
The Transcription of Transposable Elements Differentially Regulated by SVAs in the Major Histocompatibility Complex Class I Region of a Parkinson’s Progression Markers Initiative Cohort




Abstract

1. Introduction
2. Materials and Methods
3. Results
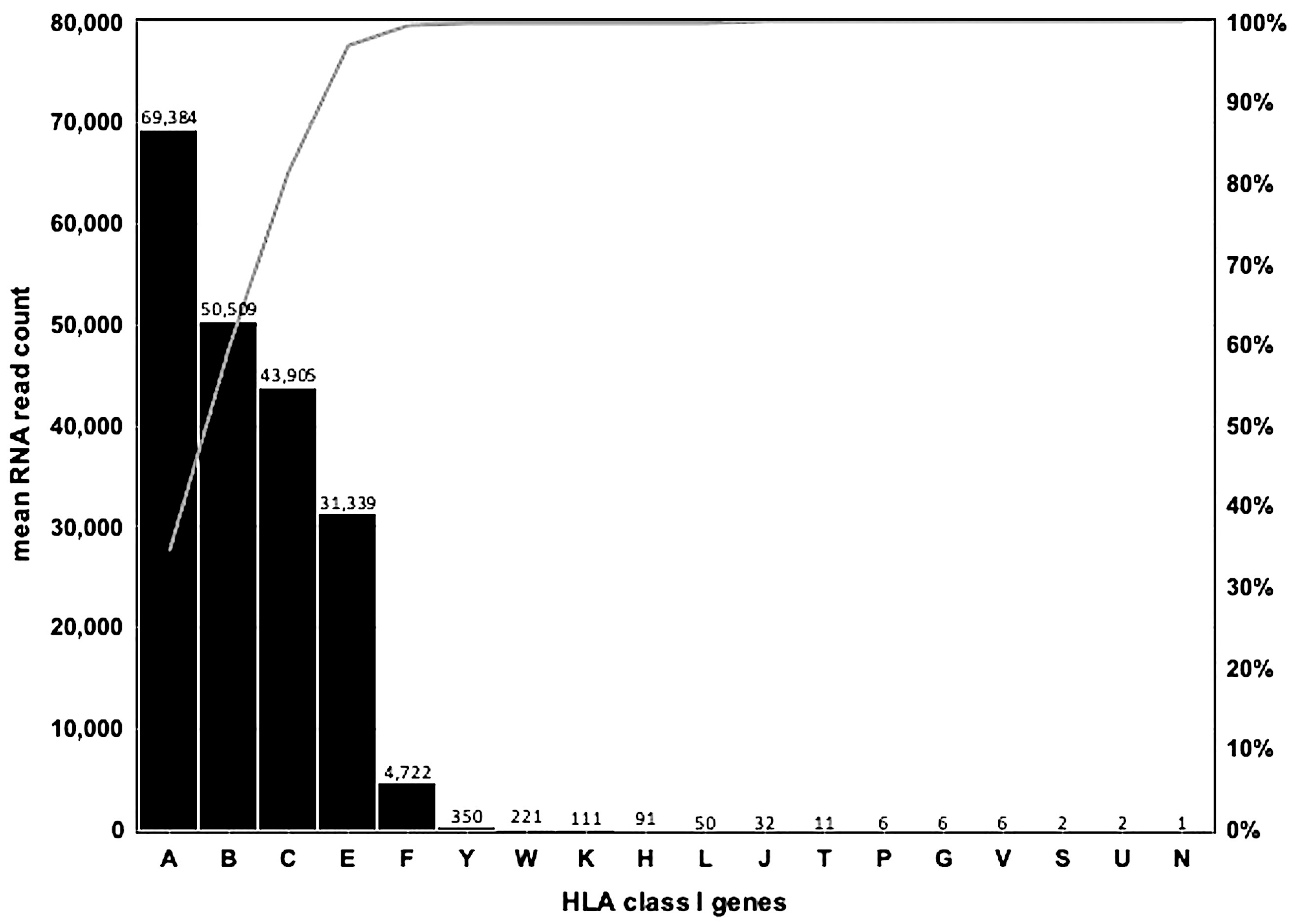
3.1. HLA Class I Gene Transcription Levels in Whole Blood Samples of 1530 PPMI Subjects
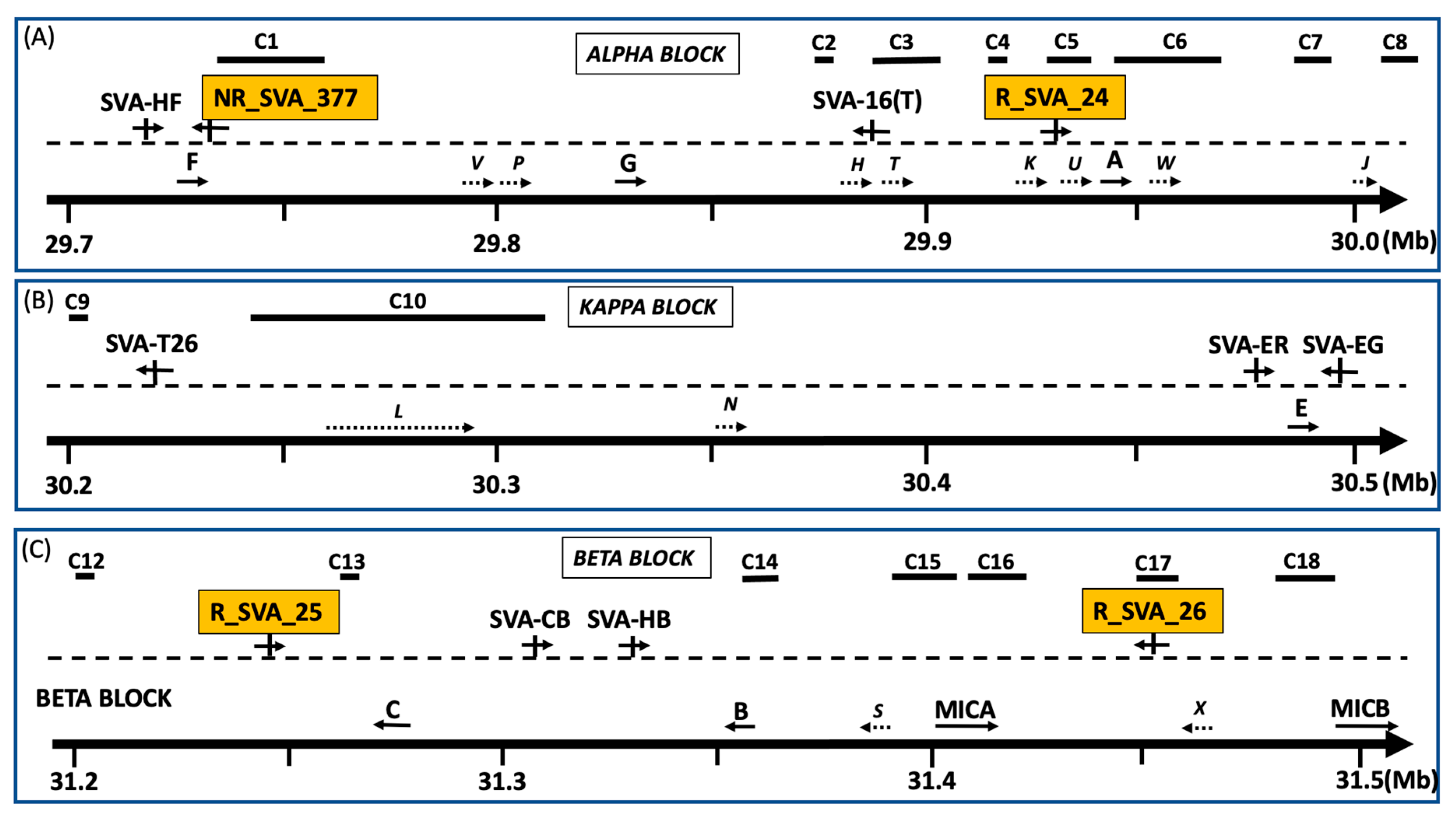
3.2. HLA and TE Clusters Regulated by Four SVA Retrotransposons Within MHC Class I Region
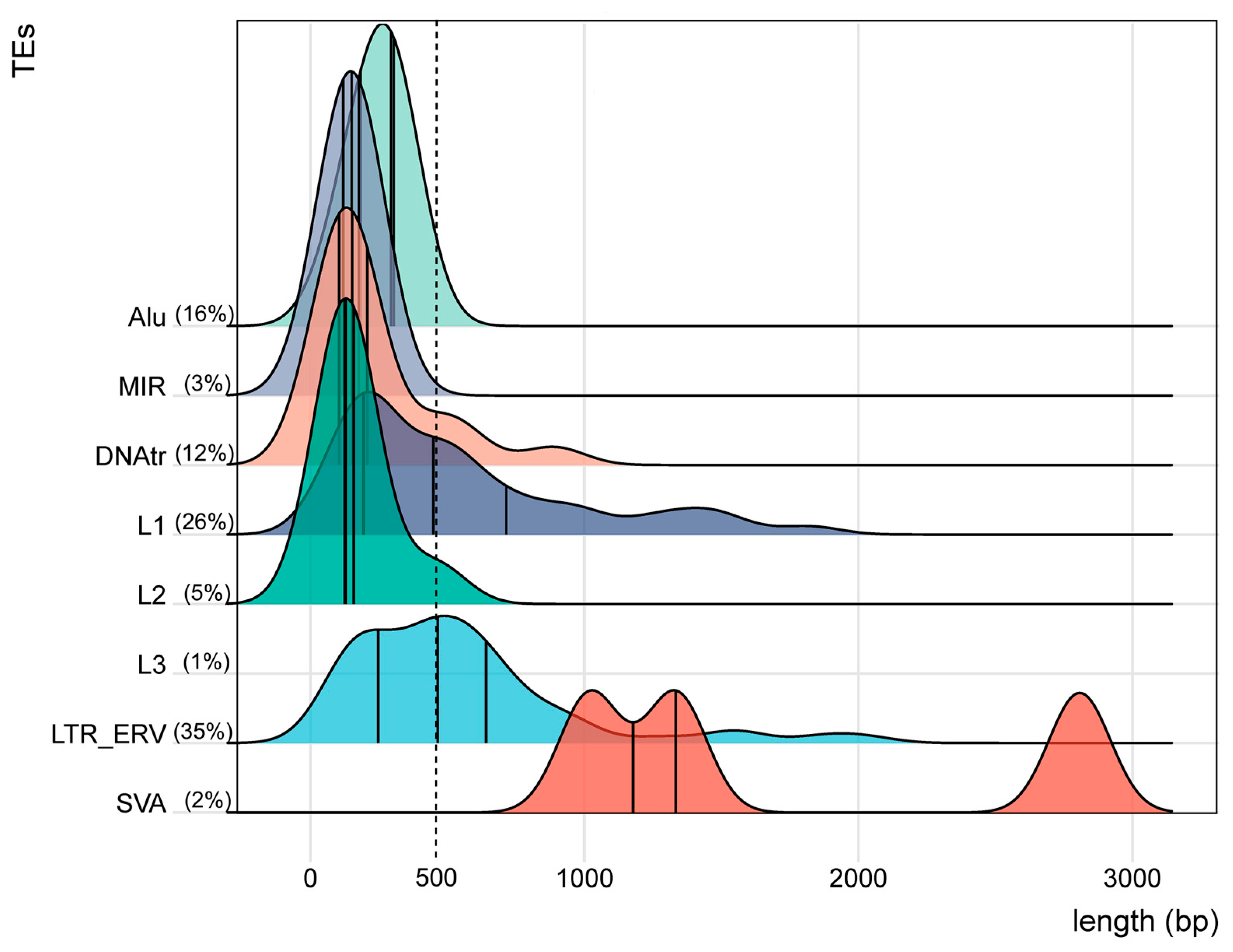
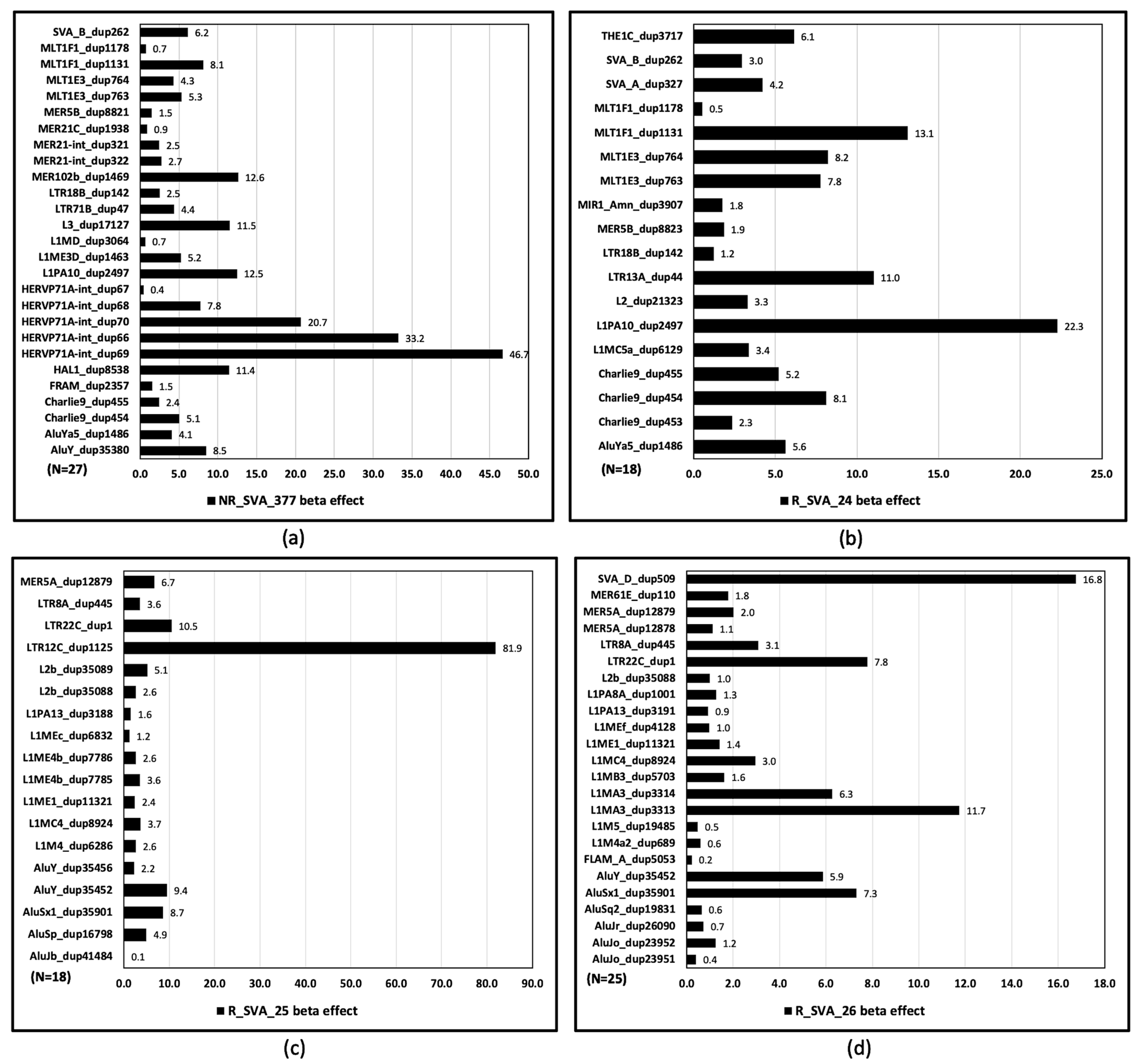
3.3. TE Families, Classes, and Individual Annotated Elements Transcribed Within Clusters
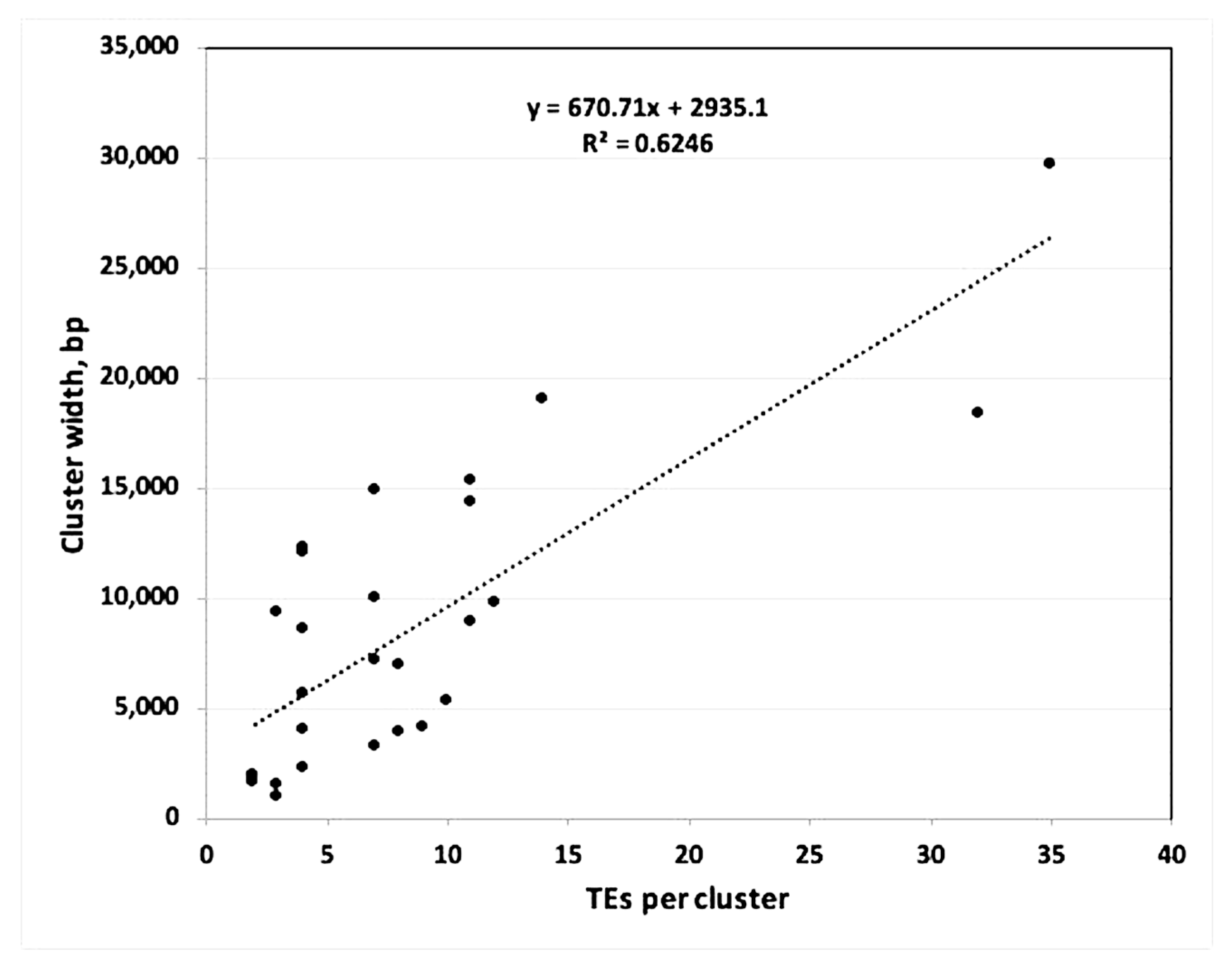
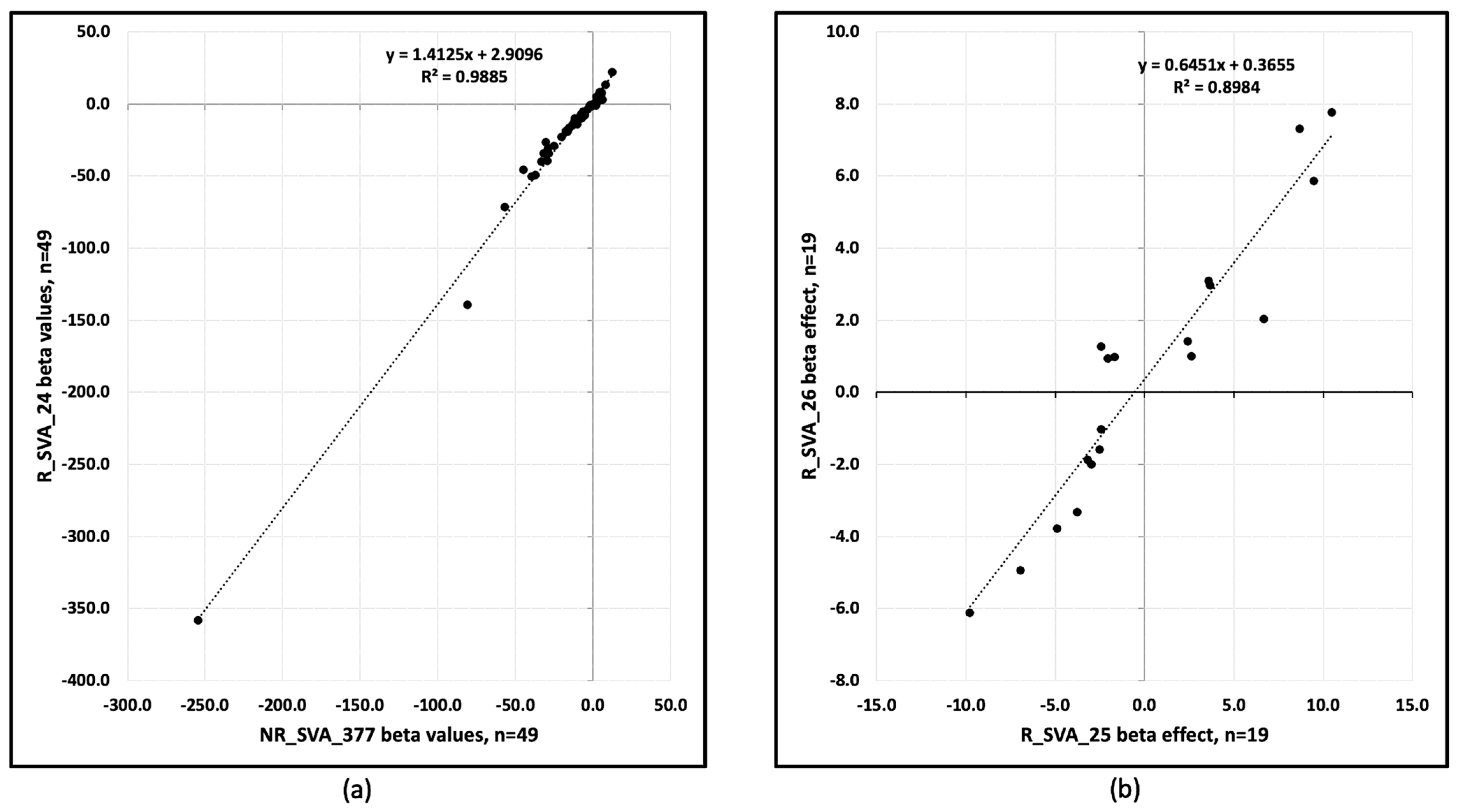
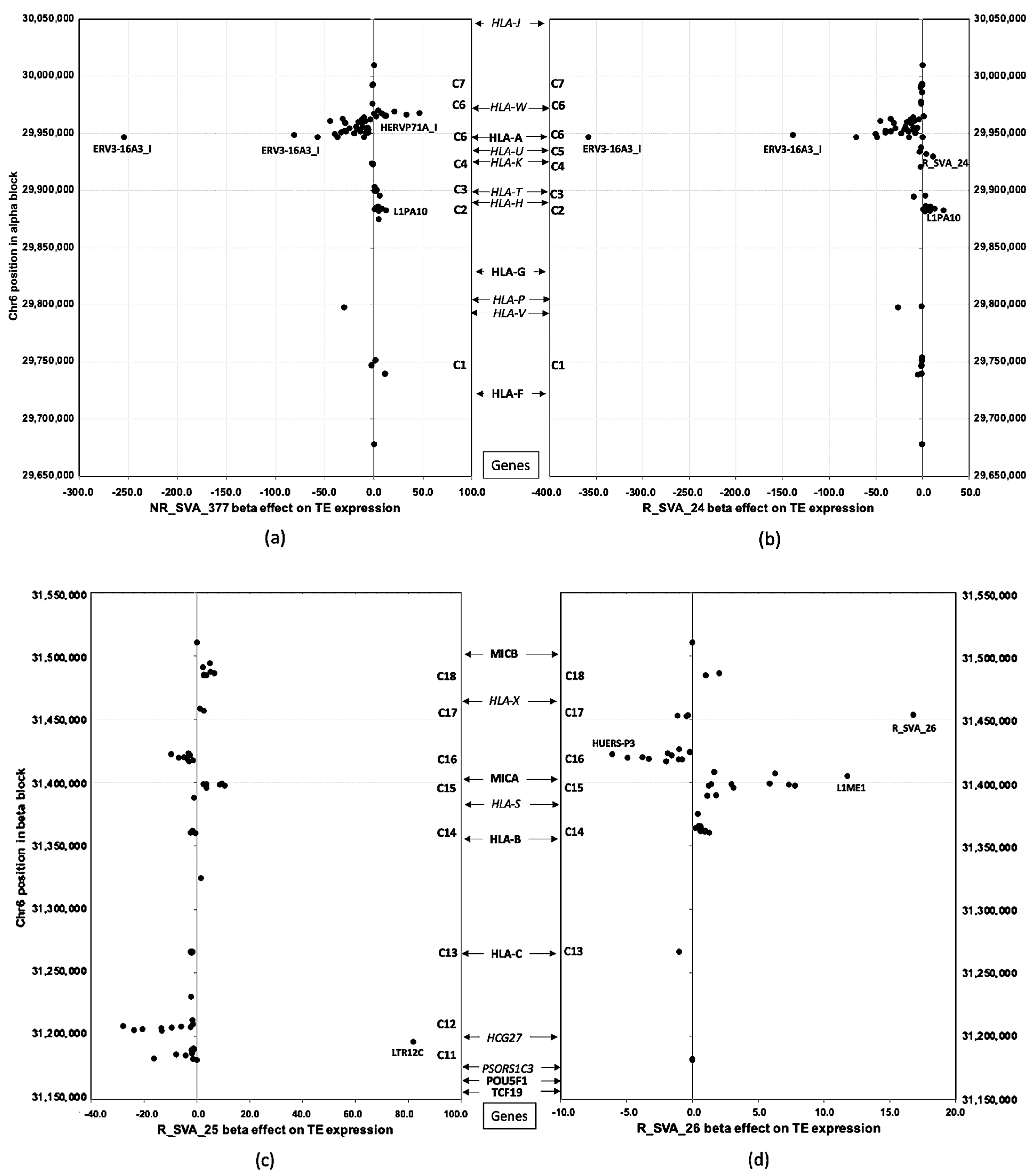
3.4. Beta Regulatory Effects of SVA on Clusters of TE Transcription Within Alpha and Beta Blocks
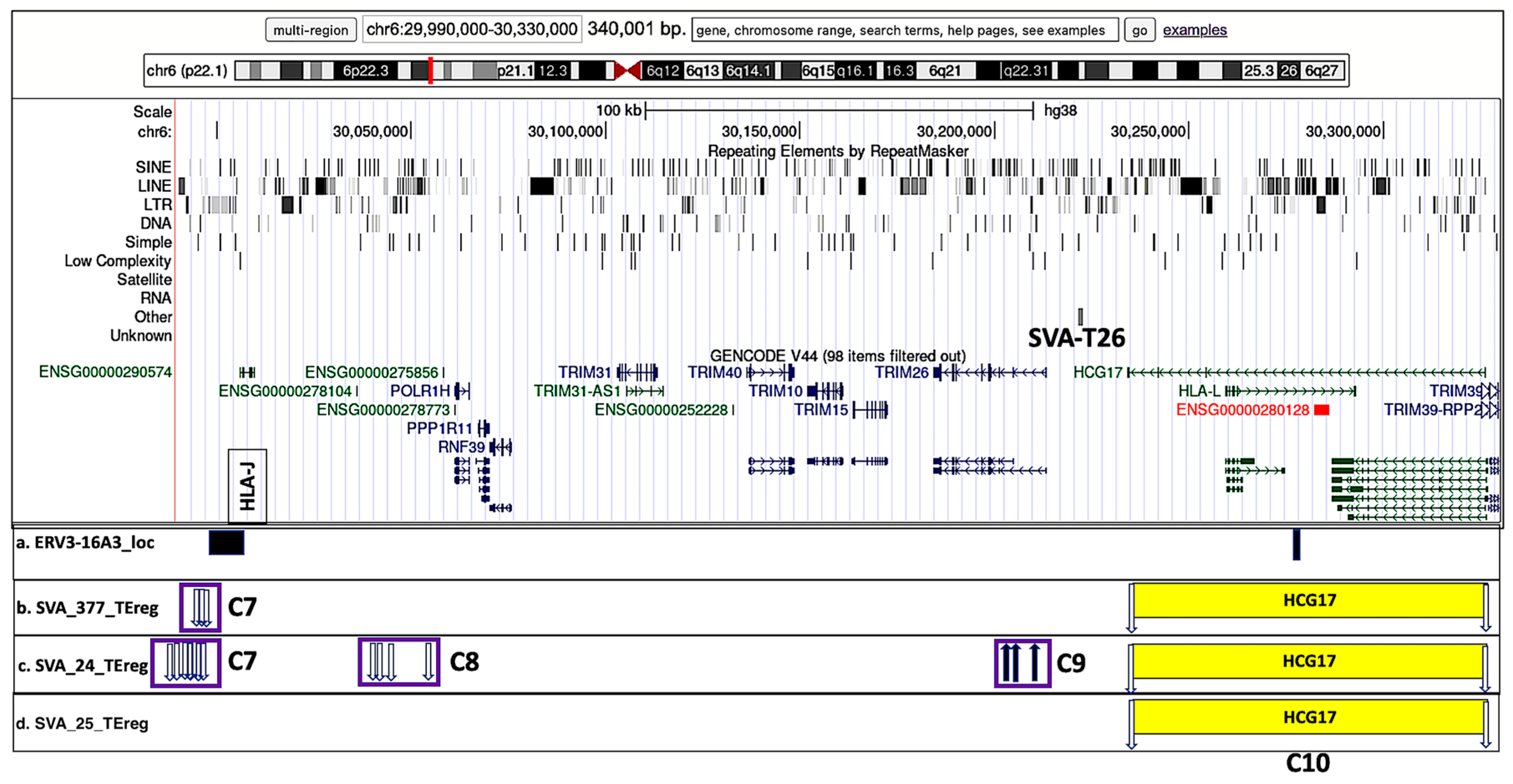
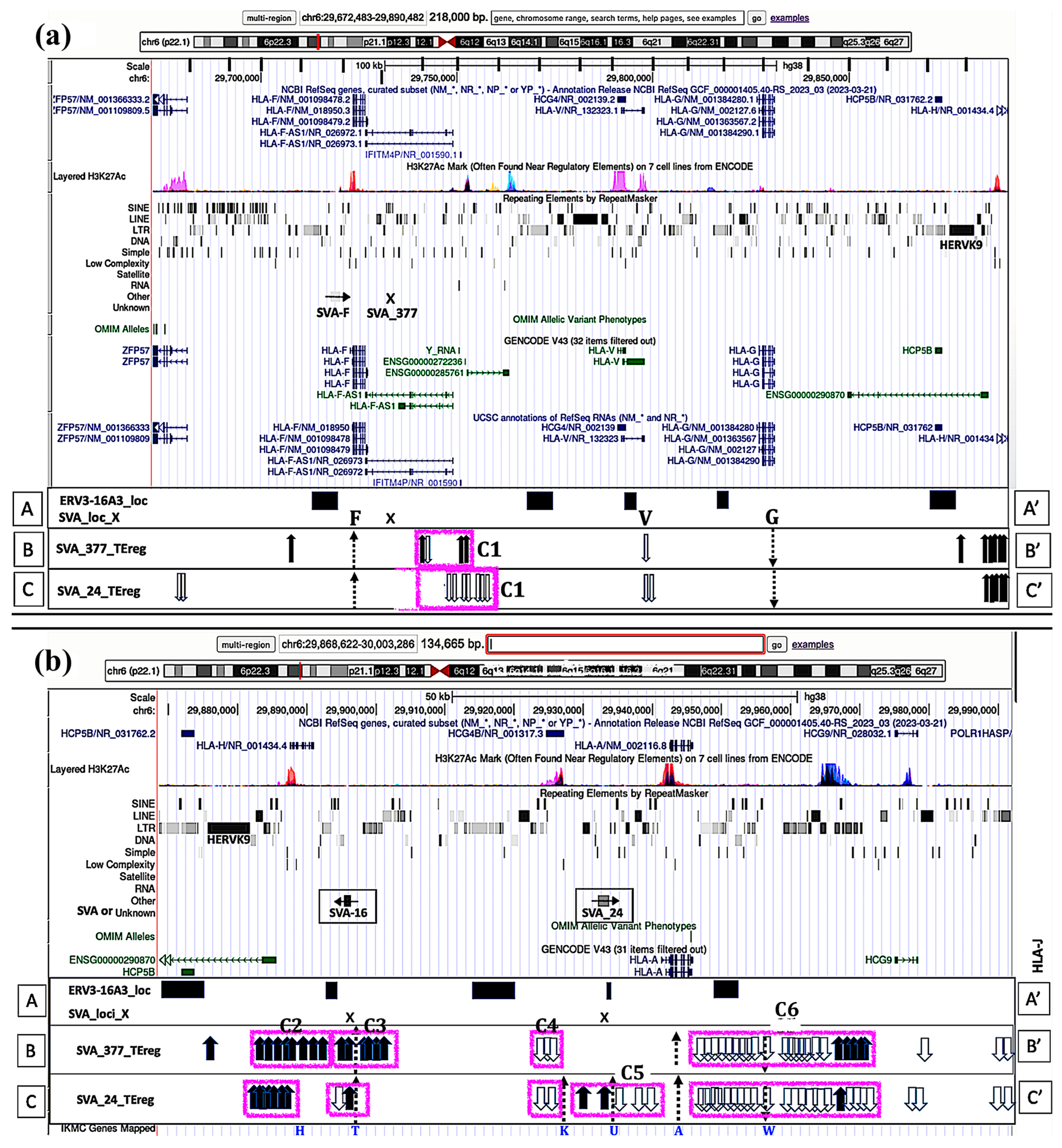
3.5. Visualization of Transcribed TE Clusters and Candidate Cis-Regulatory Elements (cCREs) Using the UCSC GRCh38 Genome Browser
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Koning, A.P.J.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive Elements May Comprise Over Two-Thirds of the Human Genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.D.; Zamudio-Hurtado, A.; Clawson, H.; Kent, W.J.; Haussler, D.; Salama, S.R.; Haeussler, M. The UCSC Repeat Browser Allows Discovery and Visualization of Evolutionary Conflict across Repeat Families. Mob. DNA 2020, 11, 13. [Google Scholar] [CrossRef]
- Piégu, B.; Bire, S.; Arensburger, P.; Bigot, Y. A Survey of Transposable Element Classification Systems—A Call for a Fundamental Update to Meet the Challenge of Their Diversity and Complexity. Mol. Phylogenetics Evol. 2015, 86, 90–109. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. A Universal Classification of Eukaryotic Transposable Elements Implemented in Repbase. Nat. Rev. Genet. 2008, 9, 411–412. [Google Scholar] [CrossRef]
- Anwar, S.; Wulaningsih, W.; Lehmann, U. Transposable Elements in Human Cancer: Causes and Consequences of Deregulation. Int. J. Mol. Sci. 2017, 18, 974. [Google Scholar] [CrossRef] [PubMed]
- Mills, R.E.; Bennett, E.A.; Iskow, R.C.; Devine, S.E. Which Transposable Elements Are Active in the Human Genome? Trends Genet. 2007, 23, 183–191. [Google Scholar] [CrossRef]
- Balachandran, P.; Walawalkar, I.A.; Flores, J.I.; Dayton, J.N.; Audano, P.A.; Beck, C.R. Transposable Element-Mediated Rearrangements Are Prevalent in Human Genomes. Nat. Commun. 2022, 13, 7115. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten Things You Should Know about Transposable Elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Bonté, P.-E.; Arribas, Y.A.; Merlotti, A.; Carrascal, M.; Zhang, J.V.; Zueva, E.; Binder, Z.A.; Alanio, C.; Goudot, C.; Amigorena, S. Single-Cell RNA-Seq-Based Proteogenomics Identifies Glioblastoma-Specific Transposable Elements Encoding HLA-I-Presented Peptides. Cell Rep. 2022, 39, 110916. [Google Scholar] [CrossRef] [PubMed]
- Larouche, J.-D.; Trofimov, A.; Hesnard, L.; Ehx, G.; Zhao, Q.; Vincent, K.; Durette, C.; Gendron, P.; Laverdure, J.-P.; Bonneil, É.; et al. Widespread and Tissue-Specific Expression of Endogenous Retroelements in Human Somatic Tissues. Genome Med. 2020, 12, 40. [Google Scholar] [CrossRef]
- Gao, Y.; Yu, X.; Chen, T. Human Endogenous Retroviruses in Cancer: Expression, Regulation and Function (Review). Oncol. Lett. 2021, 21, 121. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Bauer, J.; Hey, J.; Papageorgiou, D.N.; Stepanova, E.; Daskalakis, M.; Scheid, J.; Dubbelaar, M.; Klimovich, B.; Schwarz, D.; et al. DNMT and HDAC Inhibition Induces Immunogenic Neoantigens from Human Endogenous Retroviral Element-Derived Transcripts. Nat. Commun. 2023, 14, 6731. [Google Scholar] [CrossRef]
- Kulski, J.K. Next-Generation Sequencing—An Overview of the History, Tools, and “Omic” Applications. In Next Generation Sequencing—Advances, Applications and Challenges; Kulski, J.K., Ed.; IntechOpen: London, UK, 2016; pp. 3–60. ISBN 978-953-51-2240-1. [Google Scholar]
- Liang, G.; Bushman, F.D. The Human Virome: Assembly, Composition and Host Interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef]
- Teresi, S.J.; Teresi, M.B.; Edger, P.P. TE Density: A Tool to Investigate the Biology of Transposable Elements. Mob. DNA 2022, 13, 11. [Google Scholar] [CrossRef]
- Kulski, J.K.; Pfaff, A.L.; Koks, S. SVA Regulation of Transposable Element Clustered Transcription within the Major Histocompatibility Complex Genomic Class II Region of the Parkinson’s Progression Markers Initiative. Genes 2024, 15, 1185. [Google Scholar] [CrossRef]
- Shiina, T.; Inoko, H.; Kulski, J.K. An Update of the HLA Genomic Region, Locus Information and Disease Associations: 2004. Tissue Antigens 2004, 64, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Hosomichi, K.; Inoko, H.; Kulski, J.K. The HLA Genomic Loci Map: Expression, Interaction, Diversity and Disease. J. Hum. Genet. 2009, 54, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Kulski, J.K.; Suzuki, S.; Shiina, T. Human Leukocyte Antigen Super-Locus: Nexus of Genomic Supergenes, SNPs, Indels, Transcripts, and Haplotypes. Hum. Genome Var. 2022, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, K.; Koutsakos, M. The ABC of Major Histocompatibility Complexes and T Cell Receptors in Health and Disease. Viral Immunol. 2020, 33, 160–178. [Google Scholar] [CrossRef]
- Anaya, J.; Shoenfeld, Y.; Rojas-Villarraga, A.; Levy, R.; Cervera, R. (Eds.) Autoimmunity: From Bench to Bedside [Internet]; El Rosario University Press, 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459447/ (accessed on 1 January 2025).
- Gu, R.; Pan, J.; Awan, M.U.N.; Sun, X.; Yan, F.; Bai, L.; Bai, J. The Major Histocompatibility Complex Participates in Parkinson’s Disease. Pharmacol. Res. 2024, 203, 107168. [Google Scholar] [CrossRef]
- Kulski, J.K.; Pfaff, A.L.; Marney, L.D.; Fröhlich, A.; Bubb, V.J.; Quinn, J.P.; Koks, S. Regulation of Expression Quantitative Trait Loci by SVA Retrotransposons within the Major Histocompatibility Complex. Exp. Biol. Med. 2023, 248, 2304–2318. [Google Scholar] [CrossRef] [PubMed]
- Kulski, J.K.; Suzuki, S.; Shiina, T.; Pfaff, A.L.; Kõks, S. Regulatory SVA Retrotransposons and Classical HLA Genotyped-Transcripts Associated with Parkinson’s Disease. Front. Immunol. 2024, 15, 1349030. [Google Scholar] [CrossRef]
- Kulski, J.K.; Shigenari, A.; Inoko, H. Genetic Variation and Hitchhiking between Structurally Polymorphic Alu Insertions and HLA-A, -B, and -C Alleles and Other Retroelements within the MHC Class I Region. Tissue Antigens 2011, 78, 359–377. [Google Scholar] [CrossRef]
- Fröhlich, A.; Pfaff, A.L.; Middlehurst, B.; Hughes, L.S.; Bubb, V.J.; Quinn, J.P.; Koks, S. Deciphering the Role of a SINE-VNTR-Alu Retrotransposon Polymorphism as a Biomarker of Parkinson’s Disease Progression. Sci. Rep. 2024, 14, 10932. [Google Scholar] [CrossRef]
- Orenbuch, R.; Filip, I.; Comito, D.; Shaman, J.; Pe’er, I.; Rabadan, R. arcasHLA: High-Resolution HLA Typing from RNAseq. Bioinformatics 2020, 36, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Sepulveda, J.L. Using R and Bioconductor in Clinical Genomics and Transcriptomics. J. Mol. Diagn. 2020, 22, 3–20. [Google Scholar] [CrossRef]
- Pfaff, A.L.; Bubb, V.J.; Quinn, J.P.; Koks, S. Reference SVA Insertion Polymorphisms Are Associated with Parkinson’s Disease Progression and Differential Gene Expression. NPJ Park. Dis. 2021, 7, 44. [Google Scholar] [CrossRef]
- Kõks, S.; Pfaff, A.L.; Singleton, L.M.; Bubb, V.J.; Quinn, J.P. Non-Reference Genome Transposable Elements (TEs) Have a Significant Impact on the Progression of the Parkinson’s Disease. Exp. Biol. Med. 2022, 247, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon Provides Fast and Bias-Aware Quantification of Transcript Expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Shabalin, A.A. Matrix eQTL: Ultra Fast eQTL Analysis via Large Matrix Operations. Bioinformatics 2012, 28, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A Free Online Platform for Data Visualization and Graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef]
- Kulski, J.K.; Gaudieri, S.; Bellgard, M.; Balmer, L.; Giles, K.; Inoko, H.; Dawkins, R.L. The Evolution of MHC Diversity by Segmental Duplication and Transposition of Retroelements. J. Mol. Evol. 1997, 45, 599–609. [Google Scholar] [CrossRef]
- Kulski, J.K.; Gaudieri, S.; Dawkins, R.L. Using Alu J Elements as Molecular Clocks to Trace the Evolutionary Relationships Between Duplicated HLA Class I Genomic Segments. J. Mol. Evol. 2000, 50, 510–519. [Google Scholar] [CrossRef]
- Kulski, J.K.; Gaudieri, S.; Martin, A.; Dawkins, R.L. Coevolution of PERB11 (MIC) and HLA Class I Genes with HERV-16 and Retroelements by Extended Genomic Duplication. J. Mol. Evol. 1999, 49, 84–97. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Gea-Mallorquí, E.; Colin-York, H.; Fritzsche, M.; Gillespie, G.M.; Brackenridge, S.; Borrow, P.; McMichael, A.J. Intracellular Trafficking of HLA-E and Its Regulation. J. Exp. Med. 2023, 220, e20221941. [Google Scholar] [CrossRef] [PubMed]
- Kulski, J.K.; Shigenari, A.; Inoko, H. Polymorphic SVA Retrotransposons at Four Loci and Their Association with Classical HLA Class I Alleles in Japanese, Caucasians and African Americans. Immunogenetics 2010, 62, 211–230. [Google Scholar] [CrossRef]
- Thompson, P.J.; Macfarlan, T.S.; Lorincz, M.C. Long Terminal Repeats: From Parasitic Elements to Building Blocks of the Transcriptional Regulatory Repertoire. Mol. Cell 2016, 62, 766–776. [Google Scholar] [CrossRef]
- Gebrie, A. Transposable Elements as Essential Elements in the Control of Gene Expression. Mob. DNA 2023, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-O.; Gingeras, T.R.; Weng, Z. Genome-Wide Analysis of Polymerase III–Transcribed Alu Elements Suggests Cell-Type–Specific Enhancer Function. Genome Res. 2019, 29, 1402–1414. [Google Scholar] [CrossRef]
- Luqman-Fatah, A.; Miyoshi, T. Human LINE-1 Retrotransposons: Impacts on the Genome and Regulation by Host Factors. Genes Genet. Syst. 2023, 98, 121–154. [Google Scholar] [CrossRef]
- Zhao, K.; Du, J.; Peng, Y.; Li, P.; Wang, S.; Wang, Y.; Hou, J.; Kang, J.; Zheng, W.; Hua, S.; et al. LINE1 Contributes to Autoimmunity through Both RIG-I- and MDA5-Mediated RNA Sensing Pathways. J. Autoimmun. 2018, 90, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Boyboy, B.A.G.; Ichiyanagi, K. Insertion of Short L1 Sequences Generates Inter-Strain Histone Acetylation Differences in the Mouse. Mob. DNA 2024, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vicente-García, C.; Seruggia, D.; Moltó, E.; Fernandez-Miñán, A.; Neto, A.; Lee, E.; Gómez-Skarmeta, J.L.; Montoliu, L.; Lunyak, V.V.; et al. MIR Retrotransposon Sequences Provide Insulators to the Human Genome. Proc. Natl. Acad. Sci. USA 2015, 112, E4428–E4437. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.J.; Zovoilis, A.; Cifuentes-Rojas, C.; Han, L.; Bujisic, B.; Lee, J.T. B2 and ALU Retrotransposons Are Self-Cleaving Ribozymes Whose Activity Is Enhanced by EZH2. Proc. Natl. Acad. Sci. USA 2020, 117, 415–425. [Google Scholar] [CrossRef]
- Mandal, A.K.; Pandey, R.; Jha, V.; Mukerji, M. Transcriptome-Wide Expansion of Non-Coding Regulatory Switches: Evidence from Co-Occurrence of Alu Exonization, Antisense and Editing. Nucleic Acids Res. 2013, 41, 2121–2137. [Google Scholar] [CrossRef]
- Daniel, C.; Behm, M.; Öhman, M. The Role of Alu Elements in the Cis-Regulation of RNA Processing. Cell. Mol. Life Sci. 2015, 72, 4063–4076. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Bartholomew, B. Emerging Roles of Transcriptional Enhancers in Chromatin Looping and Promoter-Proximal Pausing of RNA Polymerase II. J. Biol. Chem. 2018, 293, 13786–13794. [Google Scholar] [CrossRef]
- Cheng, Y.; Saville, L.; Gollen, B.; Veronesi, A.A.; Mohajerani, M.; Joseph, J.T.; Zovoilis, A. Increased Alu RNA Processing in Alzheimer Brains Is Linked to Gene Expression Changes. EMBO Rep. 2021, 22, e52255. [Google Scholar] [CrossRef]
- Bai, G.-H.; Lin, S.-C.; Hsu, Y.-H.; Chen, S.-Y. The Human Virome: Viral Metagenomics, Relations with Human Diseases, and Therapeutic Applications. Viruses 2022, 14, 278. [Google Scholar] [CrossRef]
- Li, Y.; Fan, T.; Cui, J. Human Endogenous Retroviruses in Viral Disease and Therapy. Clin. Transl. Dis. 2022, 2, e38. [Google Scholar] [CrossRef]
- Suzuki, S.; Morishima, S.; Murata, M.; Tanaka, M.; Shigenari, A.; Ito, S.; Kanga, U.; Kulski, J.K.; Morishima, Y.; Shiina, T. Sequence Variations Within HLA-G and HLA-F Genomic Segments at the Human Leukocyte Antigen Telomeric End Associated With Acute Graft-Versus-Host Disease in Unrelated Bone Marrow Transplantation. Front. Immunol. 2022, 13, 938206. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sun, H.; Ma, X.; Zeng, Y.; Pan, Y.; Yu, D.; Liu, Z.; Xiang, Y. HLA-F-AS1/miR-330-3p/PFN1 Axis Promotes Colorectal Cancer Progression. Life Sci. 2020, 254, 117180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, S.; Zhang, X.; Li, C.; Zhang, J.; Zhou, W. LncRNA HLA-F-AS1 Promotes Colorectal Cancer Metastasis by Inducing PFN1 in Colorectal Cancer-Derived Extracellular Vesicles and Mediating Macrophage Polarization. Cancer Gene Ther. 2021, 28, 1269–1284. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Han, Y.; Shi, R.; Shao, X.; Sun, L.; Zhang, Y.; Huang, D.; Chen, Z.; Zhang, G.; Liang, J.; et al. ZNRD1 Gene Suppresses Cell Proliferation through Cell Cycle Arrest in G1 Phase. Cancer Biol. Ther. 2005, 4, 67–71. [Google Scholar] [CrossRef]
- Ballana, E.; Senserrich, J.; Pauls, E.; Faner, R.; Mercader, J.M.; Uyttebroeck, F.; Palou, E.; Mena, M.P.; Grau, E.; Clotet, B.; et al. ZNRD1 (Zinc Ribbon Domain–Containing 1) Is a Host Cellular Factor That Influences HIV-1 Replication and Disease Progression. Clin. Infect. Dis. 2010, 50, 1022–1032. [Google Scholar] [CrossRef]
- Zhao, G.; Li, Y.; Chen, T.; Liu, F.; Zheng, Y.; Liu, B.; Zhao, W.; Qi, X.; Sun, W.; Gao, C. TRIM26 Alleviates Fatal Immunopathology by Regulating Inflammatory Neutrophil Infiltration during Candida Infection. PLoS Pathog. 2024, 20, e1011902. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Wei, L.; Yu, Z.; Yao, Z.; Cheng, J.; Wang, Y.; Song, X.; Li, M. The Roles of TRIMs in Antiviral Innate Immune Signaling. Front. Cell. Infect. Microbiol. 2021, 11, 628275. [Google Scholar] [CrossRef]
- Zhang, M.; Tan, H.; Gong, Y.; Faleti, O.D.; Li, D.; Yang, J.; Huang, J.; Long, J.; Luo, Q.; Wu, G.; et al. TRIM26 Restricts Epstein–Barr Virus Infection in Nasopharyngeal Epithelial Cells through K48 -linked Ubiquitination of HSP-90β. FASEB J. 2024, 38, e23345. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-C. Comprehensive Analysis of Clinical and Biological Value of ING Family Genes in Liver Cancer. World J. Gastrointest. Oncol. 2024, 16, 2592–2609. [Google Scholar] [CrossRef] [PubMed]
- Salviano-Silva, A.; Becker, M.; Augusto, D.G.; Busch, H.; Adelman Cipolla, G.; Farias, T.D.-J.; Bumiller-Bini, V.; Calonga-Solís, V.; Munz, M.; Franke, A.; et al. Genetic Association and Differential Expression of HLA Complex Group lncRNAs in Pemphigus. J. Autoimmun. 2021, 123, 102705. [Google Scholar] [CrossRef]
- Mirzadeh Azad, F.; Malakootian, M.; Mowla, S.J. lncRNA PSORS1C3 Is Regulated by Glucocorticoids and Fine-Tunes OCT4 Expression in Non-Pluripotent Cells. Sci. Rep. 2019, 9, 8370. [Google Scholar] [CrossRef] [PubMed]
- Linh, N.T.T.; Giang, N.H.; Lien, N.T.K.; Trang, B.K.; Trang, D.T.; Ngoc, N.T.; Nghia, V.X.; My, L.T.; Mao, C.V.; Hoang, N.H.; et al. Association of PSORS1C3, CARD14 and TLR4 Genotypes and Haplotypes with Psoriasis Susceptibility. Genet. Mol. Biol. 2022, 45, 20220099. [Google Scholar] [CrossRef] [PubMed]
- Krönung, S.K.; Beyer, U.; Chiaramonte, M.L.; Dolfini, D.; Mantovani, R.; Dobbelstein, M. LTR12 Promoter Activation in a Broad Range of Human Tumor Cells by HDAC Inhibition. Oncotarget 2016, 7, 33484–33497. [Google Scholar] [CrossRef]
- Goerner-Potvin, P.; Bourque, G. Computational Tools to Unmask Transposable Elements. Nat. Rev. Genet. 2018, 19, 688–704. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Hammell, M. Analysis of RNA-Seq Data Using TEtranscripts. In Transcriptome Data Analysis; Wang, Y., Sun, M., Eds.; Methods in Molecular Biology; Springer New York: New York, NY, USA, 2018; Volume 1751, pp. 153–167. ISBN 978-1-4939-7709-3. [Google Scholar]
- Fröhlich, A.; Hughes, L.S.; Middlehurst, B.; Pfaff, A.L.; Bubb, V.J.; Koks, S.; Quinn, J.P. CRISPR deletion of a SINE-VNTR-Alu (SVA_67) retrotransposon demonstrates its ability to differentially modulate gene expression at the MAPT locus. Front. Neurol. 2023, 14, 1273036. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HLA Gene | Number | % Total | Mean RNA | STDEV * | Max Read | Min Read | Block Loci |
|---|---|---|---|---|---|---|---|
| Samples | Samples | Read Count | Count | ||||
| F | 1528 | 99.9 | 4723 | 1862 | 14,258 | 411 | Alpha |
| V | 1393 | 91 | 6 | 7 | 54 | 1 | Alpha |
| P | 729 | 47.6 | 6 | 8 | 45 | 1 | Alpha |
| G | 1243 | 81.2 | 6 | 10 | 73 | 1 | Alpha |
| H | 1507 | 98.5 | 91 | 50 | 358 | 1 | Alpha |
| T | 1453 | 95 | 11 | 7 | 51 | 1 | Alpha |
| K | 1507 | 98.5 | 111 | 65 | 560 | 1 | Alpha |
| U | 812 | 53.1 | 2 | 1 | 8 | 1 | Alpha |
| A | 1530 | 100 | 69,384 | 19,326 | 137,653 | 2694 | Alpha |
| W | 1530 | 100 | 221 | 127 | 1274 | 10 | Alpha |
| Y | 502 | 32.8 | 350 | 407 | 2099 | 1 | Alpha |
| J | 1529 | 99.9 | 32 | 17 | 127 | 1 | Alpha |
| L | 1530 | 100 | 50 | 24 | 199 | 2 | Kappa |
| N | 75 | 4.9 | 1 | 1 | 4 | 1 | Kappa |
| E | 1528 | 99.9 | 31,339 | 12,457 | 97,700 | 1890 | Kappa |
| C | 1528 | 99.9 | 43,905 | 20,626 | 146,659 | 5125 | Beta |
| B | 1528 | 99.9 | 50,509 | 21,624 | 151,472 | 3204 | Beta |
| S | 489 | 32 | 2 | 2 | 25 | 1 | Beta |
| Regulatory | Cluster | Number | % | % | Total No. Exp | No. TEs in | Cluster Genome Location | Cluster |
|---|---|---|---|---|---|---|---|---|
| SVA | Expressed | Expressed | Expressed | and % of | Genome | GRCh38 | Width (bp) | |
| Pos and Neg | Pos | Neg | Genome | Reference | ||||
| Strands | Strands | Strands | Reference No. | GRCh38 | ||||
| NR_SVA_377 | C1 | 3 and 1 | 75% | 25% | 4 (40%) | 10 | chr6:29,739,704–29,751,757 | 12,053 |
| chr6:29731783 | C2 | 8 and 0 | 100% | 0% | 8 (80%) | 10 | chr6:29,882,170–29,886,079 | 3909 |
| C3 | 0 and 4 | 0% | 100% | 4 (50%) | 8 | chr6:29,895,386–29,900,951 | 5565 | |
| C4 | 2 and 1 | 67% | 23% | 3 (75%) | 4 | chr6:29,922,776–29,924,284 | 1508 | |
| C6A | 26 and 6 | 81% | 19% | 32 (97%) | 33 | chr6:29,946,554–29,964,917 | 18,363 | |
| C6B | 9 and 1 | 90% | 10% | 10 (100%) | 10 | chr6:29,965,033–29,970,366 | 5333 | |
| R_SVA_24 | C1 | 5 and 2 | 71% | 29% | 7 (54%) | 13 | chr6:29,739,009–29,753,863 | 14,854 |
| chr6:29932088– | C2 | 9 and 0 | 100% | 0% | 9 (82%) | 11 | chr6:29,881,965–29,886,079 | 4114 |
| 29933753 | C3 | 1 and 1 | 50% | 50% | 2 (100%) | 2 | chr6:29,894,876–29,896,449 | 1573 |
| C5 | 3 and 1 | 75% | 25% | 4 (29%) | 14 | chr6:29,929,971–29,938,504 | 8533 | |
| C6 | 28 and 7 | 80% | 20% | 35 (70%) | 50 | chr6:29,946,554–29,976,235 | 29,681 | |
| C7 | 2 and 5 | 29% | 71% | 7 (44%) | 16 | chr6:29,986,094–29,993,212 | 7118 | |
| C8 | 4 and 0 | 100% | 0% | 4 (13%) | 31 | chr6:30,041,551–30,053,768 | 12,217 | |
| C9 | 3 and 0 | 100% | 0% | 3 (10%) | 29 | chr6:30,202,312–30,211,669 | 9357 | |
| R_SVA_25 | C11 | 2 and 9 | 29% | 71% | 11 (33%) | 33 | chr6:31,176,234–31,190,529 | 14,295 |
| chr6:31243861– | C12 | 10 and 1 | 91% | 9% | 11 (42%) | 26 | chr6:31,204,049–31,212,882 | 8833 |
| 31245322 | C13 | 3 and 0 | 100% | 0% | 3 (100%) | 3 | chr6:31,265,965–31,266,852 | 887 |
| C14 | 3 and 1 | 75% | 25% | 4 (80%) | 5 | chr6:31,360,364–31,362,611 | 2247 | |
| C15 | 4 and 3 | 57% | 43% | 7 (100%) | 7 | chr6:31,396,358–31,399,559 | 3201 | |
| C16 | 0 and 8 | 0% | 100% | 8 (47%) | 17 | chr6:31,417,163–31,424,086 | 6923 | |
| C17 | 0 and 2 | 0% | 100% | 2 (50%) | 4 | chr6:31,456,841–31,458,617 | 1776 | |
| C18 | 7 and 0 | 100% | 0% | 7 (44%) | 16 | chr6:31,484,911–31,494,850 | 9939 | |
| R_SVA_26 | C14 | 6 and 5 | 55% | 45% | 11 (44%) | 25 | chr6:31,360,458–31,375,713 | 15,255 |
| chr6:31453746– | C15 | 6 and 8 | 43% | 57% | 14 (52%) | 27 | chr6:31,389,735–31,408,725 | 18,990 |
| 31456553 | C16 | 0 and 12 | 0% | 100% | 12 (50%) | 24 | chr6:31,417,163–31,426,904 | 9741 |
| C17 | 0 and 4 | 0% | 100% | 4 (80%) | 5 | chr6:31,452,575–31,456,553 | 3978 | |
| C18 | 2 and 0 | 100% | 0% | 2 (50%) | 4 | chr6:31,484,911–31,486,837 | 1926 | |
| total | 146 and 82 | 64% | 36% | 228 (52%) | 437 |
| Adjoining | Distance | Av Distance | Distance | Av Distance | Separated Gene |
|---|---|---|---|---|---|
| Clusters | Between | Between TEs | Between | Between TEs | Regions |
| Clusters, bp | in Clusters, bp | Clusters, bp | in Clusters, bp | ||
| Alpha Block | NR_SVA_377 | R_SVA_24 | Gene1—Gene2 | ||
| C1-C2 | 130,413 | 3013–489 | 128,102 | 2122–457 | HLA-F—HLA-H |
| C2-C3 | 9307 | 489–1391 | 8797 | 457–786 | HLA-H—HLA-T |
| C3-C4 | 21,825 | 1391–503 | HLA-T—HLA-K | ||
| C3-C5 | 33,522 | 786–2133 | HLA-T—HLA-U | ||
| C4-C6 | 22,270 | 503–574 | HLA-K—HLA-A | ||
| C5-C6 | 8050 | 2133–506 | HLA-U—HLA-A | ||
| C6-C7 | 9859 | 506–1017 | HLA-A—HLA-J | ||
| C7-C8 | 48,339 | 1017–3054 | HLA-J | ||
| C8-C9 | 148,544 | 3054–3119 | TRIM26 | ||
| Beta Block | R_SVA_25 | R_SVA_26 | Gene1—Gene2 | ||
| C11-C12 | 9636 | 1300–1156 | PSORS1C3—HCG27 | ||
| C12-C13 | 53,083 | 1156–296 | HCG27—HLA-C | ||
| C13-C14 | 93,512 | 296–562 | 93,722 | 147–1387 | HLA-C—HLA-B |
| C14-C15 | 31,678 | 562–715 | 14,022 | 1387–1365 | HLA-B—MICA-AS1 |
| C15-C16 | 17,604 | 753–865 | 8438 | 1357–812 | MICA-AS1—MICA |
| C16-C17 | 32,755 | 865–888 | 25,671 | 812–995 | MICA—HLA-X |
| C17-C18 | 21,356 | 888–1242 | 28,358 | 995–963 | HLA-X—MIC-DT |
| Class or Family (%) | Name | No. | Length | No. | % on (+) |
|---|---|---|---|---|---|
| TEs | * Av bp | Clusters | Strand | ||
| DNA (11.8%) | Class | 20 | 224 | 11 | 85 |
| hAT-Charlie (95%) | 19 | 232 | 10 | ||
| Charlie | 8 | 380 | 4 | ||
| MER20, MER102 | 2 | 155 | 2 | ||
| MER30 | 2 | 46 | 1 | ||
| MER5 | 7 | 139 | 5 | ||
| TcMar-Tigger (5%) | Tigger17 | 1 | 66 | 1 | |
| LTR/ERV (34.9%) | Class | 59 | 548 | 12 | 63 |
| ERV1 (44%) | 26 | 665 | 5 | ||
| HERP71A | 7 | 735 | 1 | ||
| LTR43 | 7 | 542 | 1 | ||
| HARLEQUIN | 4 | 1100 | 1 | ||
| LTR2, LTR8, LTR9 | 3 | 602 | 2 | ||
| LTR12, LTR71 | 2 | 950 | 1 | ||
| MER41, 1 MER61 | 2 | 373 | 2 | ||
| HUERS-P3 | 1 | 792 | 1 | ||
| ERVK (3%) | 2 | 713 | 2 | ||
| LTR13. LTR22 | 2 | 713 | 2 | ||
| ERVL (22%) | 13 | 546 | 4 | ||
| ERV3-16A3 | 4 | 807 | 2 | ||
| MER21 | 5 | 537 | 2 | ||
| LTR84 | 2 | 189 | 2 | ||
| LTR16, LTR18 | 2 | 240 | 1 | ||
| ERVL-MaLR (31%) | 18 | 263 | 10 | ||
| MLT | 14 | 355 | 6 | ||
| MST | 4 | 475 | 1 | ||
| LINE (32.5%) | Class | 55 | 467 | 13 | 53 |
| L1 (80%) | 45 | 545 | 13 | ||
| L2 (16%) | 9 | 179 | 4 | ||
| L3 (4%) | 2 | 103 | 1 | ||
| SINE (18.9%) | Class | 32 | 234 | 12 | 53 |
| Alu (84%) | 27 | 251 | 11 | ||
| MIR (16%) | 5 | 178 | 3 | ||
| SVA (1.8) | 3 | 1721 | 3 | 33 | |
| * Av is average | av n, 169 | 445 |
| (a) Alpha Block (302,579 bp) 431 TE loci | |||
| TE Family (Class) | Alpha Block | TE Expressed | TE Expressed |
| Number (%) | by SVA_377 | by SVA_24 | |
| hAT-Charlie (DNA) | 49 (11%) | 16% | 20% |
| TcMar-Tigger (DNA) | 6 (1%) | 17% | |
| ERVL-MaLR (LTR) | 71 (17%) | 21% | 21% |
| ERV (LTR) | 84 (20%) | 24% | 15% |
| L1 (LINE) | 87 (20%) | 7% | 13% |
| L2 (LINE) | 39 (9%) | 10% | 18% |
| L3-CR1 (LINE) | 4 (1%) | 50% | 25% |
| Alu (SINE) | 70 (16%) | 13% | 11% |
| MIR (SINE) | 16 (4%) | 13% | 19% |
| SVA (retroposon) | 3 (<1%) | 33% | 100% |
| Total | 431 (100%) | 67 (15.5%) | 72 (16.7%) |
| (b) Beta Block (354,000 bp) 568 TE loci | |||
| TE Family (Class) | Beta Block | TE Expressed | TE Expressed |
| Number (%) | by SVA_25 | by SVA_26 | |
| hAT-Charlie (DNA) | 22 (4%) | 18% | 18% |
| MULE-MuDR (DNA) | 1 (<1%) | ||
| ERVL-MaLR (LTR) | 31 (5%) | 3% | |
| ERV (LTR) | 142 (25%) | 12% | 4% |
| L1 (LINE) | 157 (28%) | 11% | 12% |
| L2 (LINE) | 25 (4%) | 12% | 8% |
| L3-CR1 (LINE) | 1 (<1%) | ||
| Alu (SINE) | 161 (28%) | 8% | 5% |
| MIR (SINE) | 15 (3%) | 7% | |
| SSU-rRNA_Hsa (rRNA) | 1 (<1%) | ||
| Total | 568 (100%) | 55 (10%) | 40 (7%) |
| Alpha Block (302,579 bp) | ||||||||
| TE Family (Class) | NR_SVA_377: TE Number and % Regulated | NR_SVA_24: TE Number and % Regulated | ||||||
| Up | Down | Total/TE | % of 67 | Up | Down | Total/TE | % of 72 | |
| hAT-Charlie (DNA) | 4 (50%) | 4 (50%) | 8 | 12% | 2 (20%) | 8 (80%) | 10 | 14% |
| TcMar-Tigger (DNA) | 1 (100%) | 1 | 1% | |||||
| ERVL-MaLR (LTR) | 4 (27%) | 11 (73%) | 15 | 22% | 4 (36%) | 11 (64%) | 15 | 21% |
| ERV (LTR) | 9 (45%) | 11 (55%) | 20 | 30% | 2 (15%) | 11 (85%) | 13 | 18% |
| L1 (LINE) | 3 (50%) | 3 (50%) | 6 | 9% | 1 (9%) | 10 (91%) | 11 | 15% |
| L2 (LINE) | 4 (100%) | 4 | 6% | 7 (100%) | 7 | 10% | ||
| L3-CR1 (LINE) | 1 (50%) | 1 (50%) | 2 | 3% | 1 (100%) | 1 | 1% | |
| Alu (SINE) | 3 (33%) | 6 (67%) | 9 | 13% | 1 (13%) | 7 (87%) | 8 | 11% |
| MIR (SINE) | 2 (100%) | 2 | 3% | 3 (100%) | 3 | 4% | ||
| SVA (retroposon) | 1 (100%) | 1 | 2% | 3 (100%) | 3 | 4% | ||
| Total | 25 (37%) | 42 (63%) | 67 (100%) | 100% | 14 (19%) | 58 (81%) | 72 (100%) | 100% |
| Beta Block (354.000 bp) | ||||||||
| TE Family (Class) | R_SVA_25: TE Number and % Regulated | R_SVA_26: TE Number and % Regulated | ||||||
| Up | Down | Total/TE | % of 55 | Up | Down | Total/TE | % of 40 | |
| hAT-Charlie (DNA) | 1 (25%) | 3 (75%) | 4 | 7% | 2 (50%) | 2 (50%) | 4 | 10% |
| ERVL-MaLR (LTR) | 1 (100%) | 1 | 2% | |||||
| ERV (LTR) | 3 (18%) | 14 (82%) | 17 | 31% | 3 (50%) | 3 (50%) | 6 | 15% |
| L1 (LINE) | 7 (39%) | 10 (61% | 17 | 31% | 10 (53%) | 9 (47%) | 19 | 48% |
| L2 (LINE) | 2 (67%) | 1 (33%) | 3 | 5% | 1 (50%) | 1 (50%) | 2 | 5% |
| L3-CR1 (LINE) | ||||||||
| Alu (SINE) | 5 (42%) | 7 (68%) | 12 | 22% | 7 (88%) | 1 (50%) | 8 | 20% |
| MIR (SINE) | 1 (100%) | 1 | 2% | |||||
| SVA (retroposon) | 1 (100%) | 1 | 2% | |||||
| Total | 18 (33%) | 37 (67%) | 55 (100%) | 100% | 24 (60%) | 16 (40%) | 40 (100%) | 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulski, J.K.; Pfaff, A.L.; Koks, S. The Transcription of Transposable Elements Differentially Regulated by SVAs in the Major Histocompatibility Complex Class I Region of a Parkinson’s Progression Markers Initiative Cohort. J. Mol. Pathol. 2025, 6, 1. https://doi.org/10.3390/jmp6010001
Kulski JK, Pfaff AL, Koks S. The Transcription of Transposable Elements Differentially Regulated by SVAs in the Major Histocompatibility Complex Class I Region of a Parkinson’s Progression Markers Initiative Cohort. Journal of Molecular Pathology. 2025; 6(1):1. https://doi.org/10.3390/jmp6010001
Chicago/Turabian StyleKulski, Jerzy K., Abigail L. Pfaff, and Sulev Koks. 2025. "The Transcription of Transposable Elements Differentially Regulated by SVAs in the Major Histocompatibility Complex Class I Region of a Parkinson’s Progression Markers Initiative Cohort" Journal of Molecular Pathology 6, no. 1: 1. https://doi.org/10.3390/jmp6010001
APA StyleKulski, J. K., Pfaff, A. L., & Koks, S. (2025). The Transcription of Transposable Elements Differentially Regulated by SVAs in the Major Histocompatibility Complex Class I Region of a Parkinson’s Progression Markers Initiative Cohort. Journal of Molecular Pathology, 6(1), 1. https://doi.org/10.3390/jmp6010001