
An Independent Assessment of a Commercial Clinical Interpretation Software Indicates That Software Can Mitigate Variation in Human Assessment

Abstract
:1. Introduction
2. Materials and Methods
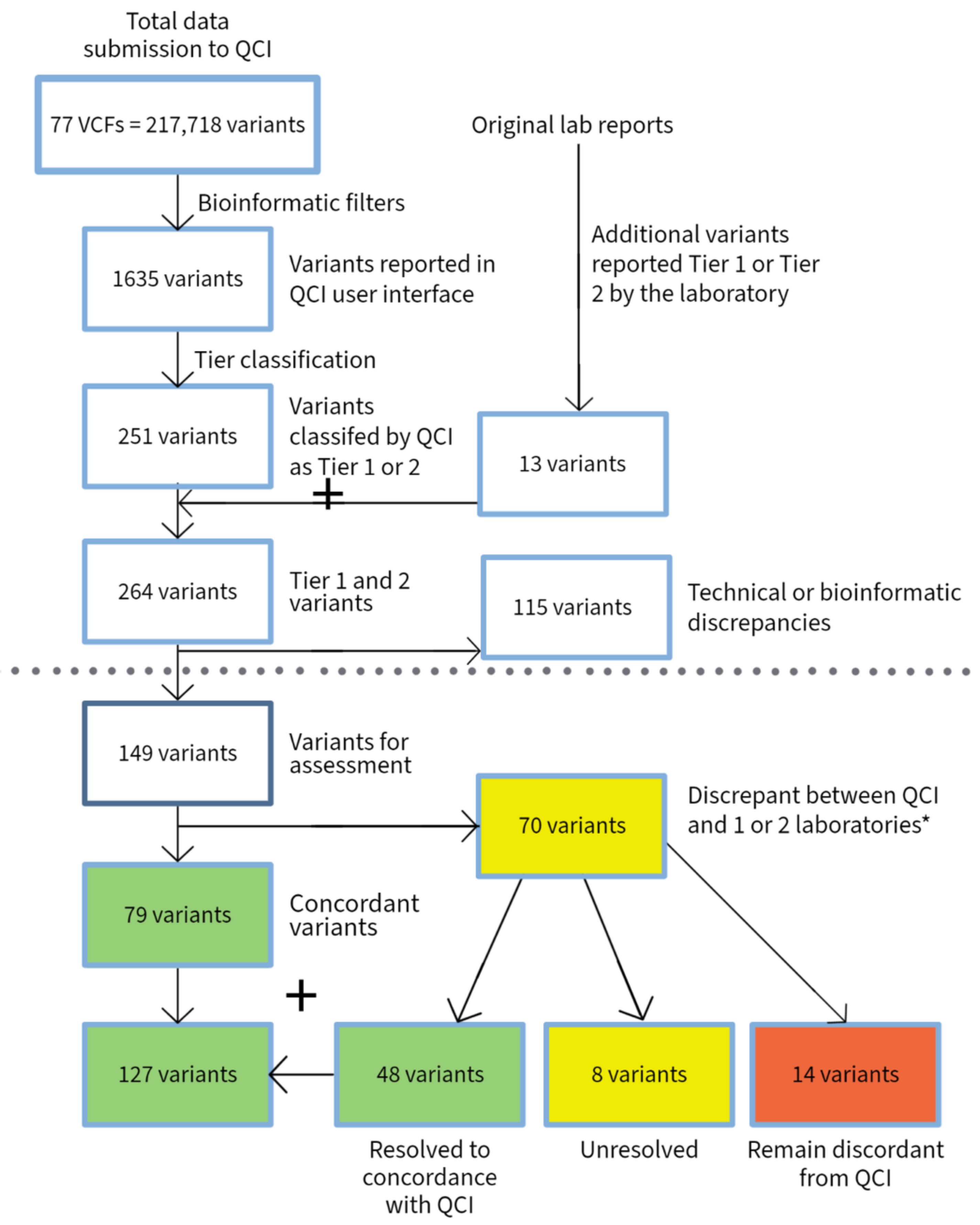
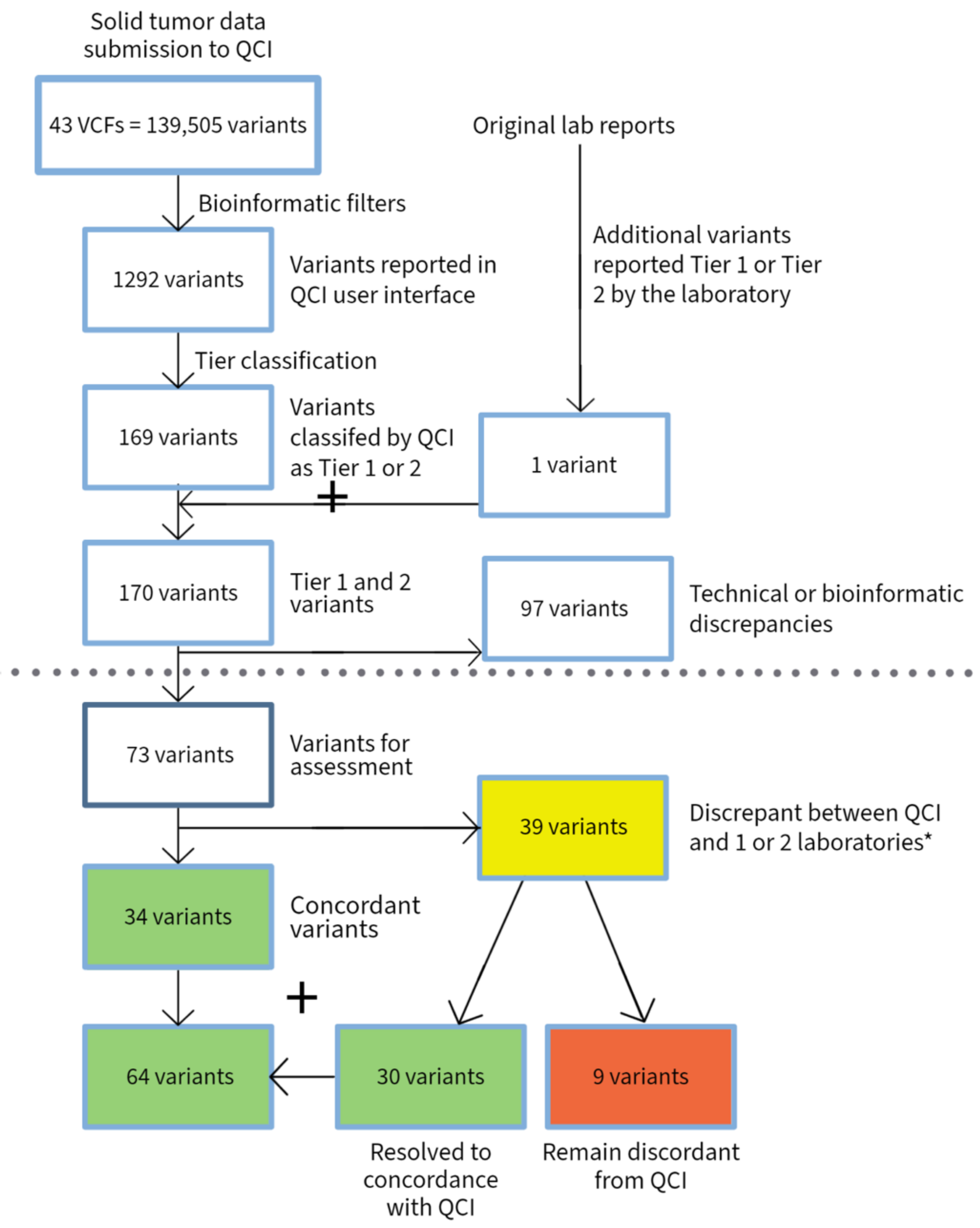
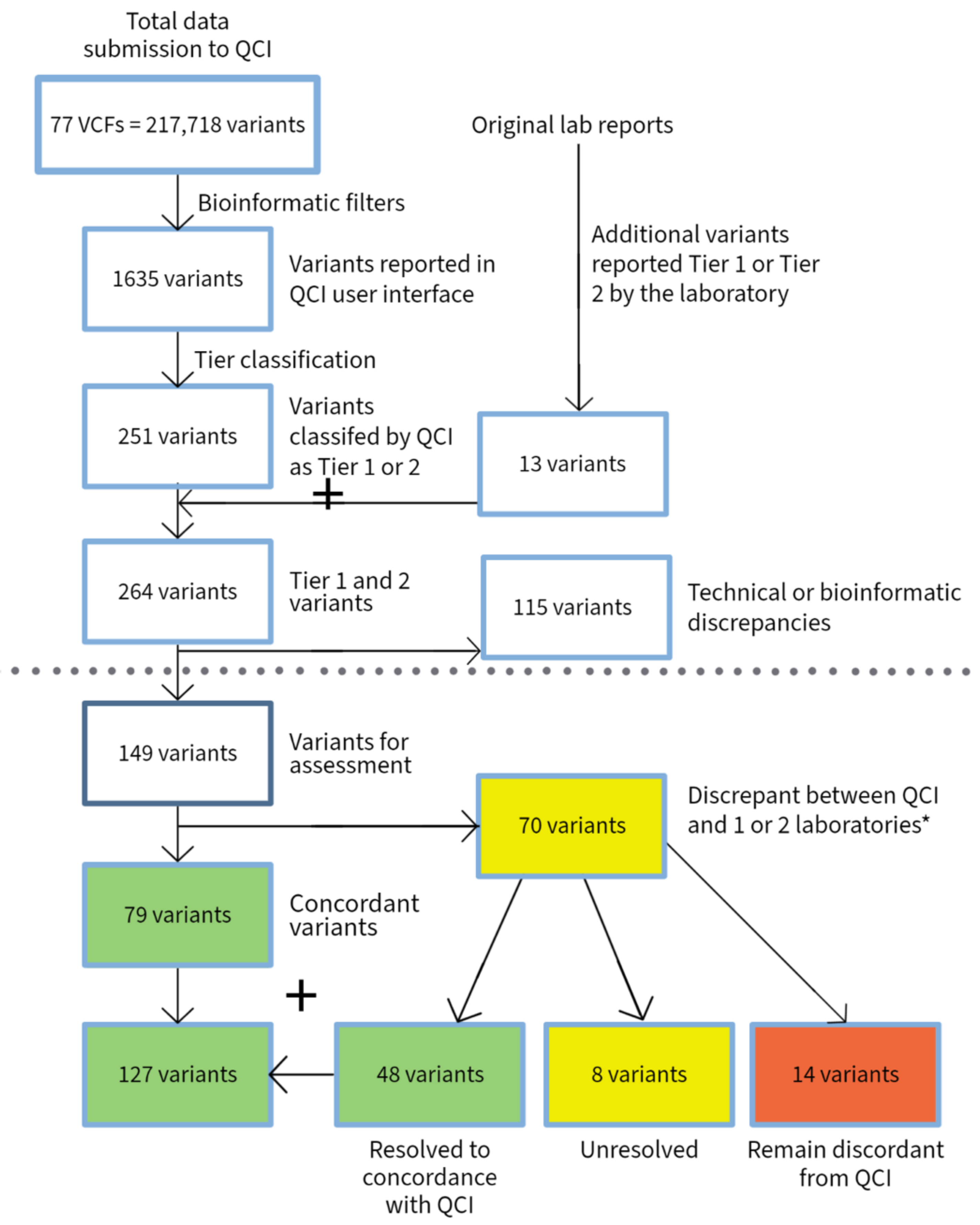
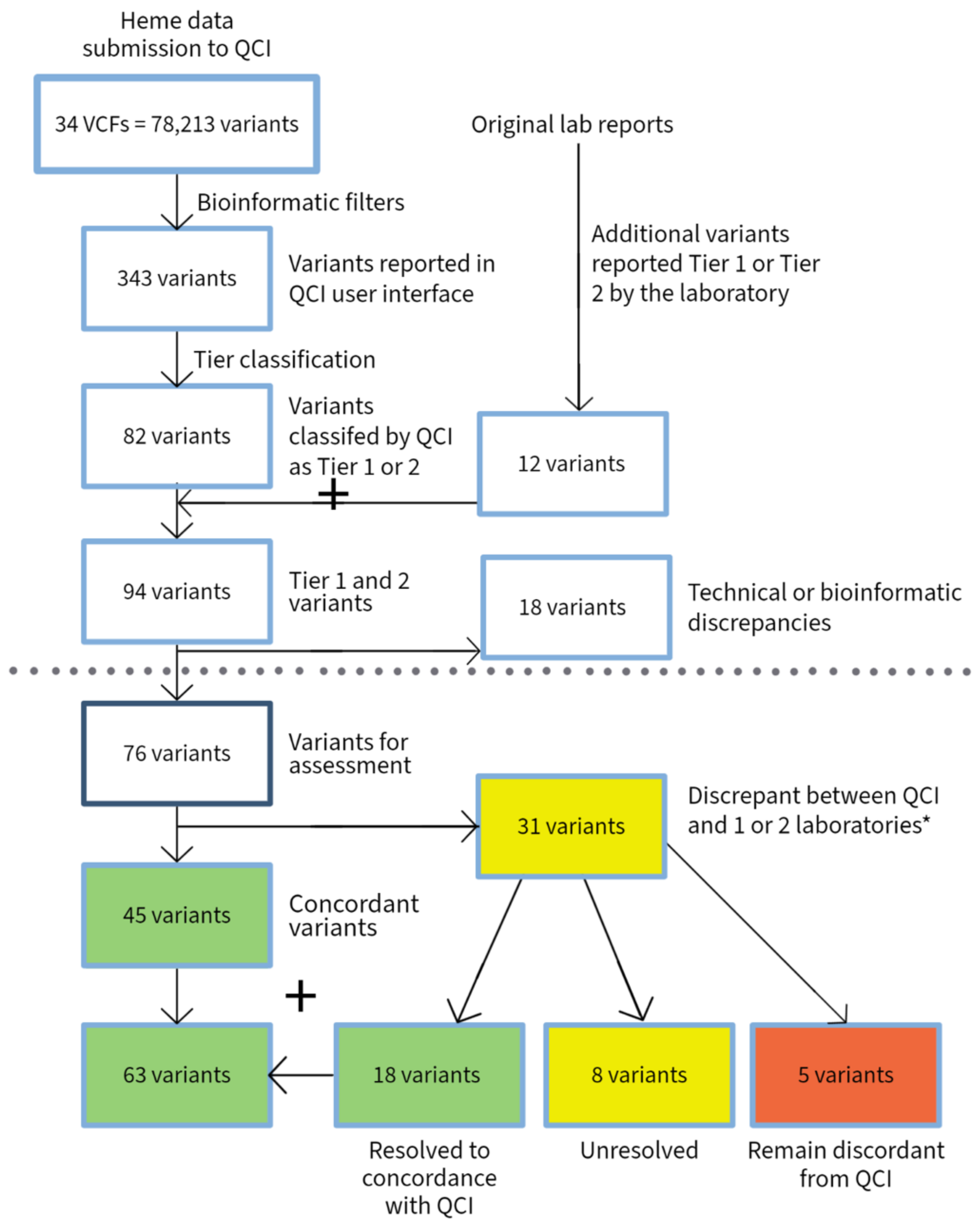
3. Results
3.1. Hematological Malignancies
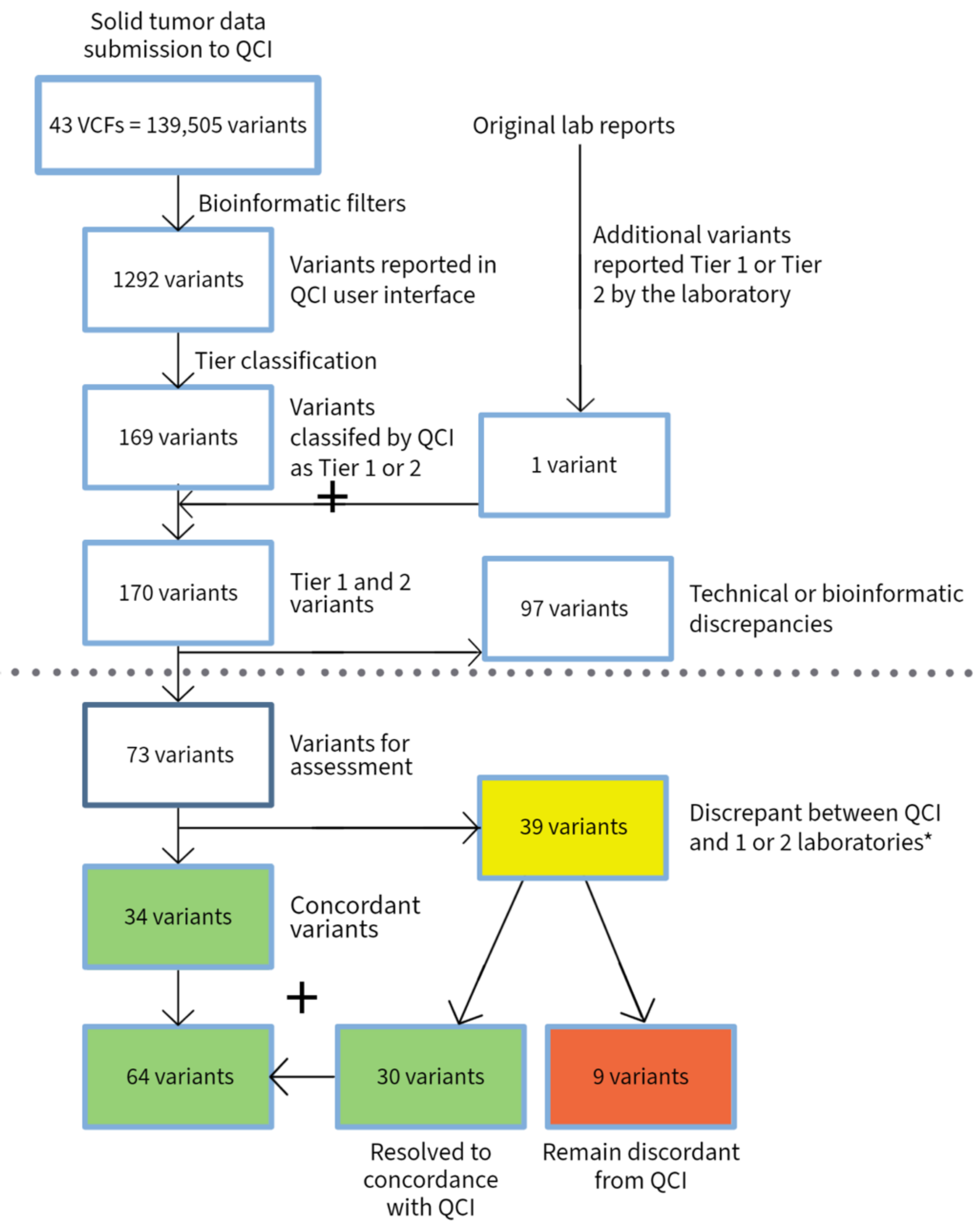
3.2. Solid Tumors
4. Discussion
4.1. Disease Specificity
4.2. Analysis of Evidence
4.3. Guidelines
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pearce, M.; Cullinan, A.; Hogg, G.; Hosseini, D.; Ehrich, M. Mutation profiling in tumor samples using the Sequenom OncoCartaTM Panel. Nat. Methods 2009, 6, vii–viii. [Google Scholar] [CrossRef]
- Patterson, S.E.; Statz, C.M.; Yin, T.; Mockus, S.M. Utility of the JAX Clinical Knowledgebase in capture and assessment of complex genomic cancer data. NPJ Precis. Oncol. 2019, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, D.; Gao, J.; Phillip, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Griffith, M.; Spies, N.C.; Krysiak, K.; McMichael, J.F.; Coffman, A.C.; Danos, A.M.; Ainscough, B.J.; Ramirez, C.A.; Rieke, D.T.; Kujan, L.; et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer. Nat. Genet. 2017, 49, 170–174. [Google Scholar] [CrossRef] [Green Version]
- Yaung, S.J.; Pek, A. From Information Overload to Actionable Insights: Digital Solutions for Interpreting Cancer Variants from Genomic Testing. J. Mol. Pathol. 2021, 2, 312–318. [Google Scholar] [CrossRef]
- PierianDx—Home. Available online: https://www.pieriandx.com (accessed on 28 April 2022).
- Bungartz, K.D.; Lalowski, K.; Elkin, S.K. Making the right calls in precision oncology. Nat. Biotechnol. 2018, 36, 692–696. [Google Scholar] [CrossRef]
- Dlamini, Z.; Francies, F.Z.; Hull, R.; Marima, R. Artificial Intelligence (AI) and big data in cancer and precision oncology. Comput. Struct. Biotechnol. J. 2020, 18, 2300–2311. [Google Scholar] [CrossRef]
- Cabitza, F.; Rasoini, R.; Gensini, G.F. Unintended Consequences of Machine Learning in Medicine. JAMA 2017, 318, 517–518. [Google Scholar] [CrossRef]
- Lohr, S. What Ever Happened to IBM’s Watson. New York Times, 16 July 2021. Available online: https://www.nytimes.com/2021/07/16/technology/what-happened-ibm-watson.html(accessed on 3 May 2022).
- Dewdney, S.B.; Lachance, J. Electronic Records, Registries, and the Development of “Big Data”: Crowd-Sourcing Quality toward Knowledge. J. Front. Oncol. 2017, 6, 268. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perakis, S.O.; Weber, S.; Zhou, Q.; Graf, R.; Hojas, S.; Riedl, J.M.; Gerger, A.; Dandachi, N.; Balic, M.; Hoefler, G.; et al. Comparison of three commercial decision support platforms for matching of next-generation sequencing results with therapies in patients with cancer. ESMO Open 2020, 5, e000872. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhang, R.; Li, Z.; Ding, J.; Xie, J.; Li, J. Challenges of Providing Concordant Interpretation of Somatic Variants in Non-Small Cell Lung Cancer: A Multicenter Study. J. Cancer 2019, 10, 1814–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirohi, D.; Schmidt, R.L.; Aisner, D.L.; Behdad, A.; Betz, B.L.; Brown, N.; Coleman, J.F.; Corless, C.L.; Deftereos, G.; Ewalt, M.D.; et al. Multi-Institutional Evaluation of Interrater Agreement of Variant Classification Based on the 2017 Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagn. 2020, 22, 284–293. [Google Scholar] [CrossRef]
- Hu, L.; Li, Z.; Cheng, J.; Rao, Q.; Gong, W.; Liu, M.; Shi, Y.G.; Zhu, J.; Wang, P.; Xu, Y. Crystal structure of TET2-DNA complex; insight into TET-mediated 5mC oxidation. Cell 2013, 155, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- Grand, F.H.; Hidalgo-Curtis, C.E.; Ernst, T.; Zoi, K.; Zoi, C.; McGuire, C.; Kreil, S.; Jones, A.; Score, J.; Metzgeroth, G.; et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 2009, 113, 6182–6192. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, S.; De Luca, A.; Stellacci, E.; Rossi, C.; Checquolo, S.; Lepri, F.; Caputo, V.; Silvano, M.; Buscherini, F.; Consoli, F.; et al. Heterozygous germline mutations in the CBL tumor suppressor gene cause a Noonan syndrome-like phenotype. Am. J. Hum. Genet. 2010, 87, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Javadi, J.; Richmond, T.D.; Huang, K.; Barber, D.L. CBL linker region and RING finger mutations lead to enhanced granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling via elevated levels of JAK2 and LYN. J. Biol. Chem. 2013, 288, 19459–19470. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, M.S.; Reddy, M.M.; Croteau, N.J.; Walz, C.; Weisbach, H.; Podar, K.; Band, H.; Carroll, M.; Reiter, A.; Larson, R.A.; et al. Novel oncogenic mutations of CBL in human acute myeloid leukemia that activate growth and survival pathways depend on increased metabolism. J. Biol. Chem. 2010, 285, 32596–32605. [Google Scholar] [CrossRef] [Green Version]
- Andoniou, C.E.; Thien, C.B.; Langdon, W.Y. Tumour induction by activated abl involves tyrosine phosphorylation of the product of the cbl oncogene. EMBO J. 1994, 13, 4515–4523. [Google Scholar] [CrossRef]
- Thien, C.B.; Walker, F.; Langdon, W.Y. RING finger mutations that abolish c-Cbl-directed polyubiquitination and downregulation of the EGF receptor are insufficient for cell transformation. Mol. Cell 2001, 7, 355–365. [Google Scholar] [CrossRef]
- Bemanian, V.; Noone, J.C.; Sauer, T.; Touma, J.; Vetvik, K.; Soderberg-Naucler, C.; Lindstrom, J.C.; Bukholm, I.R.; Kristensen, V.N.; Geisler, J. Somatic EP300-G211S mutations are associated with overall somatic mutational patterns and breast cancer specific survival in triple-negative breast cancer. Breast Cancer Res. Treat. 2018, 172, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Lin, J.; Lin, T.; Liang, W. Loss of heterozygosity: An independent prognostic factor of colorectal cancer. World J. Gastroenterol. 2005, 11, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Griffith, M.; Danos, A.M.; Pitel, B.; Madhavan, S.; Liu, X.; Chow, C.; Williams, H.; Carmody, L.; Barrow-Laing, L.; et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet. Med. 2022, 24, 986–998. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Heme | Solid Tumor | Total | |
|---|---|---|---|
| VCFs submitted | 34 | 43 | 77 |
| Total number of submitted variants | 78,213 | 139,505 | 217,718 |
| Variants reported by QCI | 343 | 1292 | 1635 |
| Variants reported as Tier 1 and 2 by QCI | 82 | 169 | 251 |
| Total variants reported as Tier 1 and 2 by QCI and submitting laboratories | 94 | 170 | 264 |
| Variants with discrepancy in reporting due to technical issue (bioinformatics or disease mapping) | 18 | 97 | 115 |
| Total variants excluding technical discrepancies | 76 | 73 | 149 |
| Variants with discrepancy in tier classification | 31 | 39 | 70 |
| Variants with discrepancies between labs or between labs and experts | 24 | 17 | 41 |
| Discrepant variants after conflict resolution | 5 | 9 | 14 |
| Group | Resolution | Resolution Description | # Discordant | # Resolved |
|---|---|---|---|---|
| A | Not resolved | Remain discordant | 5 | |
| B | Not resolved by experts | 8 | ||
| C | Resolved | QCI One expert review resolved conflict | 3 | |
| D | Expert panel agreed with QCI | 15 | ||
| E | Technical issue | QCI disease mapping issue | 6 | |
| F | Filtered out by QCI preset filters | 6 | ||
| G | Not present in submitted data | 6 | ||
| 19 | 30 | |||
| 49 | ||||
| Heme Resolutions | Solid Tumor Resolutions | |||
|---|---|---|---|---|
| Submitting laboratory and second assessing laboratory AGREE | No agreement | 2 | No agreement | 0 |
| Expert panel resolves for lab | 2 | Expert panel resolves for lab | 0 | |
| Expert panel resolves for QCI | 0 | Expert panel resolves for QCI | 3 | |
| QCI expert review resolves | 2 | QCI expert review resolves | 3 | |
| Submitting laboratory and second assessing laboratory DISAGREE | No agreement | 5 | No agreement | 0 |
| Expert panel resolves for lab | 1 | Expert panel resolves for lab | 0 | |
| Expert panel resolves for QCI | 6 | Expert panel resolves for QCI | 4 | |
| QCI expert review resolves | 0 | QCI expert review resolves | 2 | |
| Only one laboratory assessment available | No agreement | 1 | No agreement | 0 |
| Expert panel resolves for lab | 2 | Expert panel resolves for lab | 9 | |
| Expert panel resolves for QCI | 9 | Expert panel resolves for QCI | 8 | |
| QCI expert review resolves | 1 | QCI expert review resolves | 10 | |
| Group | Resolution | Resolution Description | # Discordant | # Resolved |
|---|---|---|---|---|
| A | Not resolved | Remain discordant | 9 | |
| C | Resolved | QCI One expert review resolved conflict | 15 | |
| D | Expert panel agreed with QCI | 5 | ||
| E | TP53—clinical trial inclusion led to Tier 2C from QCI—expert panel agreed | 10 | ||
| F | Technical Issue | Filtered out by QCI | 1 | |
| G | Low VAF—filtered out by QCI | 88 | ||
| H | Not reported by submitting lab | 8 | ||
| 9 | 127 | |||
| 136 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fairley, J.A.; Deans, Z.C.; Treacy, R.J.L.; Grieg, E.; Bungartz, K.; Burton, R.; Hayes, J.; Elkin, S.K. An Independent Assessment of a Commercial Clinical Interpretation Software Indicates That Software Can Mitigate Variation in Human Assessment. J. Mol. Pathol. 2022, 3, 125-139. https://doi.org/10.3390/jmp3030012
Fairley JA, Deans ZC, Treacy RJL, Grieg E, Bungartz K, Burton R, Hayes J, Elkin SK. An Independent Assessment of a Commercial Clinical Interpretation Software Indicates That Software Can Mitigate Variation in Human Assessment. Journal of Molecular Pathology. 2022; 3(3):125-139. https://doi.org/10.3390/jmp3030012
Chicago/Turabian StyleFairley, Jennifer A., Zandra C. Deans, Rebecca J. L. Treacy, Eilidh Grieg, Kathryn Bungartz, Ruth Burton, James Hayes, and Sheryl K. Elkin. 2022. "An Independent Assessment of a Commercial Clinical Interpretation Software Indicates That Software Can Mitigate Variation in Human Assessment" Journal of Molecular Pathology 3, no. 3: 125-139. https://doi.org/10.3390/jmp3030012
APA StyleFairley, J. A., Deans, Z. C., Treacy, R. J. L., Grieg, E., Bungartz, K., Burton, R., Hayes, J., & Elkin, S. K. (2022). An Independent Assessment of a Commercial Clinical Interpretation Software Indicates That Software Can Mitigate Variation in Human Assessment. Journal of Molecular Pathology, 3(3), 125-139. https://doi.org/10.3390/jmp3030012