
Genomic Characterization of Extremely Antibiotic-Resistant Strains of Pseudomonas aeruginosa Isolated from Patients of a Clinic in Sincelejo, Colombia


Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Collection and Processing of Samples
2.3. Phenotypic Tests
2.4. Genotypic Testing
2.5. Analysis of the Genomic Sequence
2.6. Analysis of Resistance Genes and Virulence Factors
2.7. Ethical Implications
3. Results
3.1. Phenotypic Characterization
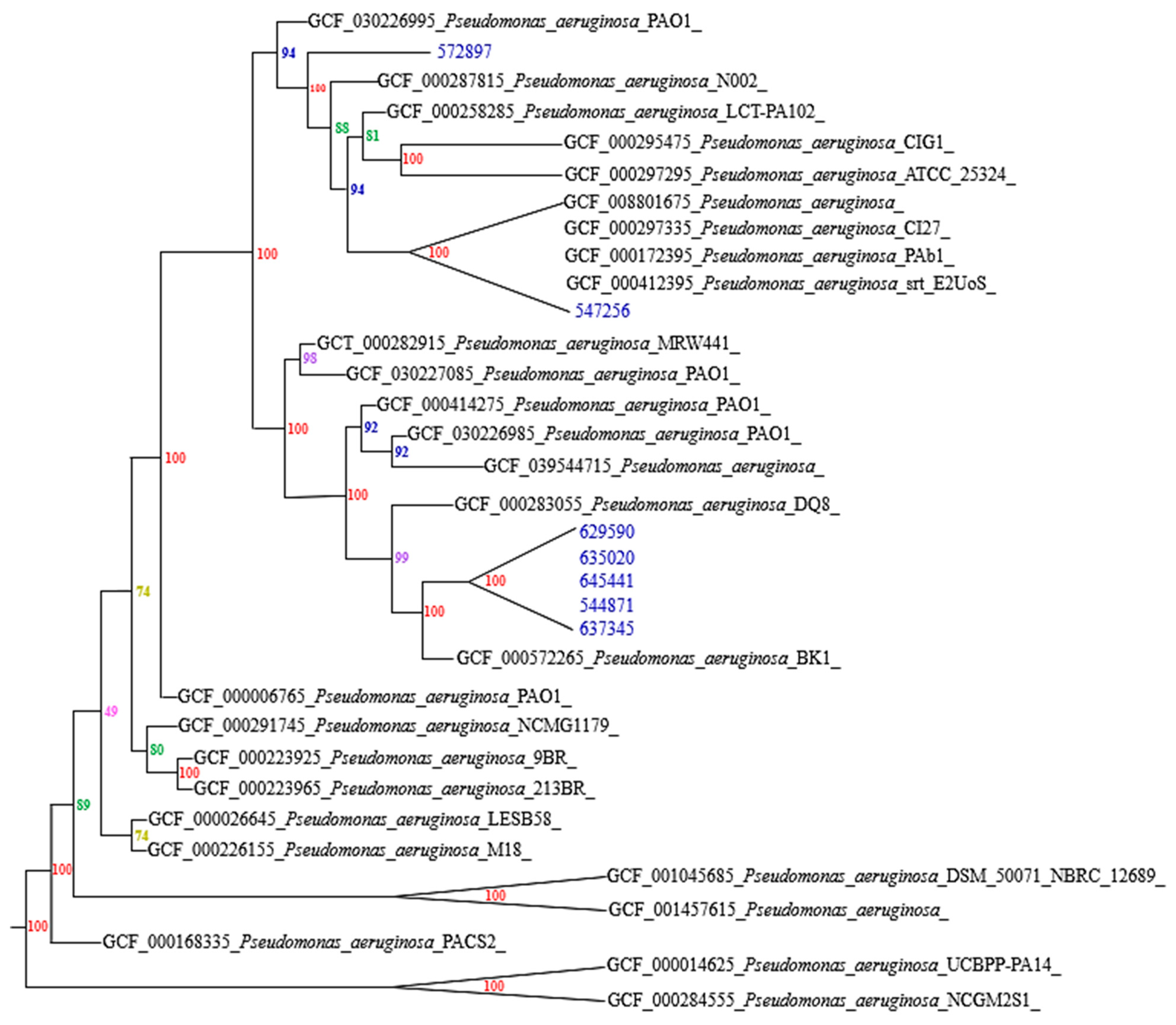
3.2. Genotypic Characterization
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANI | Average Nucleotide Identity |
| WHO | World Health Organization |
| CR-PA | Carbapenem-resistant P. aeruginosa |
| MDR | Multidrug-resistant |
| XDR | Extremely drug-resistant |
| MIC | Minimum inhibitory concentration |
| CNSG | National Center for Genomic Sequencing |
| RGI | Resistance Gene Identifier |
| CARD | Comprehensive Antibiotic Resistance Database |
| VFDB | Virulence Factor Database |
| SNPs | Single nucleotide polymorphisms |
References
- Krell, T.; Matilla, M.A. Pseudomonas aeruginosa. Trends Microbiol. 2024, 32, 216–218. [Google Scholar] [CrossRef]
- Botelho, J.; Grosso, F.; Peixe, L. Antibiotic resistance in Pseudomonas aeruginosa—Mechanisms, epidemiology and evolution. Drug Resist. Updat. 2019, 44, 100640. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Roberts, J.A.; Walker, M.M.; Aslan, A.T.; Harris, P.N.A.; Sime, F.B. The global epidemiology of ventilator-associated pneumonia caused by multi-drug resistant Pseudomonas aeruginosa: A systematic review and meta-analysis. Int. J. Infect. Dis. 2024, 139, 78–85. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Publishes List of Bacteria for Which New Antibiotics Are Urgently Needed. 2017. Available online: https://www.who.int/es/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 12 November 2024).
- Horcajada, J.P.; Montero, M.; Oliver, A.; Sorli, L.; Luque, S.; Gómez, S.; Benito, N.; Grau, S. Epidemiology and Treatment of Multidrug-Resistant and Extensively Drug-Resistant Pseudomonas aeruginosa Infections. Clin. Microbiol. Rev. 2019, 32, e00031-19. [Google Scholar] [CrossRef] [PubMed]
- Shortridge, D.; Gales, A.C.; Streit, J.M.; Huband, M.D.; Tsakris, A.; Jones, R.N. Geographic and Temporal Patterns of Antimicrobial Resistance in Pseudomonas aeruginosa over 20 Years from the SENTRY Antimicrobial Surveillance Program, 1997–2016. Open Forum Infect. Dis. 2019, 6, S63–S68. [Google Scholar] [CrossRef]
- Giovagnorio, F.; De Vito, A.; Madeddu, G.; Parisi, S.G.; Geremia, N. Resistance in Pseudomonas aeruginosa: A Narrative Review of Antibiogram Interpretation and Emerging Treatments. Antibiotics 2023, 12, 1621. [Google Scholar] [CrossRef]
- Tabak, Y.P.; Merchat, S.; Ye, G.; Vankeepuram, L.; Gupta, V.; Kurtz, S.G.; Puzniak, L.A. Incremental clinical and economic burden of suspected respiratory infections due to multi-drug-resistant Pseudomonas aeruginosa in the United States. J. Hosp. Infect. 2019, 103, 134–141. [Google Scholar] [CrossRef]
- Morata, L.; Cobos, N.; Martinez, J.; Soriano, A.; Almela, M.; Marco, D.; Sterzik, H.; Nuñez, R.; Hernandez, C.; Mensa, J. Influence of Multidrug Resistance and Appropriate Empirical Therapy on the 30-Day Mortality Rate of Pseudomonas aeruginosa Bacteremia. Antimicrob. Agents Chemother. 2012, 56, 4833–4837. [Google Scholar] [CrossRef]
- Castanheira, M.; Deshpande, L.M.; Costello, A.; Davies, T.A.; Jones, R.N. Epidemiology and carbapenem resistance mechanisms of carbapenem-non-susceptible Pseudomonas aeruginosa collected during 2009–11 in 14 European and Mediterranean countries. J. Antimicrob. Chemother. 2014, 69, 1804–1814. [Google Scholar] [CrossRef]
- Qin, S.; Xiao, W.; Zhou, C.; Pu, Q.; Deng, X.; Lan, L.; Liang, H.; Song, X.; Wu, M. Pseudomonas aeruginosa: Pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct. Target. Ther. 2022, 7, 199. [Google Scholar] [CrossRef]
- Alshammari, H.O.; Somily, A.; Qattan, M.Y.; Alsubki, R.A.; Moussa, I.M. Susceptibility pattern of multi-drug resistance Pseudomonas aeruginosa isolates from tertiary care hospital in Riyadh, KSA. J. King Saud Univ. Sci. 2023, 35, 102702. [Google Scholar] [CrossRef]
- Cuello, M. Seguimiento Epidemiológico de los Perfiles Genómicos de Resistencia a Antibióticos, en Aislamientos Clínicos de Pseudomonas aeruginosa Mediante Secuenciación de Genoma Completo (WGS). Ph.D. Thesis, Universidad Nacional de Colombia, Bogotá, Colombia, 2022. Available online: https://repositorio.unal.edu.co/bitstream/handle/unal/84156/1065651232.2023.pdf?sequence=4&isAllowed=y (accessed on 23 November 2024).
- Osorio, N. Caracterización Genómica de Factores de Virulencia de Aislados Clínicos de Pseudomonas aeruginosa Basados en WGS Provenientes de un Hospital de Bogotá, Colombia. Ph.D. Thesis, Universidad Nacional de Colombia, Bogotá, Colombia, 2022. Available online: https://repositorio.unal.edu.co/handle/unal/84264 (accessed on 13 November 2024).
- Rocha, A.J.; Barsottini, M.R.D.O.; Rocha, R.R.; Laurindo, M.V.; Moraes, F.L.L.D.; Rocha, S.L.D. Pseudomonas Aeruginosa: Virulence Factors and Antibiotic Resistance Genes. Braz. Arch. Biol. Technol. 2019, 62, e19180503. [Google Scholar] [CrossRef]
- Hu, Y.; Peng, W.; Wu, Y.; Li, H.; Wang, Q.; Yi, H.; Zhang, R.; Shao, B.; Zhu, K. A Potential High-Risk Clone of Pseudomonas aeruginosa ST463. Front. Microbiol. 2021, 12, 670202. [Google Scholar] [CrossRef] [PubMed]
- Telling, K.; Laht, M.; Brauer, A.; Remm, M.; Kisand, V.; Maimets, M.; Tenson, T.; Lutsar, I. Multidrug resistant Pseudomonas aeruginosa in Estonian hospitals. BMC Infect. Dis. 2018, 18, 513. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, D.; Ji, B.; Zhang, X.; Anbo, M.; Jelsbak, L. Whole-genome sequencing reveals high-risk clones of Pseudomonas aeruginosa in Guangdong, China. Front. Microbiol. 2023, 14, 1117017. [Google Scholar] [CrossRef]
- Adenipekun, E.O.; Akinleye, E.F.; Tewogbade, O.A.; Iwalokun, B.A. Detection of virulence genes and multidrug resistance in Pseudomonas aeruginosa clinical isolates from a public hospital in Lagos, Nigeria. Sci. Afr. 2023, 22, e01950. [Google Scholar] [CrossRef]
- Patil, S.; Chavan, S.; Patil, S.; Patil, S. Antimicrobial Susceptibility Pattern of Pseudomonas aeruginosa isolates from Different Clinical samples at a Tertiary care hospital. Int. J. Life Sci. 2023, 12. [Google Scholar]
- Clinical and Laboratory Standards Institute. CLSI M100 Performance Standards for Antimicrobial Susceptibility Testing, 34th ed.; CLSI supplement M100 (ISBN 978-1-68440-221-2 [Electronic]); Clinical and Laboratory Standards Institute: Malvern, PA, USA, 2024. [Google Scholar]
- Universidad El Bosque. Pseudomonas aeruginosa Genome Assembly ASM2309393v1. 2022. Available online: https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_023093935.1/ (accessed on 24 November 2024).
- Berger, O.; Lurie-Weinberger, M.N.; Tsyba, E.; Talisman, R. ST773 Pseudomonas aeruginosa wound infection as a result of medical tourism to Turkey. J. Travel Med. 2024, 31, taad097. [Google Scholar] [CrossRef]
- Jeganathan, L.P.; Prakash, L.; Sivakumar, N.; Antony, A.; Alqarawi, S.; Prajna, L.; Devarajan, B.; Mohankumar, V. Draft Genome Sequence of an Invasive Multidrug-Resistant Strain, Pseudomonas aeruginosa BK1, Isolated from a Keratitis Patient. Genome Announc. 2014, 2, e00153-14. [Google Scholar] [CrossRef]
- Technical University of Denmark. Pseudomonas aeruginosa PAO1 Genome Assembly ASM3022699v1. 2023. Available online: https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_030226995.1/ (accessed on 13 December 2024).
- Lorusso, A.B.; Carrara, J.A.; Barroso, C.D.N.; Tuon, F.F.; Faoro, H. Role of Efflux Pumps on Antimicrobial Resistance in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2022, 23, 15779. [Google Scholar] [CrossRef]
- Husna, A.; Rahman, M.M.; Badruzzaman, A.T.; Sikder, M.H.; Islam, M.R.; Rahman, M.T.; Alam, J.; Ashour, H.M. Extended-Spectrum β-Lactamases (ESBL): Challenges and Opportunities. Biomedicines 2023, 11, 2937. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, A.; Llamas, M.A.; Marcos-Torres, F.J. Transcriptional Regulators Controlling Virulence in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2023, 24, 11895. [Google Scholar] [CrossRef] [PubMed]
- Blanc, D.S.; Francioli, P.; Zanetti, G. Molecular Epidemiology of Pseudomonas aeruginosa in the Intensive Care Units—A Review. Open Microbiol. J. 2007, 1, 8–11. [Google Scholar] [CrossRef]
- Savaş, L.; Duran, N.; Savaş, N.; Önlen, Y.; Ocak, S. The Prevalence and Resistance Patterns of Pseudomonas aeruginosa in Intensive Care Units in a University Hospital. Turk. J. Med. Sci. 2005, 35, 317–322. [Google Scholar]
- World Health Organization. WHO Bacterial Priority Pathogens List, 2024: Bacterial Pathogens of Public Health Importance to Guide Research, Development and Strategies to Prevent and Control Antimicrobial Resistance. 2024. Available online: https://iris.who.int/bitstream/handle/10665/376776/9789240093461-eng.pdf?sequence=1 (accessed on 13 December 2024).
- Folic, M.M.; Djordjevic, Z.; Folic, N.; Radojevic, M.Z.; Jankovic, S.M. Epidemiology and risk factors for healthcare-associated infections caused by Pseudomonas aeruginosa. J. Chemother. 2021, 33, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Kos, V.N.; Déraspe, M.; McLaughlin, R.E.; Whiteaker, J.D.; Roy, P.H.; Alm, R.A.; Corbeil, J.; Gardner, H. The Resistome of Pseudomonas aeruginosa in Relationship to Phenotypic Susceptibility. Antimicrob. Agents Chemother. 2015, 59, 427–436. [Google Scholar] [CrossRef]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.L.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef]
- Brinkman, F.S.; Winsor, G.L.; Done, R.E.; Filloux, A.; Francis, V.I.; Goldberg, J.B.; Greenberg, E.P.; Han, K.; Hancock, R.E.; Haney, C.H.; et al. The Pseudomonas aeruginosa whole genome sequence: A 20th anniversary celebration. Adv. Microb. Physiol. 2021, 79, 25–88. [Google Scholar] [CrossRef]
- Shaaban, M.T.; Abdel-Raouf, M.; Zayed, M.; Emara, M.A. Microbiological and molecular studies on a multidrug-resistant Pseudomonas aeruginosa from a liver transplant patient with urinary tract infection in Egypt. BMC Microbiol. 2024, 24, 184. [Google Scholar] [CrossRef]
- Klockgether, J.; Cramer, N.; Wiehlmann; Davenport, C.F.; Tümmler, B. Pseudomonas aeruginosa Genomic Structure and Diversity. Front. Microbiol. 2011, 2, 150. [Google Scholar] [CrossRef]
- Boukerb, A.M.; Simon, M.; Pernet, E.; Jouault, A.; Portier, E.; Persyn, E.; Bouffartigues, E.; Bazire, A.; Chevalier, S.; Feuilloley, M.G.J.; et al. Draft Genome Sequences of Four Pseudomonas aeruginosa Clinical Strains with Various Biofilm Phenotypes. Microbiol. Resour. Announc. 2020, 9, e01286-19. [Google Scholar] [CrossRef]
- Castañeda, F. Genética de Poblaciones y Perfiles de Resistencia a Antibióticos de Una Colección Clínica de Pseudomonas aeruginosa; Instituto Politécnico Nacional: Mexico City, México, 2018; Available online: https://repositorio.cinvestav.mx/handle/cinvestav/3548 (accessed on 13 October 2024).
- Quick, J.; Cumley, N.; Wearn, C.M.; Niebel, M.; Constantinidou, C.; Thomas, C.M.; Pallen, M.J.; Moiemen, N.S.; Bamford, A.; Oppenheim, B.; et al. Seeking the source of Pseudomonas aeruginosa infections in a recently opened hospital: An observational study using whole-genome sequencing. BMJ Open 2014, 4, e006278. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, B.; Valot, B.; Abdelbary, M.M.H.; Prod’hom, G.; Greub, G.; Senn, L.; Blanc, D.S. Combining Standard Molecular Typing and Whole Genome Sequencing to Investigate Pseudomonas aeruginosa Epidemiology in Intensive Care Units. Front. Public Health 2020, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Kas, A.; Smith, E.E.; Raymond, C.K.; Sims, E.H.; Hastings, M.; Burns, J.L.; Kaul, R.; Olson, M.V. Whole-Genome Sequence Variation among Multiple Isolates of Pseudomonas aeruginosa. J. Bacteriol. 2003, 185, 1316–1325. [Google Scholar] [CrossRef] [PubMed]
- Kiyaga, S.; Kyany’a, C.; Muraya, A.W.; Smith, H.J.; Mills, E.G.; Kibet, C.; Mboowa, G.; Musila, L. Genetic Diversity, Distribution, and Genomic Characterization of Antibiotic Resistance and Virulence of Clinical Pseudomonas aeruginosa Strains in Kenya. Front. Microbiol. 2022, 13, 835403. [Google Scholar] [CrossRef]
- Baquero, F. From pieces to patterns: Evolutionary engineering in bacterial pathogens. Nat. Rev. Microbiol. 2004, 2, 510–518. [Google Scholar] [CrossRef]
- Kocsis, B.; Gulyás, D.; Szabó, D. Diversity and Distribution of Resistance Markers in Pseudomonas aeruginosa International High-Risk Clones. Microorganisms 2021, 9, 359. [Google Scholar] [CrossRef]
- Jung, H.; Pitout, J.D.D.; Matsumura, Y.; Strydom, K.A.; Kingsburgh, C.; Ehlers, M.M.; Kock, M.M. Genomic epidemiology and molecular characteristics of blaNDM-1-positive carbapenem-resistant Pseudomonas aeruginosa belonging to international high-risk clone ST773 in the Gauteng region, South Africa. Eur. J. Clin. Microbiol. Infect. Dis. 2024, 43, 627–640. [Google Scholar] [CrossRef]
- Singh, S.; Pulusu, C.P.; Pathak, A.; Pradeep, B.E.; Prasad, K.N. Complete genome sequence of an extensively drug-resistant Pseudomonas aeruginosa ST773 clinical isolate from North India. J. Glob. Antimicrob. Resist. 2021, 27, 244–246. [Google Scholar] [CrossRef]

{kind=link}
| Strains | Class | Antibiotic | MIC | Interpretation |
|---|---|---|---|---|
| 544871 572897 629590 635020 637345 645441 5472561 | Beta-lactam/betalactamase inhibitor | Piperacillin/Tazobactam | ≥128 | R |
| Ceftazidime/Avibactam | ≥16 | R | ||
| Cephalosporins | Cefazolin | ≥64 | R | |
| Ceftazidime | ≥64 | R | ||
| Cefepime | ≥64 | R | ||
| Monobactam | Aztreonam | ≥64 | R | |
| Carbapenems | Meropenem | ≥16 | R | |
| Aminoglycosides | Amikacin | ≥64 | R | |
| Gentamicin | ≥ 16 | R | ||
| Fluoroquinolones | Ciprofloxacin | ≥4, 2 1 | R, I 1 | |
| Glycylcyclines | Tigecycline | ≥8 | R |
| Strains | Total Number of Sequences | Average Depth | Total Readings | Total Sequence Length (bp) | Q30(%) | GC% | N50 STATISTICS (bp) | Number of CDSs | Integrity Percentage |
|---|---|---|---|---|---|---|---|---|---|
| 544871 | 228 | 367,544 | 36,511,708 | 7,304,619 bp | 94.53 | 65.69% | 265,943 | 6838 | 99.86% |
| 547256 | 134 | 339,786 | 32,642,984 | 7,046,366 | 93.97 | 65.7% | 271,048 | 6594 | 99.79% |
| 572897 | 233 | 283,907 | 27,412,470 | 7,073,876 | 93.94 | 66.0% | 205,279 | 6580 | 99.86% |
| 629590 | 206 | 484,218 | 46,881,312 | 7,109,622 | 94.07 | 65.8% | 284,837 | 6651 | 99.86% |
| 635020 | 182 | 426,345 | 41,478,374 | 7,104,378 | 92.59 | 65.8% | 363,875 | 6621 | 99.86% |
| 637345 | 168 | 207,998 | 19,829,196 | 6,982,541 | 93.11 | 65.6% | 258,373 | 6580 | 97.94% |
| 645441 | 184 | 299,912 | 28,952,368 | 7,041,958 | 92.44 | 65.9% | 265,943 | 6546 | 99.86% |
| Strains 1 | Strains 2 | Aligned Bases % | Avg Identity % | Total SNPs |
|---|---|---|---|---|
| 544871 | 34Pae36 1 | 97.0824 | 99.9785 | 156 |
| 547256 | ST773 2 | 91.8498 | 99.9663 | 1379 |
| 572897 | 34Pae36 | 99.2537 | 99.9828 | 98 |
| 629590 | 34Pae36 | 99.5708 | 99.9859 | 78 |
| 635020 | 34Pae36 | 99.1635 | 99.9788 | 196 |
| 637345 | 34Pae36 | 98.7693 | 99.9829 | 89 |
| 645441 | 34Pae36 | 99.1467 | 99.9747 | 336 |
| Strains 1 | Strains 2 | Aligned Bases % | Avg Identity % | Total SNPs |
|---|---|---|---|---|
| 572897 | 645441 | 99.0469 | 99.9871 | 269 |
| 635020 | 645441 | 99.0203 | 99.9918 | 109 |
| 572897 | 629590 | 98.9943 | 99.9945 | 56 |
| 572897 | 635020 | 98.9639 | 99.988 | 103 |
| 629590 | 635020 | 98.9449 | 99.9874 | 61 |
| 629590 | 645441 | 98.5915 | 99.9866 | 282 |
| 637345 | 645441 | 97.4426 | 99.9858 | 291 |
| 544871 | 635020 | 96.8277 | 99.989 | 125 |
| 629590 | 637345 | 96.7057 | 99.9903 | 44 |
| 572897 | 637345 | 96.6922 | 99.987 | 78 |
| 544871 | 572897 | 96.6521 | 99.9896 | 92 |
| 635020 | 637345 | 96.6505 | 99.9863 | 47 |
| 544871 | 629590 | 96.4404 | 99.9907 | 102 |
| 544871 | 645441 | 96.2189 | 99.9872 | 290 |
| 544871 | 637345 | 95.0555 | 99.9897 | 103 |
| 547256 | 635020 | 88.6768 | 98.8711 | 64,848 |
| 547256 | 572897 | 88.1132 | 98.8737 | 64,629 |
| 547256 | 645441 | 88.1131 | 98.8686 | 64,874 |
| 547256 | 629590 | 87.6866 | 98.8643 | 64,474 |
| 547256 | 637345 | 86.3051 | 98.897 | 61,629 |
| 544871 | 547256 | 85.685 | 98.8757 | 64,831 |
| Class | Antibiotic | Related Gene |
|---|---|---|
| Beta-lactam with Beta-lactamase inhibitor | Piperacillin/Tazobactam | mexY, mexX, APH(3′)-IIb, OpmH, MexB, mexM, OprM (Resistance: Efflux pump), OXA-395, bcr-1 (Resistance: Beta-lactamases), ArmR (Regulation) |
| Ceftazidime/Avibactam | ||
| Cephalosporins | Cefazolin | KPC-2, VIM-2 (Resistance: Beta-lactamases), MexB, OprJ, MexD, opmE, mexN (Resistance: Efflux pump) |
| Ceftazidime | ||
| Cefepime | ||
| Monobactam | Aztreonam | MuxC, MuxA (Virulence: Efflux pump), OpmB, MuxB (Resistance: Efflux pump) |
| Carbapenems | Meropenem | MexB, opmE, mexN (Resistance: Efflux pump) |
| Aminoglycosides | Amikacin | mexY, mexX, APH(3′)-IIb, opmE, mexQ, mexN, mexP (Resistance: Efflux pump) |
| Gentamicin | ||
| Fluoroquinolones | Ciprofloxacin | MexA, MexE, OprN, MexW, MexV, MexC, MexF, MexI, MexB, MexG, mexM, OprJ, MexH, OprM, MexD, opmE, mexQ, mexN, mexP, MexL (Resistance: Efflux pump), ArmR (Regulation) |
| Glycylcyclines | Tigecycline | MexX, mexY, OprM (Resistance: Efflux pump) |
| Quaternary compounds | Benzalkonium chloride | emrE (Resistance: Efflux pump) |
| Inhibitor of bacterial protein synthesis | Chloramphenicol | catB7, MexI, MexB, OprM, mexQ, mexP, MexL (Resistance: Efflux pump) |
| Macrolides | Erythromycin | MexB, MexG, MexI, OprJ, MexH, OpmB, MexD, MuxB, OprM, mexQ, mexP, MexL, MexK, MexJ (Resistencia:Bomba de eflujo), MuxC, MuxA (Virulence: Efflux pump), ArmR (Regulation) |
| Polymyxins | Colistin | arnA (Resistance: Lipid A modification), ParS, basS (Resistance: Two-component systems) |
| Sulfonamides | Sulfamethoxazole, Sulfadiazine, Sulfisoxazole | sul1 (Resistance: Sulfonamines) |
| Antiseptic | Triclosan | TriB, TriA, TriC (Resistance: Efflux pump) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pajaro-Castro, N.; Diaz-Morales, E.; Hoyos, K. Genomic Characterization of Extremely Antibiotic-Resistant Strains of Pseudomonas aeruginosa Isolated from Patients of a Clinic in Sincelejo, Colombia. BioTech 2025, 14, 21. https://doi.org/10.3390/biotech14010021
Pajaro-Castro N, Diaz-Morales E, Hoyos K. Genomic Characterization of Extremely Antibiotic-Resistant Strains of Pseudomonas aeruginosa Isolated from Patients of a Clinic in Sincelejo, Colombia. BioTech. 2025; 14(1):21. https://doi.org/10.3390/biotech14010021
Chicago/Turabian StylePajaro-Castro, Nerlis, Erick Diaz-Morales, and Kenia Hoyos. 2025. "Genomic Characterization of Extremely Antibiotic-Resistant Strains of Pseudomonas aeruginosa Isolated from Patients of a Clinic in Sincelejo, Colombia" BioTech 14, no. 1: 21. https://doi.org/10.3390/biotech14010021
APA StylePajaro-Castro, N., Diaz-Morales, E., & Hoyos, K. (2025). Genomic Characterization of Extremely Antibiotic-Resistant Strains of Pseudomonas aeruginosa Isolated from Patients of a Clinic in Sincelejo, Colombia. BioTech, 14(1), 21. https://doi.org/10.3390/biotech14010021