
Impact of Fatty Acid-Binding Proteins in α-Synuclein-Induced Mitochondrial Injury in Synucleinopathy




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. α-Synuclein
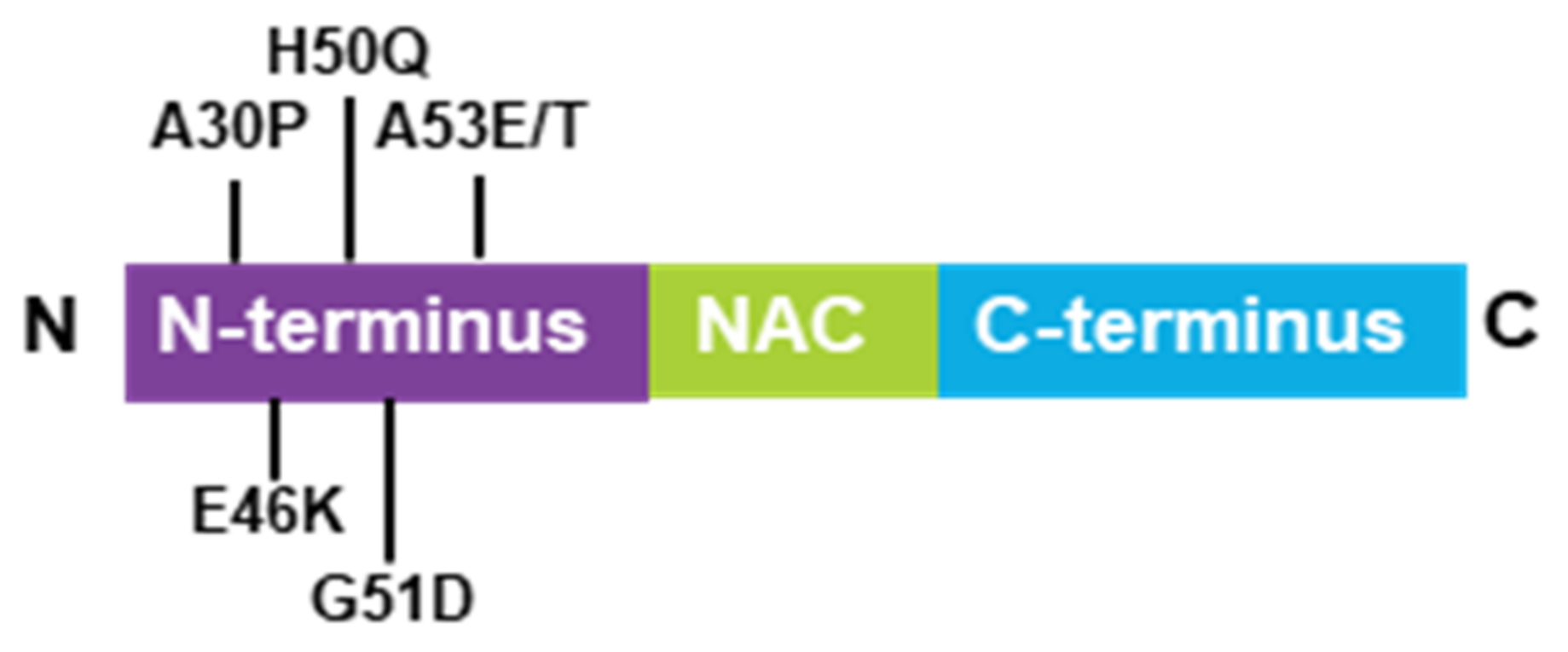
2.1. α-Synuclein Structure and Mutations
2.2. α-Synuclein Oligomers and Neuronal Toxicity
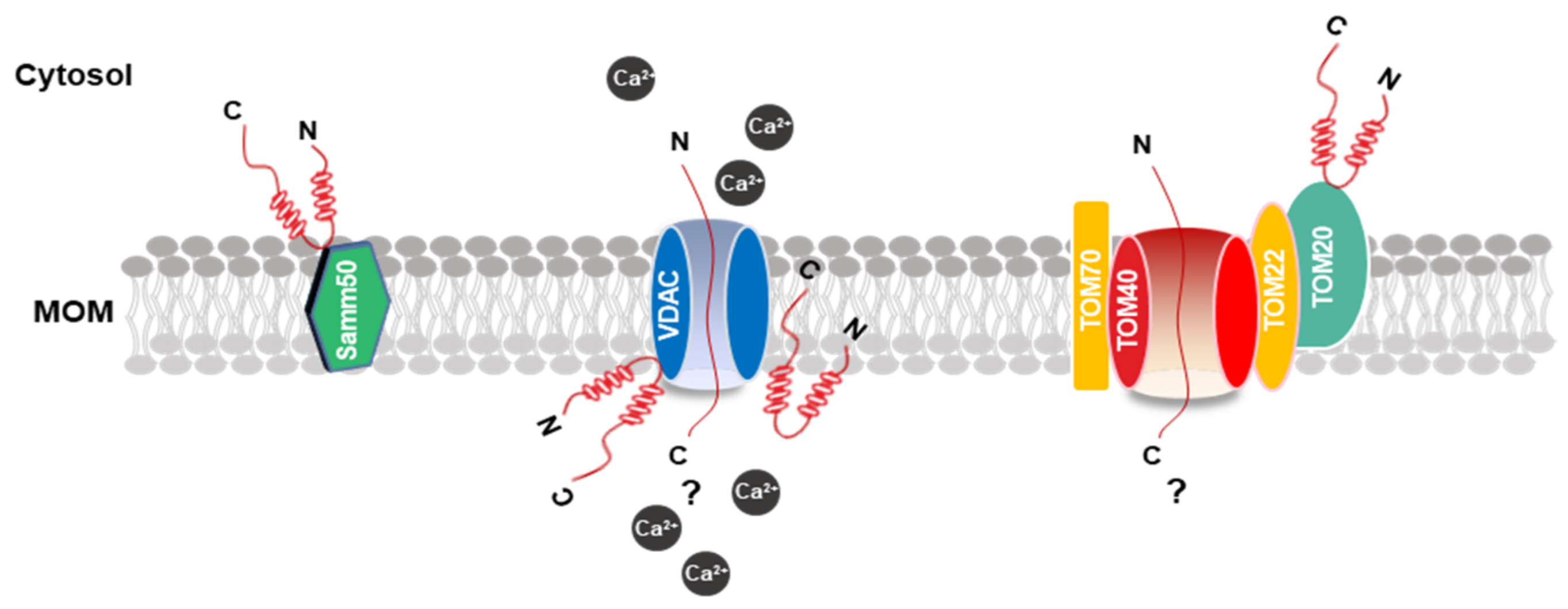
2.3. α-Synuclein and the Mitochondrial Surface
2.4. α-Synuclein in Mitochondrial Injury
3. FABPs
3.1. Expression and Functions of FABPs in the Brain
3.2. Functions of FABPs in Neurodegenerations
3.2.1. FABP3 in the Dopamine Nervous System
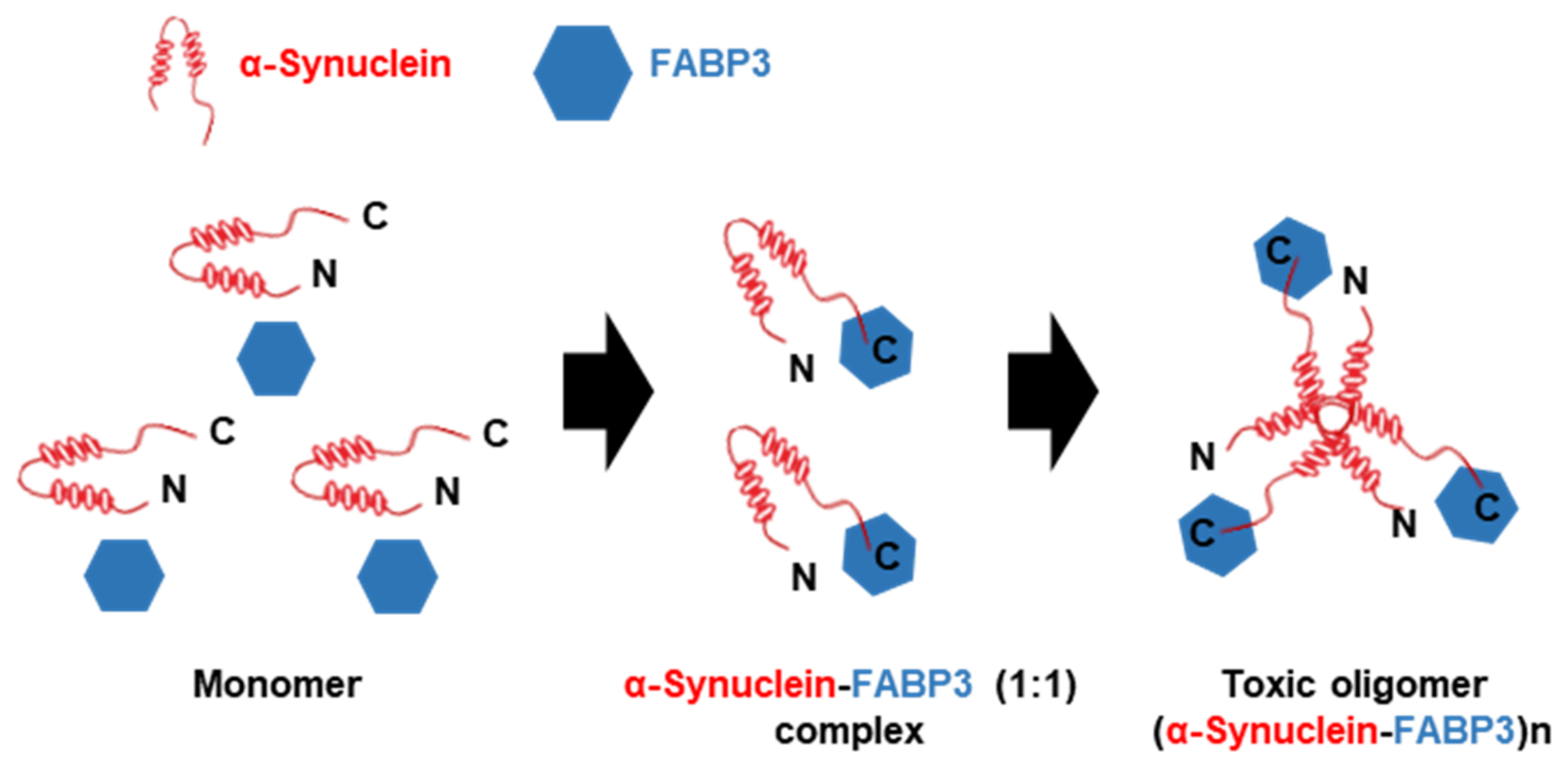
3.2.2. FABP3 in α-Synuclein Oligomerization and Migration
3.2.3. FABP5 in Cognitive Deficits
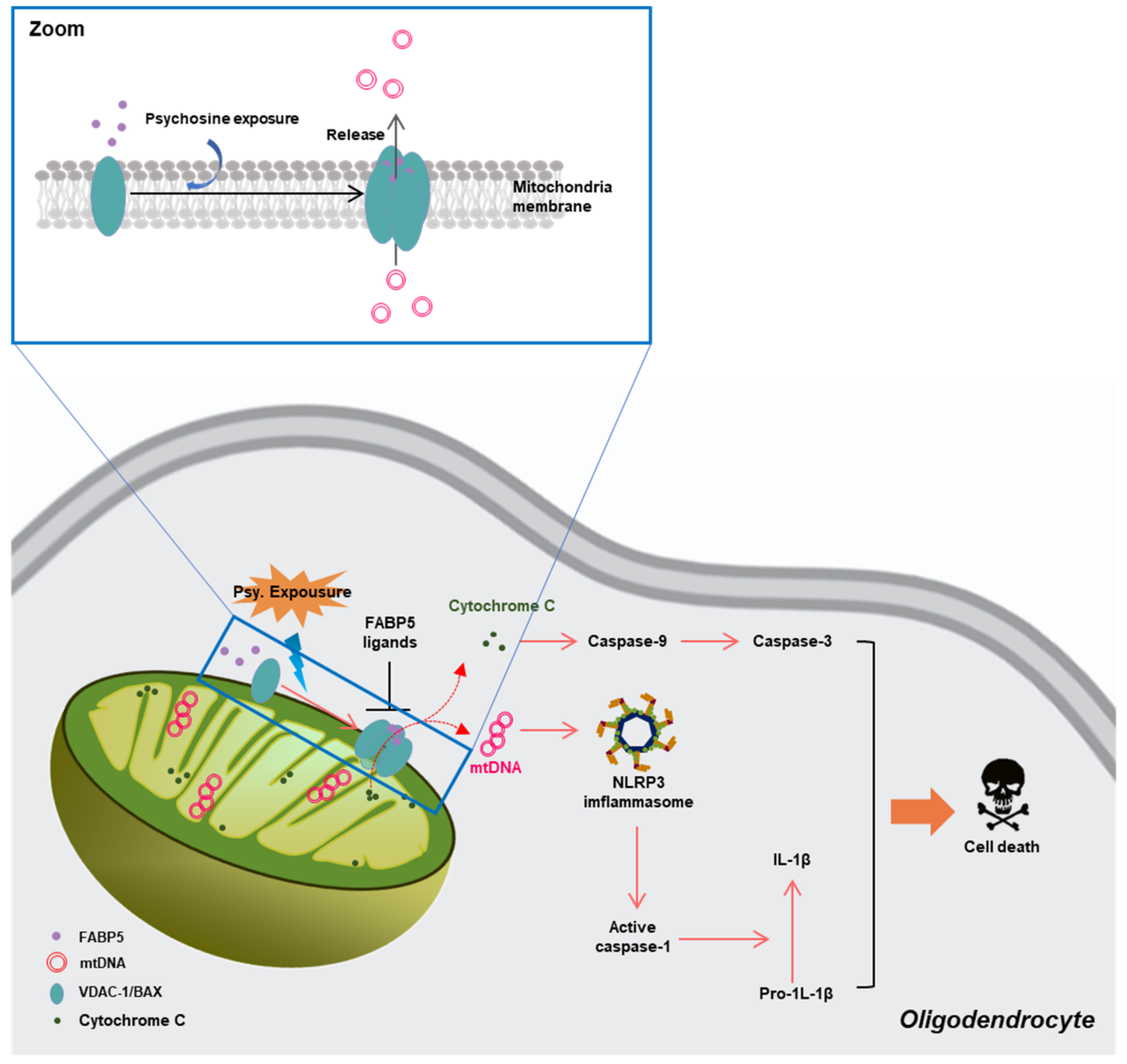
3.2.4. FABP5 in Mitochondrial Injury
3.2.5. FABP7 in Neurodegenerations
4. Novel Therapeutic Target to Mitochondrial Dysfunction via FABP Inhibition
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Przedborski, S.; Vila, M.; Jackson-Lewis, V. Series Introduction: Neurodegeneration: What is it and where are we? J. Clin. Investig. 2003, 111, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Hayes, M.T. Parkinson’s Disease and Parkinsonism. Am. J. Med. 2019, 132, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Kawamoto, Y.; Ayaki, T.; Urushitani, M.; Ito, H.; Takahashi, R. Activated caspase-9 immunoreactivity in glial and neuronal cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 2016, 628, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.E.; Markus, M.; Seetulsingh, S.P.; Wrigglesworth, J.M. Kinetics of inhibition of purified and mitochondrial cytochrome c oxidase by psychosine (beta-galactosylsphingosine). Biochem. J. 1993, 290 (Pt 1), 139–144. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.; Brustovetsky, T.; Brustovetsky, N. Oxidative metabolism and Ca2+ handling in striatal mitochondria from YAC128 mice, a model of Huntington’s disease. Neurochem. Int. 2017, 109, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Shavali, S.; Brown-Borg, H.M.; Ebadi, M.; Porter, J. Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci. Lett. 2008, 439, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol. Life Sci. 2008, 65, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Shioda, N.; Yabuki, Y.; Kobayashi, Y.; Onozato, M.; Owada, Y.; Fukunaga, K. FABP3 protein promotes alpha-synuclein oligomerization associated with 1-methyl-1,2,3,6-tetrahydropiridine-induced neurotoxicity. J. Biol. Chem. 2014, 289, 18957–18965. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, I.; Bousset, L.; Melki, R.; Fukunaga, K. Fatty Acid-Binding Protein 3 is Critical for alpha-Synuclein Uptake and MPP(+)-Induced Mitochondrial Dysfunction in Cultured Dopaminergic Neurons. Int. J. Mol. Sci. 2019, 20, 5358. [Google Scholar] [CrossRef]
- Cheng, A.; Wang, Y.-f.; Shinoda, Y.; Kawahata, I.; Yamamoto, T.; Jia, W.-b.; Yamamoto, H.; Mizobata, T.; Kawata, Y.; Fukunaga, K. Fatty acid-binding protein 7 triggers α-synuclein oligomerization in glial cells and oligodendrocytes associated with oxidative stress. Acta Pharmacol. Sin. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kamijo-Ikemori, A.; Ichikawa, D.; Matsui, K.; Yokoyama, T.; Sugaya, T.; Kimura, K. [Urinary L-type fatty acid binding protein (L-FABP) as a new urinary biomarker promulgated by the Ministry of Health, Labour and Welfare in Japan]. Rinsho Byori. Jpn. J. Clin. Pathol. 2013, 61, 635–640. [Google Scholar]
- Li, H.; Xiao, Y.; Tang, L.; Zhong, F.; Huang, G.; Xu, J.M.; Xu, A.M.; Dai, R.P.; Zhou, Z.G. Adipocyte Fatty Acid-Binding Protein Promotes Palmitate-Induced Mitochondrial Dysfunction and Apoptosis in Macrophages. Front. Immunol. 2018, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Kawahata, I.; Fukunaga, K. Fatty Acid Binding Protein 5 Mediates Cell Death by Psychosine Exposure through Mitochondrial Macropores Formation in Oligodendrocytes. Biomedicines 2020, 8, 635. [Google Scholar] [CrossRef] [PubMed]
- Bayer, T.A.; Jäkälä, P.; Hartmann, T.; Havas, L.; McLean, C.; Culvenor, J.G.; Li, Q.-X.; Masters, C.L.; Falkai, P.; Beyreuther, K. α-Synuclein accumulates in Lewy bodies in Parkinson’s disease and dementia with Lewy bodies but not in Alzheimer’s disease β-amyloid plaque cores. Neurosci. Lett. 1999, 266, 213–216. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys. J. 2007, 92, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Prots, I.; Veber, V.; Brey, S.; Campioni, S.; Buder, K.; Riek, R.; Böhm, K.J.; Winner, B. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 2013, 288, 21742–21754. [Google Scholar] [CrossRef] [PubMed]
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Pöyhönen, M.; Paetau, A. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1–2180.e5. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Harper, J.D.; Williamson, R.E.; Lansbury, P.T., Jr. Accelerated oligomerization by Parkinson’s disease linked alpha-synuclein mutants. Ann. N. Y. Acad. Sci. 2000, 920, 42–45. [Google Scholar] [CrossRef]
- Ghosh, D.; Singh, P.K.; Sahay, S.; Jha, N.N.; Jacob, R.S.; Sen, S.; Kumar, A.; Riek, R.; Maji, S.K. Structure based aggregation studies reveal the presence of helix-rich intermediate during α-Synuclein aggregation. Sci. Rep. 2015, 5, 9228. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef]
- Villar-Pique, A.; Lopes da Fonseca, T.; Outeiro, T.F. Structure, function and toxicity of alpha-synuclein: The Bermuda triangle in synucleinopathies. J. Neurochem. 2016, 139 (Suppl. 1), 240–255. [Google Scholar] [CrossRef]
- Bussell, R., Jr.; Eliezer, D. Residual structure and dynamics in Parkinson’s disease-associated mutants of alpha-synuclein. J. Biol. Chem. 2001, 276, 45996–46003. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Sharikov, Y.; Kouznetsova, V.L.; Greenberg, J.P.; Wrasidlo, W.; Overk, C.; Gonzalez, T.; Trejo, M.; Spencer, B.; Kosberg, K.; et al. Molecular determinants of α-synuclein mutants’ oligomerization and membrane interactions. ACS Chem. Neurosci. 2015, 6, 403–416. [Google Scholar] [CrossRef]
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J. Mol. Biol. 2002, 315, 799–807. [Google Scholar] [CrossRef]
- Fares, M.B.; Ait-Bouziad, N.; Dikiy, I.; Mbefo, M.K.; Jovičić, A.; Kiely, A.; Holton, J.L.; Lee, S.J.; Gitler, A.D.; Eliezer, D.; et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 2014, 23, 4491–4509. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Sahay, S.; Ranjan, P.; Salot, S.; Mohite, G.M.; Singh, P.K.; Dwivedi, S.; Carvalho, E.; Banerjee, R.; Kumar, A.; et al. The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry 2014, 53, 6419–6421. [Google Scholar] [CrossRef] [PubMed]
- Bertoncini, C.W.; Fernandez, C.O.; Griesinger, C.; Jovin, T.M.; Zweckstetter, M. Familial mutants of alpha-synuclein with increased neurotoxicity have a destabilized conformation. J. Biol. Chem. 2005, 280, 30649–30652. [Google Scholar] [CrossRef] [PubMed]
- Sahay, S.; Ghosh, D.; Dwivedi, S.; Anoop, A.; Mohite, G.M.; Kombrabail, M.; Krishnamoorthy, G.; Maji, S.K. Familial Parkinson disease-associated mutations alter the site-specific microenvironment and dynamics of α-synuclein. J. Biol. Chem. 2015, 290, 7804–7822. [Google Scholar] [CrossRef]
- Karpinar, D.P.; Balija, M.B.; Kugler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H.; et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 2009, 28, 3256–3268. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schutz, O.; et al. alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef]
- Rockenstein, E.; Nuber, S.; Overk, C.R.; Ubhi, K.; Mante, M.; Patrick, C.; Adame, A.; Trejo-Morales, M.; Gerez, J.; Picotti, P.; et al. Accumulation of oligomer-prone alpha-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 2014, 137, 1496–1513. [Google Scholar] [CrossRef]
- Choi, B.K.; Choi, M.G.; Kim, J.Y.; Yang, Y.; Lai, Y.; Kweon, D.H.; Lee, N.K.; Shin, Y.K. Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 2013, 110, 4087–4092. [Google Scholar] [CrossRef]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, C.R.; Morales, C.N.; Ramirez, A.E.; Munoz, F.J.; Gallegos, S.S.; Caviedes, P.A.; Aguayo, L.G.; Opazo, C.M. Extracellular alpha-synuclein alters synaptic transmission in brain neurons by perforating the neuronal plasma membrane. J. Neurochem. 2015, 132, 731–741. [Google Scholar] [CrossRef]
- Volles, M.J.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar] [CrossRef]
- Van Rooijen, B.D.; Claessens, M.M.; Subramaniam, V. Membrane Permeabilization by Oligomeric α-Synuclein: In Search of the Mechanism. PLoS ONE 2010, 5, e14292. [Google Scholar] [CrossRef]
- Melachroinou, K.; Xilouri, M.; Emmanouilidou, E.; Masgrau, R.; Papazafiri, P.; Stefanis, L.; Vekrellis, K. Deregulation of calcium homeostasis mediates secreted α-synuclein-induced neurotoxicity. Neurobiol. Aging 2013, 34, 2853–2865. [Google Scholar] [CrossRef]
- Stöckl, M.; Claessens, M.M.; Subramaniam, V. Kinetic measurements give new insights into lipid membrane permeabilization by α-synuclein oligomers. Mol. Biosyst. 2012, 8, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Tosatto, L.; Andrighetti, A.O.; Plotegher, N.; Antonini, V.; Tessari, I.; Ricci, L.; Bubacco, L.; Dalla Serra, M. Alpha-synuclein pore forming activity upon membrane association. Biochim. Biophys. Acta 2012, 1818, 2876–2883. [Google Scholar] [CrossRef]
- Chen, L.; Jin, J.; Davis, J.; Zhou, Y.; Wang, Y.; Liu, J.; Lockhart, P.J.; Zhang, J. Oligomeric alpha-synuclein inhibits tubulin polymerization. Biochem. Biophys. Res. Commun. 2007, 356, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Khoshaghideh, F.; Lee, S.; Lee, S.J. Impairment of microtubule-dependent trafficking by overexpression of alpha-synuclein. Eur. J. Neurosci. 2006, 24, 3153–3162. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. α-Synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef]
- Crews, L.; Spencer, B.; Desplats, P.; Patrick, C.; Paulino, A.; Rockenstein, E.; Hansen, L.; Adame, A.; Galasko, D.; Masliah, E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS ONE 2010, 5, e9313. [Google Scholar] [CrossRef]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoL ONE 2009, 4, e5515. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194. [Google Scholar] [CrossRef]
- McNaught, K.S.; Belizaire, R.; Jenner, P.; Olanow, C.W.; Isacson, O. Selective loss of 20S proteasome alpha-subunits in the substantia nigra pars compacta in Parkinson’s disease. Neurosci. Lett. 2002, 326, 155–158. [Google Scholar] [CrossRef]
- Furukawa, Y.; Vigouroux, S.; Wong, H.; Guttman, M.; Rajput, A.H.; Ang, L.; Briand, M.; Kish, S.J.; Briand, Y. Brain proteasomal function in sporadic Parkinson’s disease and related disorders. Ann. Neurol. 2002, 51, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.N.; Hanni, K.B.; Markesbery, W.R. Impaired proteasome function in Alzheimer’s disease. J. Neurochem. 2000, 75, 436–439. [Google Scholar] [CrossRef]
- McNaught, K.S.; Olanow, C.W.; Halliwell, B.; Isacson, O.; Jenner, P. Failure of the ubiquitin-proteasome system in Parkinson’s disease. Nat. Rev. Neurosci. 2001, 2, 589–594. [Google Scholar] [CrossRef]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome impairment by α-synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Valina, L.D.; Dawson, T.M.; Ross, C.A. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Lindersson, E.; Beedholm, R.; Højrup, P.; Moos, T.; Gai, W.; Hendil, K.B.; Jensen, P.H. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 2004, 279, 12924–12934. [Google Scholar] [CrossRef]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Deger, J.M.; Gerson, J.E.; Kayed, R. The interrelationship of proteasome impairment and oligomeric intermediates in neurodegeneration. Aging Cell 2015, 14, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Shvadchak, V.V.; Yushchenko, D.A.; Pievo, R.; Jovin, T.M. The mode of α-synuclein binding to membranes depends on lipid composition and lipid to protein ratio. FEBS Lett. 2011, 585, 3513–3519. [Google Scholar] [CrossRef] [PubMed]
- Rhoades, E.; Ramlall, T.F.; Webb, W.W.; Eliezer, D. Quantification of alpha-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys. J. 2006, 90, 4692–4700. [Google Scholar] [CrossRef]
- Maekawa, H.; Inoue, T.; Ouchi, H.; Jao, T.M.; Inoue, R.; Nishi, H.; Fujii, R.; Ishidate, F.; Tanaka, T.; Tanaka, Y.; et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019, 29, 1261–1273.e6. [Google Scholar] [CrossRef]
- Perrin, R.J.; Woods, W.S.; Clayton, D.F.; George, J.M. Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J. Biol. Chem. 2000, 275, 34393–34398. [Google Scholar] [CrossRef]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef]
- McFarland, M.A.; Ellis, C.E.; Markey, S.P.; Nussbaum, R.L. Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol. Cell. Proteom. MCP 2008, 7, 2123–2137. [Google Scholar] [CrossRef]
- Martin, L.J.; Semenkow, S.; Hanaford, A.; Wong, M. Mitochondrial permeability transition pore regulates Parkinson’s disease development in mutant α-synuclein transgenic mice. Neurobiol. Aging 2014, 35, 1132–1152. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. α-Synuclein Shows High Affinity Interaction with Voltage-dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef] [PubMed]
- Telford, J.E.; Kilbride, S.M.; Davey, G.P. Complex I is rate-limiting for oxygen consumption in the nerve terminal. J. Biol. Chem. 2009, 284, 9109–9114. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra378. [Google Scholar] [CrossRef]
- Miraglia, F.; Ricci, A.; Rota, L.; Colla, E. Subcellular localization of alpha-synuclein aggregates and their interaction with membranes. Neural Regen. Res. 2018, 13, 1136–1144. [Google Scholar] [CrossRef]
- Vicario, M.; Cieri, D.; Brini, M.; Cali, T. The Close Encounter Between Alpha-Synuclein and Mitochondria. Front. Neurosci. 2018, 12, 388. [Google Scholar] [CrossRef]
- Hosios, A.M.; Manning, B.D. Lysosomal catch-and-release controls mTORC1. Nat. Cell Biol. 2018, 20, 996–997. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed]
- Veerkamp, J.H.; Peeters, R.A.; Maatman, R.G. Structural and functional features of different types of cytoplasmic fatty acid-binding proteins. Biochim. Biophys. Acta 1991, 1081, 1–24. [Google Scholar] [CrossRef]
- Coe, N.R.; Bernlohr, D.A. Physiological properties and functions of intracellular fatty acid-binding proteins. Biochim. Biophys. Acta 1998, 1391, 287–306. [Google Scholar] [CrossRef]
- Spener, F.; Börchers, T.; Mukherjea, M. On the role of fatty acid binding proteins in fatty acid transport and metabolism. FEBS Lett. 1989, 244, 1–5. [Google Scholar] [CrossRef]
- Glatz, J.F.; Vork, M.M.; Cistola, D.P.; van der Vusse, G.J. Cytoplasmic fatty acid binding protein: Significance for intracellular transport of fatty acids and putative role on signal transduction pathways. Prostaglandins Leukot. Essent. Fat. Acids 1993, 48, 33–41. [Google Scholar] [CrossRef]
- Owada, Y.; Abdelwahab, S.A.; Kitanaka, N.; Sakagami, H.; Takano, H.; Sugitani, Y.; Sugawara, M.; Kawashima, H.; Kiso, Y.; Mobarakeh, J.I.; et al. Altered emotional behavioral responses in mice lacking brain-type fatty acid-binding protein gene. Eur. J. Neurosci. 2006, 24, 175–187. [Google Scholar] [CrossRef]
- Storch, J.; Corsico, B. The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu. Rev. Nutr. 2008, 28, 73–95. [Google Scholar] [CrossRef]
- Owada, Y.; Yoshimoto, T.; Kondo, H. Spatio-temporally differential expression of genes for three members of fatty acid binding proteins in developing and mature rat brains. J. Chem. Neuroanat. 1996, 12, 113–122. [Google Scholar] [CrossRef]
- Liu, R.Z.; Mita, R.; Beaulieu, M.; Gao, Z.; Godbout, R. Fatty acid binding proteins in brain development and disease. Int. J. Dev. Biol. 2010, 54, 1229–1239. [Google Scholar] [CrossRef]
- Umaru, B.A.; Kagawa, Y.; Shil, S.K.; Arakawa, N.; Pan, Y.; Miyazaki, H.; Kobayashi, S.; Yang, S.; Cheng, A.; Wang, Y.; et al. Ligand Bound Fatty Acid Binding Protein 7 (FABP7) Drives Melanoma Cell Proliferation Via Modulation of Wnt/β-Catenin Signaling. Pharm. Res. 2021, 38, 479–490. [Google Scholar] [CrossRef]
- Liu, Y.; Longo, L.D.; De León, M. In situ and immunocytochemical localization of E-FABP mRNA and protein during neuronal migration and differentiation in the rat brain. Brain Res. 2000, 852, 16–27. [Google Scholar] [CrossRef]
- Myers-Payne, S.C.; Hubbell, T.; Pu, L.; Schnütgen, F.; Börchers, T.; Wood, W.G.; Spener, F.; Schroeder, F. Isolation and characterization of two fatty acid binding proteins from mouse brain. J. Neurochem. 1996, 66, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Widstrom, R.L.; Norris, A.W.; Van Der Veer, J.; Spector, A.A. Fatty acid-binding proteins inhibit hydration of epoxyeicosatrienoic acids by soluble epoxide hydrolase. Biochemistry 2003, 42, 11762–11767. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Steinacker, P.; Bahn, E.; Bibl, M.; Brechlin, P.; Schlossmacher, M.G.; Locascio, J.J.; Wiltfang, J.; Kretzschmar, H.A.; Poser, S.; et al. Serum heart-type fatty acid-binding protein and cerebrospinal fluid tau: Marker candidates for dementia with Lewy bodies. Neuro-Degener. Dis. 2007, 4, 366–375. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Veerhuis, R.; De Vente, J.; Verhey, F.R.; Vreeling, F.; van Boxtel, M.P.; Glatz, J.F.; Pelsers, M.A. Brain-specific fatty acid-binding protein is elevated in serum of patients with dementia-related diseases. Eur. J. Neurol. 2011, 18, 865–871. [Google Scholar] [CrossRef]
- Grace, A.A. Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat. Rev. Neurosci. 2016, 17, 524–532. [Google Scholar] [CrossRef]
- Vosberg, D.E.; Leyton, M.; Flores, C. The Netrin-1/DCC guidance system: Dopamine pathway maturation and psychiatric disorders emerging in adolescence. Mol. Psychiatry 2020, 25, 297–307. [Google Scholar] [CrossRef]
- Correll, C.U.; Kane, J.M.; Citrome, L.L. Epidemiology, Prevention, and Assessment of Tardive Dyskinesia and Advances in Treatment. J. Clin. Psychiatry 2017, 78, 1136–1147. [Google Scholar] [CrossRef]
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine receptors: From structure to function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Fukunaga, K. Different activation of NF-kappaB by stimulation of dopamine D2L and D2S receptors through calcineurin activation. J. Neurochem. 2004, 90, 155–163. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Fukunaga, K. Dopamine D2 receptor activates extracellular signal-regulated kinase through the specific region in the third cytoplasmic loop. J. Neurochem. 2004, 89, 1498–1507. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, Y.; Fukunaga, K. Differential subcellular localization of two dopamine D2 receptor isoforms in transfected NG108-15 cells. J. Neurochem. 2003, 85, 1064–1074. [Google Scholar] [CrossRef] [PubMed]
- Shioda, N.; Yamamoto, Y.; Watanabe, M.; Binas, B.; Owada, Y.; Fukunaga, K. Heart-type fatty acid binding protein regulates dopamine D2 receptor function in mouse brain. J. Neurosci. 2010, 30, 3146–3155. [Google Scholar] [CrossRef]
- Jia, W.; Wilar, G.; Kawahata, I.; Cheng, A.; Fukunaga, K. Impaired Acquisition of Nicotine-Induced Conditioned Place Preference in Fatty Acid-Binding Protein 3 Null Mice. Mol. Neurobiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Kawahata, I.; Cheng, A.; Fukunaga, K. The Role of CaMKII and ERK Signaling in Addiction. Int. J. Mol. Sci. 2021, 22, 3189. [Google Scholar] [CrossRef]
- Fukui, N.; Yamamoto, H.; Miyabe, M.; Aoyama, Y.; Hongo, K.; Mizobata, T.; Kawahata, I.; Yabuki, Y.; Shinoda, Y.; Fukunaga, K.; et al. An alpha-synuclein decoy peptide prevents cytotoxic alpha-synuclein aggregation caused by fatty acid binding protein 3. J. Biol. Chem. 2021. [Google Scholar] [CrossRef]
- Cheng, A.; Shinoda, Y.; Yamamoto, T.; Miyachi, H.; Fukunaga, K. Development of FABP3 ligands that inhibit arachidonic acid-induced α-synuclein oligomerization. Brain Res. 2019, 1707, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Cheng, A.; Yabuki, Y.; Takahata, I.; Miyachi, H.; Fukunaga, K. Inhibition of MPTP-induced alpha-synuclein oligomerization by fatty acid-binding protein 3 ligand in MPTP-treated mice. Neuropharmacology 2019, 150, 164–174. [Google Scholar] [CrossRef]
- Kawahata, I.; Sekimori, T.; Wang, H.; Wang, Y.; Sasaoka, T.; Bousset, L.; Melki, R.; Mizobata, T.; Kawata, Y.; Fukunaga, K. Dopamine D2 Long Receptors Are Critical for Caveolae-Mediated alpha-Synuclein Uptake in Cultured Dopaminergic Neurons. Biomedicines 2021, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Celver, J.; Octeau, J.C.; Kovoor, A. Plasma membrane compartmentalization of D2 dopamine receptors. J. Biol. Chem. 2013, 288, 12554–12568. [Google Scholar] [CrossRef]
- Cho, D.I.; Min, C.; Jung, K.S.; Cheong, S.Y.; Zheng, M.; Cheong, S.J.; Oak, M.H.; Cheong, J.H.; Lee, B.K.; Kim, K.M. The N-terminal region of the dopamine D2 receptor, a rhodopsin-like GPCR, regulates correct integration into the plasma membrane and endocytic routes. Br. J. Pharmacol. 2012, 166, 659–675. [Google Scholar] [CrossRef]
- Young, C.; Gean, P.W.; Chiou, L.C.; Shen, Y.Z. Docosahexaenoic acid inhibits synaptic transmission and epileptiform activity in the rat hippocampus. Synapse 2000, 37, 90–94. [Google Scholar] [CrossRef]
- Itokazu, N.; Ikegaya, Y.; Nishikawa, M.; Matsuki, N. Bidirectional actions of docosahexaenoic acid on hippocampal neurotransmissions in vivo. Brain Res. 2000, 862, 211–216. [Google Scholar] [CrossRef]
- Rapoport, S.I.; Chang, M.C.; Spector, A.A. Delivery and turnover of plasma-derived essential PUFAs in mammalian brain. J. Lipid Res. 2001, 42, 678–685. [Google Scholar] [CrossRef]
- Rapoport, S.I.; Rao, J.S.; Igarashi, M. Brain metabolism of nutritionally essential polyunsaturated fatty acids depends on both the diet and the liver. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 251–261. [Google Scholar] [CrossRef]
- Pan, Y.; Scanlon, M.J.; Owada, Y.; Yamamoto, Y.; Porter, C.J.; Nicolazzo, J.A. Fatty Acid-Binding Protein 5 Facilitates the Blood-Brain Barrier Transport of Docosahexaenoic Acid. Mol. Pharm. 2015, 12, 4375–4385. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Short, J.L.; Choy, K.H.; Zeng, A.X.; Marriott, P.J.; Owada, Y.; Scanlon, M.J.; Porter, C.J.; Nicolazzo, J.A. Fatty Acid-Binding Protein 5 at the Blood-Brain Barrier Regulates Endogenous Brain Docosahexaenoic Acid Levels and Cognitive Function. J. Neurosci. 2016, 36, 11755–11767. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Levi, L.; Casadesus, G.; Kunos, G.; Noy, N. Fatty acid-binding protein 5 (FABP5) regulates cognitive function both by decreasing anandamide levels and by activating the nuclear receptor peroxisome proliferator-activated receptor beta/delta (PPARbeta/delta) in the brain. J. Biol. Chem. 2014, 289, 12748–12758. [Google Scholar] [CrossRef]
- Li, B.; Reynolds, J.M.; Stout, R.D.; Bernlohr, D.A.; Suttles, J. Regulation of Th17 differentiation by epidermal fatty acid-binding protein. J. Immunol. Baltim. Md. 1950 2009, 182, 7625–7633. [Google Scholar] [CrossRef]
- Reynolds, J.M.; Liu, Q.; Brittingham, K.C.; Liu, Y.; Gruenthal, M.; Gorgun, C.Z.; Hotamisligil, G.S.; Stout, R.D.; Suttles, J. Deficiency of fatty acid-binding proteins in mice confers protection from development of experimental autoimmune encephalomyelitis. J. Immunol. Baltim. Md. 1950 2007, 179, 313–321. [Google Scholar] [CrossRef]
- Rao, E.; Singh, P.; Li, Y.; Zhang, Y.; Chi, Y.I.; Suttles, J.; Li, B. Targeting epidermal fatty acid binding protein for treatment of experimental autoimmune encephalomyelitis. BMC Immunol. 2015, 16, 28. [Google Scholar] [CrossRef]
- Wang, Y.; Shinoda, Y.; Cheng, A.; Kawahata, I.; Fukunaga, K. Epidermal Fatty Acid-Binding Protein 5 (FABP5) Involvement in Alpha-Synuclein-Induced Mitochondrial Injury Under Oxidative Stress. Biomedicines 2021, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Bernlohr, D.A. Metabolic functions of FABPs--mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 2015, 11, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Field, C.S.; Baixauli, F.; Kyle, R.L.; Puleston, D.J.; Cameron, A.M.; Sanin, D.E.; Hippen, K.L.; Loschi, M.; Thangavelu, G.; Corrado, M.; et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab. 2020, 31, 422–437.e5. [Google Scholar] [CrossRef]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef]
- Ma, D.; Zhang, M.; Mori, Y.; Yao, C.; Larsen, C.P.; Yamashima, T.; Zhou, L. Cellular localization of epidermal-type and brain-type fatty acid-binding proteins in adult hippocampus and their response to cerebral ischemia. Hippocampus 2010, 20, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Boneva, N.B.; Kaplamadzhiev, D.B.; Sahara, S.; Kikuchi, H.; Pyko, I.V.; Kikuchi, M.; Tonchev, A.B.; Yamashima, T. Expression of fatty acid-binding proteins in adult hippocampal neurogenic niche of postischemic monkeys. Hippocampus 2011, 21, 162–171. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; McTigue, D.M.; Jakeman, L.B. Regional heterogeneity in astrocyte responses following contusive spinal cord injury in mice. J. Comp. Neurol. 2010, 518, 1370–1390. [Google Scholar] [CrossRef]
- Bannerman, P.; Hahn, A.; Soulika, A.; Gallo, V.; Pleasure, D. Astrogliosis in EAE spinal cord: Derivation from radial glia, and relationships to oligodendroglia. Glia 2007, 55, 57–64. [Google Scholar] [CrossRef]
- Kamizato, K.; Sato, S.; Shil, S.K.; Umaru, B.A.; Kagawa, Y.; Yamamoto, Y.; Ogata, M.; Yasumoto, Y.; Okuyama, Y.; Ishii, N.; et al. The role of fatty acid binding protein 7 in spinal cord astrocytes in a mouse model of experimental autoimmune encephalomyelitis. Neuroscience 2019, 409, 120–129. [Google Scholar] [CrossRef]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Cole, N.B.; Dieuliis, D.; Leo, P.; Mitchell, D.C.; Nussbaum, R.L. Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp. Cell Res. 2008, 314, 2076–2089. [Google Scholar] [CrossRef] [PubMed]
- Lagadic-Gossmann, D.; Huc, L.; Lecureur, V. Alterations of intracellular pH homeostasis in apoptosis: Origins and roles. Cell Death Differ. 2004, 11, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.L.; Wang, S.M.; Chen, C.C.; Fong, T.H.; Wu, M.L. Mechanism of oxidative stress-induced intracellular acidosis in rat cerebellar astrocytes and C6 glioma cells. J. Physiol. 1997, 502, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Marini, A.M.; Nowak, T.S., Jr. Metabolic effects of 1-methyl-4-phenylpyridinium (MPP(+)) in primary neuron cultures. J. Neurosci. Res. 2000, 62, 814–820. [Google Scholar] [CrossRef]
- Seo, J.H.; Kang, S.W.; Kim, K.; Wi, S.; Lee, J.W.; Cho, S.R. Environmental Enrichment Attenuates Oxidative Stress and Alters Detoxifying Enzymes in an A53T α-Synuclein Transgenic Mouse Model of Parkinson’s Disease. Antioxidants 2020, 9, 928. [Google Scholar] [CrossRef]
- Wi, S.; Lee, J.W.; Kim, M.; Park, C.H.; Cho, S.R. An Enriched Environment Ameliorates Oxidative Stress and Olfactory Dysfunction in Parkinson’s Disease with α-Synucleinopathy. Cell Transplant. 2018, 27, 831–839. [Google Scholar] [CrossRef]
- Zhu, Z.J.; Wu, K.C.; Yung, W.H.; Qian, Z.M.; Ke, Y. Differential interaction between iron and mutant alpha-synuclein causes distinctive Parkinsonian phenotypes in Drosophila. Biochim. Biophys. Acta 2016, 1862, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Ostrerova-Golts, N.; Petrucelli, L.; Hardy, J.; Lee, J.M.; Farer, M.; Wolozin, B. The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci. 2000, 20, 6048–6054. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, A.; Jia, W.; Kawahata, I.; Fukunaga, K. Impact of Fatty Acid-Binding Proteins in α-Synuclein-Induced Mitochondrial Injury in Synucleinopathy. Biomedicines 2021, 9, 560. https://doi.org/10.3390/biomedicines9050560
Cheng A, Jia W, Kawahata I, Fukunaga K. Impact of Fatty Acid-Binding Proteins in α-Synuclein-Induced Mitochondrial Injury in Synucleinopathy. Biomedicines. 2021; 9(5):560. https://doi.org/10.3390/biomedicines9050560
Chicago/Turabian StyleCheng, An, Wenbin Jia, Ichiro Kawahata, and Kohji Fukunaga. 2021. "Impact of Fatty Acid-Binding Proteins in α-Synuclein-Induced Mitochondrial Injury in Synucleinopathy" Biomedicines 9, no. 5: 560. https://doi.org/10.3390/biomedicines9050560
APA StyleCheng, A., Jia, W., Kawahata, I., & Fukunaga, K. (2021). Impact of Fatty Acid-Binding Proteins in α-Synuclein-Induced Mitochondrial Injury in Synucleinopathy. Biomedicines, 9(5), 560. https://doi.org/10.3390/biomedicines9050560