. Hum. Genet. 2001, 68, 238–240. [Google Scholar] [CrossRef]
- Hardy, S.A.; Blakely, E.L.; Purvis, A.I.; Rocha, M.C.; Ahmed, S.; Falkous, G.; Poulton, J.; Rose, M.R.; O’Mahony, O.; Bermingham, N.; et al. Pathogenic mtDNA mutations causing mitochondrial myopathy: The need for muscle biopsy. Neurol. Genet. 2017, 3, e196. [Google Scholar] [CrossRef]
- Moraes, C.T.; Ricci, E.; Petruzzella, V.; Shanske, S.; DiMauro, S.; Schon, E.A.; Bonilla, E. Molecular analysis of the muscle pathology associated with mitochondrial DNA deletions. Nat. Genet. 1992, 1, 359–367. [Google Scholar] [CrossRef]
- Mavraki, E.; Labrum, R.; Sergeant, K.; Alston, C.L.; Woodward, C.; Smith, C.; Knowles, C.V.Y.; Patel, Y.; Hodsdon, P.; Baines, J.P.; et al. Genetic testing for mitochondrial disease: The United Kingdom best practice guidelines. Eur. J. Hum. Genet. 2023, 31, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Gusic, M.; Prokisch, H. Genetic basis of mitochondrial diseases. FEBS Lett. 2021, 595, 1132–1158. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; McCormick, E.M.; Clarke Muraresku, C.; Falk, M.J.; Gai, X. Clinical Bioinformatics in Precise Diagnosis of Mitochondrial Disease. Bioinformatics 2020, 36, 3016–3024. [Google Scholar] [CrossRef] [PubMed]
- Legati, A.; Zanetti, N.; Nasca, A.; Peron, C.; Lamperti, C.; Lamantea, E.; Ghezzi, D. Current and New Next-Generation Sequencing Approaches to Study Mitochondrial DNA. J. Mol. Diagn. 2021, 23, 732–741. [Google Scholar] [CrossRef]
- Pereira, R.; Oliveira, J.; Sousa, M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J. Clin. Med. 2020, 9, 132. [Google Scholar] [CrossRef]
- Bris, C.; Goudenege, D.; Desquiret-Dumas, V.; Charif, M.; Colin, E.; Bonneau, D.; Amati-Bonneau, P.; Lenaers, G.; Reynier, P.; Procaccio, V. Bioinformatics Tools and Databases to Assess the Pathogenicity of Mitochondrial DNA Variants in the Field of Next Generation Sequencing. Front. Genet. 2018, 9, 632. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Forer, L.; Fuchsberger, C.; Schöpf, B.; Kloss-Brandstätter, A.; Specht, G.; Kronenberg, F.; Schönherr, S. mtDNA-Server: Next-Generation Sequencing Data Analysis of Human Mitochondrial DNA in the Cloud. Nucleic Acids Res. 2016, 44, W64–W69. [Google Scholar] [CrossRef]
- Battle, S.L.; Puiu, D.; Verlouw, J.; Broer, L.; Boerwinkle, E.; Taylor, K.D.; Rotter, J.I.; Rich, S.S.; Grove, M.L.; Pankratz, N.; et al. A Bioinformatics Pipeline for Estimating Mitochondrial DNA Copy Number and Heteroplasmy Levels from Whole Genome Sequencing Data. PLoS Comput. Biol. 2022, 18, e1009976. [Google Scholar] [CrossRef]
- Puttick, C.; Davis, R.L.; Kumar, K.R.; Quinn, J.M.W.; Zeng, T.; Fares, C.; Pinese, M.; Thomas, D.M.; Dinger, M.E.; Sue, C.M.; et al. mity: A Highly Sensitive Mitochondrial Variant Analysis Pipeline for Whole Genome Sequencing Data. J. Bioinform. Syst. Biol. 2024, 7, 05–16. [Google Scholar] [CrossRef]
- Goldstein, A.; Falk, M.J. Single Large-Scale Mitochondrial DNA Deletion Syndromes. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Eds.; University of Washington: Seattle, WA, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1203/ (accessed on 20 September 2024).
- Frascarelli, C.; Zanetti, N.; Nasca, A.; Izzo, R.; Lamperti, C.; Lamantea, E.; Legati, A.; Ghezzhi, D. Nanopore long-read next-generation sequencing for detection of mitochondrial DNA large-scale deletions. Front. Genet. 2023, 14, 1089956. [Google Scholar] [CrossRef]
- Basu, S.; Xie, X.; Uhler, J.P.; Hedberg-Oldfors, C.; Milenkovic, D.; Baris, O.R.; Kimoloi, S.; Matic, S.; Stewart, J.B.; Larsson, N.-G.; et al. Accurate Mapping of Mitochondrial DNA Deletions and Duplications Using Deep Sequencing. PLoS Genet. 2020, 16, e1009242. [Google Scholar] [CrossRef] [PubMed]
- Dobner, J.; Nguyen, T.; Pavez-Giani, M.G.; Cyganek, L.; Distelmaier, F.; Krutmann, J.; Prigione, A.; Rossi, A. mtDNA Analysis Using Mitopore. Mol. Ther. Methods Clin. Dev. 2024, 32, 2. [Google Scholar] [CrossRef] [PubMed]
- Macken, W.L.; Falabella, M.; Pizzamiglio, C.; Woodward, C.E.; Scotchman, E.; Chitty, L.S.; Pitceathly, R.D.S. Enhanced mitochondrial genome analysis: Bioinformatic and long-read sequencing advances and their diagnostic implications. Expert Rev. Mol. Diagn. 2023, 23, 797–814. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Deletion Size | Heteroplasmy | Deletion Breakpoints (Deletion Size in bp) |
|---|---|---|---|
| MT11227M | ~5 kb | ~60% 1 | m.8466_13463del (4980) |
| MT11267M | ~3 kb | ~50% 1 | m.11025_14302del (3270) |
| MT6297M | ~3 kb | ~30% 1 | m.10917_14498del (3555) |
| MT6885M | ~3 kb | ~10% 1 | m.9420_13679del (4259) |
| MT7073M | ~1 kb | ~50% 1 | m.13681_15420del (1732) |
| MT7093M | ~3 kb | ~70% 1 | m.10934_15376del (4425) |
| MT7321M | ~5 kb | ~40% 1 | m.8467_13461del (4980) |
| MT8585M | ~4 kb | ~40% 2 | m.9185_12914del (3721) |
| MT8903M | ~5 kb | ~50% 1 | m.10932_15537del (4594) |
| MT9384M | ~5 kb | ~50% 1 | m.8472_1362del (4980) |
| Sample 1 | Pathogenic Variant (Gene) | Heteroplasmy (%) | Heteroplasmy Mitopore (%) |
|---|---|---|---|
| MT11883L | m.8993T>G (MT-ATP6) | >95 *–98 ** | 97 |
| MT8221M | m.9907G>A (MT-CO3) | 99 ** | 100 |
| MT10304M | m.3890G>A (MT-ND1) | 100 ** | 100 |
| MT9797L | m.3243A>G (MT-TL1) | 10 *–31 ** | 31 |
| MT3582M | m.642T>C (MT-TF) | 90 *–67 ** | 67 |
| MT7095M | m.4301A>G (MT-TI) | 50 ** | 51 |
| MT8636M | m.13513G>A (MT-ND5) | 80 *–88 ** | 88 |
| MT8722M | m.3249G>A (MT-TL1) | 65 ** | 65 |
| MT10618L | m.11778G>A (MT-ND4) | 100 **–100 *** | 100 |
| MT3289L | m.8993T>C (MT-ATP6) | 87 **–90 *** | 90 |
| Mitochondrial Features | Scoring and Filtering Features | Steps | |||
|---|---|---|---|---|---|
| refchr | MT | score_threshold | 80 | dna | yes |
| msize | 16,569 | evalue_threshold | 0.00001 | enriched | yes |
| exclude | 5 | split_length | 15 | nu_mt | no |
| orihs | 16,081 | paired_distance | 1000 | rmtmp | yes |
| orihe | 407 | deletion_threshold_min | 30 | o_mt | yes |
| orils | 5730 | deletion_threshold_max | 30,000 | i_del | yes |
| orile | 5763 | breakthreshold | −2 | cn_mt | no |
| cluster_threshold | 2 | ||||
| Breakspan | 15 | ||||
| Sizelimit | 10,000 | ||||
| Hplimit | 0.01 | ||||
| Flank | 15 | ||||
| split_distance_threshold | 5 | ||||
| Mitopore Run Parameters | |
|---|---|
| Type of Analysis | SNV & INDEL |
| Sequencing data | Illumina (short read) |
| Genome | Human hg38 (Homo Sapiens) |
| Haplotree | phylotree-rcrs@17.2 |
| Variance threshold (%) | 5 |
| Minimum sequence depth | 1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barresi, M.; Dal Santo, G.; Izzo, R.; Zauli, A.; Lamantea, E.; Caporali, L.; Ghezzi, D.; Legati, A. Bioinformatics Tools for NGS-Based Identification of Single Nucleotide Variants and Large-Scale Rearrangements in Mitochondrial DNA. BioTech 2025, 14, 9. https://doi.org/10.3390/biotech14010009
Barresi M, Dal Santo G, Izzo R, Zauli A, Lamantea E, Caporali L, Ghezzi D, Legati A. Bioinformatics Tools for NGS-Based Identification of Single Nucleotide Variants and Large-Scale Rearrangements in Mitochondrial DNA. BioTech. 2025; 14(1):9. https://doi.org/10.3390/biotech14010009
Chicago/Turabian StyleBarresi, Marco, Giulia Dal Santo, Rossella Izzo, Andrea Zauli, Eleonora Lamantea, Leonardo Caporali, Daniele Ghezzi, and Andrea Legati. 2025. "Bioinformatics Tools for NGS-Based Identification of Single Nucleotide Variants and Large-Scale Rearrangements in Mitochondrial DNA" BioTech 14, no. 1: 9. https://doi.org/10.3390/biotech14010009
APA StyleBarresi, M., Dal Santo, G., Izzo, R., Zauli, A., Lamantea, E., Caporali, L., Ghezzi, D., & Legati, A. (2025). Bioinformatics Tools for NGS-Based Identification of Single Nucleotide Variants and Large-Scale Rearrangements in Mitochondrial DNA. BioTech, 14(1), 9. https://doi.org/10.3390/biotech14010009