
Using Pharmacogenetics of Direct Oral Anticoagulants to Predict Changes in Their Pharmacokinetics and the Risk of Adverse Drug Reactions

 ,
,  , ,
, , 
Abstract
1. Introduction
2. Dabigatran
3. Rivaroxaban
4. Apixaban
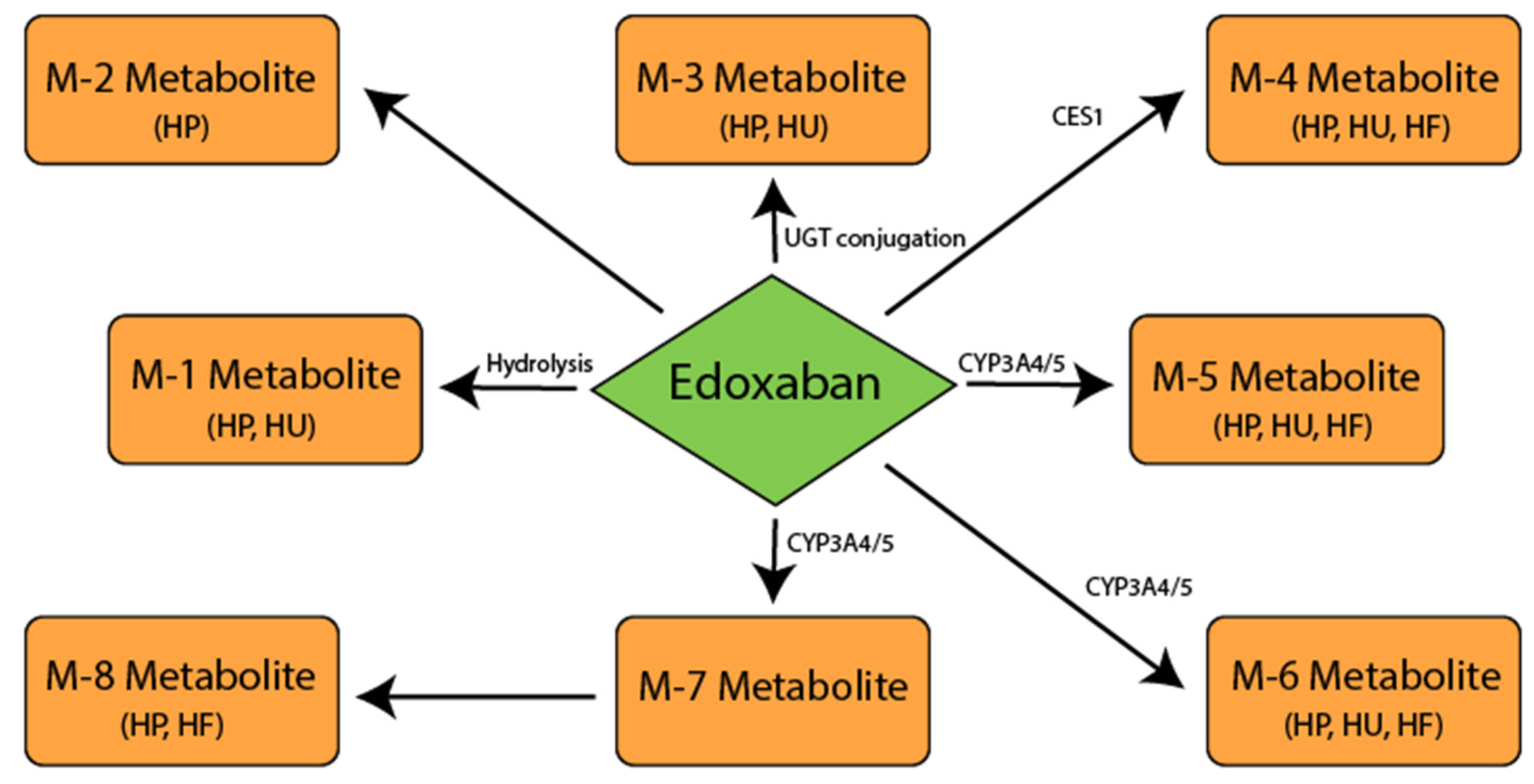
5. Edoxaban
6. Discussion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heit, J.A. Venous thromboembolism: Disease burden, outcomes and risk factors. J. Thromb. Haemost. 2005, 3, 1611–1617. [Google Scholar] [CrossRef]
- Feigin, V.L.; Lawes, C.M.; A Bennett, D.; Barker-Collo, S.L.; Parag, V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: A systematic review. Lancet Neurol. 2009, 8, 355–369. [Google Scholar] [CrossRef]
- Falck-Ytter, Y.; Francis, C.W.; Johanson, N.A.; Curley, C.; Dahl, O.E.; Schulman, S.; Ortel, T.L.; Pauker, S.G.; Colwell, C.W. Prevention of VTE in Orthopedic Surgery Patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141 (Suppl. 2), e278S–e325S. [Google Scholar] [CrossRef]
- Pedersen, A.B.; Mehnert, F.; Sorensen, H.T.; Emmeluth, C.; Overgaard, S.; Johnsen, S.P. The risk of venous thromboembolism, myocardial infarction, stroke, major bleeding and death in patients undergoing total hip and knee replacement. Bone Jt. J. 2014, 96-B, 479–485. [Google Scholar] [CrossRef]
- Ezekowitz, M.D.; Bridgers, S.L.; James, K.E.; Carliner, N.H.; Colling, C.L.; Gornick, C.C.; Krause-Steinrauf, H.; Kurtzke, J.F.; Nazarian, S.M.; Radford, M.J.; et al. Warfarin in the Prevention of Stroke Associated with Nonrheumatic Atrial Fibrillation. N. Engl. J. Med. 1992, 327, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Sors, H.; Charbonnier, B.; Page, Y.; Laaban, J.-P.; Azarian, R.; Laurent, M.; Hirsch, J.-L.; Ferrari, E.; Bosson, J.-L.; et al. A Comparison of Low-Molecular-Weight Heparin with Unfractionated Heparin for Acute Pulmonary Embolism. The THESEE Study Group. Tinzaparine ou Heparine Standard: Evaluations dans l’Embolie Pulmonaire. N. Engl. J. Med. 1997, 337, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Holford, N.H.G. Clinical Pharmacokinetics and Pharmacodynamics of Warfarin. Understanding the dose-effect relationship. Clin. Pharmacokinet. 1986, 11, 483–504. [Google Scholar] [CrossRef]
- Shendre, A.; Dillon, C.; Beasley, T.M.; Parmar, G.M.; A Limdi, N. Influence of Age on Warfarin Dose, Anticoagulation Control, and Risk of Hemorrhage. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2018, 38, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Shatzel, J.J.; Daughety, M.M.; Prasad, V.; Deloughery, T.G. Reversal of warfarin era thinking. J. Intern. Med. 2017, 283, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Barnes, G.D. Predicting the Quality of Warfarin Therapy: Reframing the Question. Thromb. Haemost. 2019, 119, 509–511. [Google Scholar] [CrossRef]
- Wu, A.H. Pharmacogenomic-guided dosing for warfarin: Too little too late? Pers. Med. 2018, 15, 71–73. [Google Scholar] [CrossRef]
- Schulman, S.; Kearon, C.; Kakkar, A.K.; Mismetti, P.; Schellong, S.; Eriksson, H.; Baanstra, D.; Schnee, J.; Goldhaber, S.Z. Dabigatran versus Warfarin in the Treatment of Acute Venous Thromboembolism. N. Engl. J. Med. 2009, 361, 2342–2352. [Google Scholar] [CrossRef]
- Stangier, J.; Rathgen, K.; Stähle, H.; Gansser, D.; Roth, W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br. J. Clin. Pharmacol. 2007, 64, 292–303. [Google Scholar] [CrossRef]
- Hankey, G.J.; Eikelboom, J.W. Dabigatran Etexilate: A new oral thrombin inhibitor. Circulation 2011, 123, 1436–1450. [Google Scholar] [CrossRef]
- Goldsack, N.R.; Chambers, R.C.; Dabbagh, K.; Laurent, G.J. Molecules in focus Thrombin. Int. J. Biochem. Cell Biol. 1998, 30, 641–646. [Google Scholar] [CrossRef]
- Davie, E.W.; Kulman, J.D. An Overview of the Structure and Function of Thrombin. Semin. Thromb. Hemost. 2006, 32, 003–015. [Google Scholar] [CrossRef]
- Comin, J.; Kallmes, D. Dabigatran (Pradaxa). Am. J. Neuroradiol. 2012, 33, 426–428. [Google Scholar] [CrossRef]
- Gelosa, P.; Castiglioni, L.; Tenconi, M.; Baldessin, L.; Racagni, G.; Corsini, A.; Bellosta, S. Pharmacokinetic drug interactions of the non-vitamin K antagonist oral anticoagulants (NOACs). Pharmacol. Res. 2018, 135, 60–79. [Google Scholar] [CrossRef]
- Comuth, W.J.; Henriksen, L.Ø.; van de Kerkhof, D.; Husted, S.E.; Kristensen, S.D.; de Maat, M.P.; Münster, A.-M.B. Comprehensive characteristics of the anticoagulant activity of dabigatran in relation to its plasma concentration. Thromb. Res. 2018, 164, 32–39. [Google Scholar] [CrossRef]
- Antonijevic, N.M.; Zivkovic, I.D.; Jovanovic, L.M.; Matic, D.M.; Kocica, M.J.; Mrdovic, I.B.; Kanjuh, V.I.; Culafic, M.D. Dabigatran—Metabolism, Pharmacologic Properties and Drug Interactions. Curr. Drug Metab. 2017, 18, 622–635. [Google Scholar] [CrossRef]
- Fawzy, A.M.; Lip, G.Y.H. Pharmacokinetics and pharmacodynamics of oral anticoagulants used in atrial fibrillation. Expert Opin. Drug Metab. Toxicol. 2019, 15, 381–398. [Google Scholar] [CrossRef]
- Satoh, T.; Hosokawa, M. Structure, function and regulation of carboxylesterases. Chem. Interact. 2006, 162, 195–211. [Google Scholar] [CrossRef]
- Ghosh, S.; Natarajan, R. Cloning of the Human Cholesteryl Ester Hydrolase Promoter: Identification of Functional Peroxisomal Proliferator-Activated Receptor Responsive Elements. Biochem. Biophys. Res. Commun. 2001, 284, 1065–1070. [Google Scholar] [CrossRef]
- Shi, J.; Wang, X.; Nguyen, J.-H.; Bleske, B.E.; Liang, Y.; Liu, L.; Zhu, H.-J. Dabigatran etexilate activation is affected by the CES1 genetic polymorphism G143E (rs71647871) and gender. Biochem. Pharmacol. 2016, 119, 76–84. [Google Scholar] [CrossRef]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef]
- Gouin-Thibault, I.; Delavenne, X.; Blanchard, A.; Siguret, V.; Salem, J.E.; Narjoz, C.; Gaussem, P.; Beaune, P.; Funck-Brentano, C.; Azizi, M.; et al. Interindividual variability in dabigatran and rivaroxaban exposure: Contribution of ABCB 1 genetic polymorphisms and interaction with clarithromycin. J. Thromb. Haemost. 2017, 15, 273–283. [Google Scholar] [CrossRef]
- Bernier, M.; Lancrerot, S.L.; Rocher, F.; Van-Obberghen, E.K.; Olivier, P.; Lavrut, T.; Parassol-Girard, N.; Drici, M.D. Major bleed-ing events in octagenarians associated with drug interactions between dabigatran and P-gp inhibitors. J. Geriatr. Cardiol. 2019, 16, 806–811. [Google Scholar] [CrossRef]
- Daud, A.N.A.; Bergman, J.E.H.; Bakker, M.K.; Wang, H.; Kerstjens-Frederikse, W.S.; De Walle, H.E.K.; Groen, H.; Bos, J.H.J.; Hak, E.; Wilffert, B. P-Glycoprotein-Mediated Drug Interactions in Pregnancy and Changes in the Risk of Congenital Anomalies: A Case-Reference Study. Drug Saf. 2015, 38, 651–659. [Google Scholar] [CrossRef]
- Ebner, T.; Wagner, K.; Wienen, W. Dabigatran Acylglucuronide, the Major Human Metabolite of Dabigatran: In Vitro Formation, Stability, and Pharmacological Activity. Drug Metab. Dispos. 2010, 38, 1567–1575. [Google Scholar] [CrossRef]
- UniProt. UDP-glucuronosyltransferase 2B15. UniProt Knowledgebase. Available online: www.uniprot.org/uniprot/P54855 (accessed on 20 December 2020).
- Chung, J.; Cho, J.; Yu, K.-S.; Kim, J.; Jung, H.-R.; Lim, K.; Jang, I.-J.; Shin, S.-G. Effect of the genotype on the pharmacokinetics, pharmacodynamics, and drug interactions of intravenous lorazepam in healthy volunteers. Clin. Pharmacol. Ther. 2005, 77, 486–494. [Google Scholar] [CrossRef]
- Savinova, A.V.; Shnayder, N.A.; Petrova, M.M.; Nasyrova, R.F. Promising areas of research on the pharmacogenetics of dabigatran etexilate. Pharmacokinet. Pharm. 2020, 1, 35–41. [Google Scholar] [CrossRef]
- Paré, G.; Eriksson, N.; Lehr, T.; Connolly, S.; Eikelboom, J.; Ezekowitz, M.D.; Axelsson, T.; Haertter, S.; Oldgren, J.; Reilly, P.; et al. Genetic Determinants of Dabigatran Plasma Levels and Their Relation to Bleeding. Circulation 2013, 127, 1404–1412. [Google Scholar] [CrossRef]
- Dimatteo, C.; D’Andrea, G.; Vecchione, G.; Paoletti, O.; Cappucci, F.; Tiscia, G.L.; Buono, M.; Grandone, E.; Testa, S.; Margaglione, M. Pharmacogenetics of dabigatran etexilate interindividual variability. Thromb. Res. 2016, 144, 1–5. [Google Scholar] [CrossRef]
- He, X.; Hesse, L.M.; Hazarika, S.; Masse, G.; Harmatz, J.S.; Greenblatt, D.J.; Court, M.H. Evidence for oxazepam as aninvivoprobe of UGT2B15: Oxazepam clearance is reduced byUGT2B15D85Y polymorphism but unaffected byUGT2B17deletion. Br. J. Clin. Pharmacol. 2009, 68, 721–730. [Google Scholar] [CrossRef]
- Court, M.H.; Zhu, Z.; Masse, G.; Duan, S.X.; James, L.P.; Harmatz, J.S.; Greenblatt, D.J. Race, Gender, and Genetic Polymorphism Contribute to Variability in Acetaminophen Pharmacokinetics, Metabolism, and Protein-Adduct Concentrations in Healthy African-American and European-American Volunteers. J. Pharmacol. Exp. Ther. 2017, 362, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Savelyeva, M.I.; Urvantseva, I.A.; Ignatova, A.K.; Panchenko, J.S.; Poddubnaya, I.V. Pharmacogenetic features of the phase II bio-transformation of tamoxifen: A systematic review. Pharmacogenomics 2017, 1, 10–15. [Google Scholar]
- Ethell, B.T.; Anderson, G.D.; Burchell, B. The effect of valproic acid on drug and steroid glucuronidation by expressed human UDP-glucuronosyltransferases. Biochem. Pharmacol. 2003, 65, 1441–1449. [Google Scholar] [CrossRef]
- Stringer, F.; Ploeger, B.A.; De Jongh, J.; Scott, G.; Urquhart, R.; Karim, A.; Danhof, M. Evaluation of the Impact of UGT Polymorphism on the Pharmacokinetics and Pharmacodynamics of the Novel PPAR Agonist Sipoglitazar. J. Clin. Pharmacol. 2013, 53, 256–263. [Google Scholar] [CrossRef]
- Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.; Mueck, W.; Laux, V. Rivaroxaban: A New Oral Factor Xa Inhibitor. Arter. Thromb. Vasc. Biol. 2010, 30, 376–381. [Google Scholar] [CrossRef]
- Turpie, A.G. Oral, Direct Factor Xa Inhibitors in Development for the Prevention and Treatment of Thromboembolic Diseases. Arter. Thromb. Vasc. Biol. 2007, 27, 1238–1247. [Google Scholar] [CrossRef]
- Kubitza, D.; Becka, M.; Voith, B.; Zuehlsdorf, M.; Wensing, G. Safety, pharmacodynamics, and pharmacokinetics of single doses of BAY 59-7939, an oral, direct factor Xa inhibitor. Clin. Pharmacol. Ther. 2005, 78, 412–421. [Google Scholar] [CrossRef]
- Kubitza, D.; Becka, M.; Wensing, G.; Voith, B.; Zuehlsdorf, M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—An oral, direct Factor Xa inhibitor—After multiple dosing in healthy male subjects. Eur. J. Clin. Pharmacol. 2005, 61, 873–880. [Google Scholar] [CrossRef]
- Weinz, C.; Buetehorn, U.; Daehler, H.-P.; Kohlsdorfer, C.; Pleiss, U.; Sandmann, S.; Schlemmer, K.-H.; Schwarz, T.; Steinke, W. Pharmacokinetics of BAY 59-7939—An oral, direct Factor Xa inhibitor—In rats and dogs. Xenobiotica 2005, 35, 891–910. [Google Scholar] [CrossRef] [PubMed]
- Weinz, C.; Schwarz, T.; Kubitza, D.; Mueck, W.; Lang, D. Metabolism and Excretion of Rivaroxaban, an Oral, Direct Factor Xa Inhibitor, in Rats, Dogs, and Humans. Drug Metab. Dispos. 2009, 37, 1056–1064. [Google Scholar] [CrossRef]
- CHMP Assessment Report For Xarelto. Available online: https://www.ema.europa.eu/en/documents/assessment-report/xarelto-epar-public-assessment-report_en.pdf (accessed on 20 December 2020).
- Mueck, W.; Kubitza, D.; Becka, M. Co-administration of rivaroxaban with drugs that share its elimination pathways: Pharmacokinetic effects in healthy subjects. Br. J. Clin. Pharmacol. 2013, 76, 455–466. [Google Scholar] [CrossRef]
- Xie, Q.; Xiang, Q.; Mu, G.; Ma, L.; Chen, S.; Zhou, S.; Hu, K.; Zhang, Z.; Cui, Y.; Jiang, J. Effect of ABCB1 Genotypes on the Pharmacokinetics and Clinical Outcomes of New Oral Anticoagulants: A Systematic Review and Meta-analysis. Curr. Pharm. Des. 2018, 24, 3558–3565. [Google Scholar] [CrossRef]
- Sychev, D.; Minnigulov, R.; Bochkov, P.; Ryzhikova, K.; Yudina, I.; Lychagin, A.; Morozova, T. Effect of CYP3A4, CYP3A5, ABCB1 Gene Polymorphisms on Rivaroxaban Pharmacokinetics in Patients Undergoing Total Hip and Knee Replacement Surgery. High Blood Press. Cardiovasc. Prev. 2019, 26, 413–420. [Google Scholar] [CrossRef]
- Sennesael, A.-L.; Panin, N.; Vancraeynest, C.; Pochet, L.; Spinewine, A.; Haufroid, V.; Elens, L. Effect of ABCB1 genetic polymorphisms on the transport of rivaroxaban in HEK293 recombinant cell lines. Sci. Rep. 2018, 8, 1–6. [Google Scholar] [CrossRef]
- Kanuri, S.H.; Kreutz, R.P. Pharmacogenomics of Novel Direct Oral Anticoagulants: Newly Identified Genes and Genetic Variants. J. Pers. Med. 2019, 9, 7. [Google Scholar] [CrossRef]
- Cusatis, G.; Sparreboom, A. Pharmacogenomic importance of ABCG2. Pharmacogenomics 2008, 9, 1005–1009. [Google Scholar] [CrossRef]
- Cusatis, G.; Gregorc, V.; Li, J.; Spreafico, A.; Ingersoll, R.G.; Verweij, J.; Ludovini, V.; Villa, E.; Hidalgo, M.; Sparreboom, A.; et al. Pharmacogenetics of ABCG2 and Adverse Reactions to Gefitinib. J. Natl. Cancer Inst. 2006, 98, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Tukaye, D.N.; Cui, J.; Greenwell, P.; Constantoulakis, L.M.; Parker, B.S.; Rao, A.; Köttgen, M.; Maloney, P.C.; Guggino, W.B. Gout-causing Q141K mutation in ABCG2 leads to instability of the nucleotide-binding domain and can be corrected with small molecules. Proc. Natl. Acad. Sci. USA 2013, 110, 5223–5228. [Google Scholar] [CrossRef]
- O’Connor, C.T.; Kiernan, T.J.; Yan, B.P. The genetic basis of antiplatelet and anticoagulant therapy: A pharmacogenetic review of newer antiplatelets (clopidogrel, prasugrel and ticagrelor) and anticoagulants (dabigatran, rivaroxaban, apixaban and edoxaban). Expert Opin. Drug Metab. Toxicol. 2017, 13, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Solus, J.F.; Arietta, B.J.; Harris, J.R.; Sexton, D.P.; Steward, J.Q.; McMUNN, C.; Ihrie, P.; Mehall, J.M.; Edwards, T.L.; Dawson, E.P. Genetic variation in eleven phase I drug metabolism genes in an ethnically diverse population. Pharmacogenomics 2004, 5, 895–931. [Google Scholar] [CrossRef]
- Galgani, A.; Palleria, C.; Iannone, L.F.; De Sarro, G.; Giorgi, F.S.; Maschio, M.; Russo, E. Pharmacokinetic Interactions of Clinical Interest Between Direct Oral Anticoagulants and Antiepileptic Drugs. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef]
- Brings, A.; Lehmann, M.; Foerster, K.I.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Czock, D. Perpetrator effects of ciclosporin (P-glycoprotein inhibitor) and its combination with fluconazole (CYP3A inhibitor) on the pharmacokinetics of rivaroxaban in healthy volunteers. Br. J. Clin. Pharmacol. 2019, 85, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Harskamp, R.E.; Teichert, M.; Lucassen, W.A.M.; Van Weert, H.C.P.M.; Lopes, R.D. Impact of Polypharmacy and P-Glycoprotein- and CYP3A4-Modulating Drugs on Safety and Efficacy of Oral Anticoagulation Therapy in Patients with Atrial Fibrillation. Cardiovasc. Drugs Ther. 2019, 33, 615–623. [Google Scholar] [CrossRef]
- Wu, S.-N.; Zhang, Y.; Gardner, C.O.; Chen, Q.; Li, Y.; Wang, G.-L.; Gao, P.-J.; Zhu, D.-L. Evidence for Association of Polymorphisms in CYP2J2 and Susceptibility to Essential Hypertension. Ann. Hum. Genet. 2007, 71, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Luettgen, J.M.; Knabb, R.M.; He, K.; Pinto, D.J.P.; Rendina, A.R. Apixaban inhibition of factor Xa: Microscopic rate constants and inhibition mechanism in purified protein systems and in human plasma. J. Enzym. Inhib. Med. Chem. 2010, 26, 514–526. [Google Scholar] [CrossRef]
- Ansell, J. Factor Xa or thrombin: Is factor Xa a better target? J. Thromb. Haemost. 2007, 5, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Crain, E.J.; Luettgen, J.M.; Schumacher, W.A.; Wong, P.C. Apixaban, an oral direct factor Xa inhibitor, inhibits human clot-bound factor Xa activity in vitro. Thromb. Haemost. 2009, 101, 780–782. [Google Scholar] [CrossRef]
- Savinova, A.V.; Petrova, M.M.; Shnayder, N.A.; Bochanova, E.N.; Nasyrova, R.F. Pharmacokinetics and Pharmacogenetics of Apixaban. Ration. Pharmacother. Cardiol. 2020, 16, 852–860. [Google Scholar] [CrossRef]
- Byon, W.; Nepal, S.; Schuster, A.E.; Shenker, A.; Frost, C.E. Regional Gastrointestinal Absorption of Apixaban in Healthy Subjects. J. Clin. Pharmacol. 2018, 58, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Vakkalagadda, B.; Frost, C.; Byon, W.; Boyd, R.A.; Wang, J.; Zhang, D.; Yu, Z.; Dias, C.; Shenker, A.; LaCreta, F. Effect of Rifampin on the Pharmacokinetics of Apixaban, an Oral Direct Inhibitor of Factor Xa. Am. J. Cardiovasc. Drugs 2016, 16, 119–127. [Google Scholar] [CrossRef]
- Raghavan, N.; Frost, C.E.; Yu, Z.; He, K.; Zhang, H.; Humphreys, W.G.; Pinto, D.; Chen, S.; Bonacorsi, S.; Wong, P.C.; et al. Apixaban Metabolism and Pharmacokinetics after Oral Administration to Humans. Drug Metab. Dispos. 2008, 37, 74–81. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, D.; Raghavan, N.; Yao, M.; Nirmala, R.; Frost, C.A.; Maxwell, B.D.; Chen, S.-Y.; He, K.; Goosen, T.C.; et al. In Vitro Assessment of Metabolic Drug-Drug Interaction Potential of Apixaban through Cytochrome P450 Phenotyping, Inhibition, and Induction Studies. Drug Metab. Dispos. 2009, 38, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; He, K.; Herbst, J.J.; Kolb, J.; Shou, W.; Wang, L.; Balimane, P.V.; Han, Y.-H.; Gan, J.; Frost, C.E.; et al. Characterization of Efflux Transporters Involved in Distribution and Disposition of Apixaban. Drug Metab. Dispos. 2013, 41, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Frost, C.; Wang, J.; Nepal, S.; Schuster, A.; Barrett, Y.C.; Mosqueda-Garcia, R.; Reeves, R.A.; LaCreta, F. Apixaban, an oral, direct factor X a inhibitor: Single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br. J. Clin. Pharmacol. 2013, 75, 476–487. [Google Scholar] [CrossRef]
- Frost, C.; Nepal, S.; Wang, J.; Schuster, A.; Byon, W.; Boyd, R.A.; Yu, Z.; Shenker, A.; Barrett, Y.C.; Mosqueda-Garcia, R.; et al. Safety, pharmacokinetics and pharmacodynamics of multiple oral doses of apixaban, a factor X a inhibitor, in healthy subjects. Br. J. Clin. Pharmacol. 2013, 76, 776–786. [Google Scholar] [CrossRef]
- He, K.; Luettgen, J.M.; Zhang, D.; He, B.; Grace, J.E.; Xin, B.; Pinto, D.J.P.; Wong, P.C.; Knabb, R.M.; Lam, P.Y.S.; et al. Preclinical pharmacokinetics and pharmacodynamics of apixaban, a potent and selective factor Xa inhibitor. Eur. J. Drug Metab. Pharmacokinet. 2011, 36, 129–139. [Google Scholar] [CrossRef]
- Wang, X.; Mondal, S.; Wang, J.; Tirucherai, G.; Zhang, D.; Boyd, R.A.; Frost, C. Effect of Activated Charcoal on Apixaban Pharmacokinetics in Healthy Subjects. Am. J. Cardiovasc. Drugs 2014, 14, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Ueshima, S.; Hira, D.; Fujii, R.; Kimura, Y.; Tomitsuka, C.; Yamane, T.; Tabuchi, Y.; Ozawa, T.; Itoh, H.; Horie, M.; et al. Impact of ABCB1, ABCG2, and CYP3A5 polymorphisms on plasma trough concentrations of apixaban in Japanese patients with atrial fibrillation. Pharm. Genom. 2017, 27, 329–336. [Google Scholar] [CrossRef]
- SNPedia. CYP3A5. Available online: https://www.snpedia.com/index.php/CYP3A5 (accessed on 20 December 2020).
- Kang, R.-H.; Jung, S.-M.; Kim, K.-A.; Lee, D.-K.; Cho, H.-K.; Jung, B.-J.; Kim, Y.-K.; Kim, S.-H.; Han, C.; Lee, M.-S.; et al. Effects of CYP2D6 and CYP3A5 Genotypes on the Plasma Concentrations of Risperidone and 9-Hydroxyrisperidone in Korean Schizophrenic Patients. J. Clin. Psychopharmacol. 2009, 29, 272–277. [Google Scholar] [CrossRef]
- Canonico, M.; Bouaziz, E.; Carcaillon, L.; Verstuyft, C.; Guiochon-Mantel, A.; Becquemont, L.; Scarabin, P.-Y. Synergism between Oral Estrogen Therapy and Cytochrome P450 3A5*1 Allele on the Risk of Venous Thromboembolism among Postmenopausal Women. J. Clin. Endocrinol. Metab. 2008, 93, 3082–3087. [Google Scholar] [CrossRef] [PubMed]
- SNPedia. CYP1A2. Available online: https://www.snpedia.com/index.php/CYP1A2 (accessed on 20 December 2020).
- Sweezy, T.; A Mousa, S. Genotype-guided use of oral antithrombotic therapy: A pharmacoeconomic perspective. Pers. Med. 2014, 11, 223–235. [Google Scholar] [CrossRef]
- Carlini, E.J.; Raftogianis, R.B.; Wood, T.C.; Jin, F.; Zheng, W.; Rebbeck, T.R.; Weinshilboum, R.M. Sulfation pharmacogenetics:SULT1A1 and SULT1A2 allele frequencies in Caucasian, Chinese and African-American subjects. Pharmacogenetics 2001, 11, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Raghavan, N.; He, K.; Luettgen, J.M.; Humphreys, W.G.; Knabb, R.M.; Pinto, D.J.; Zhang, D. Sulfation of O-Demethyl Apixaban: Enzyme Identification and Species Comparison. Drug Metab. Dispos. 2009, 37, 802–808. [Google Scholar] [CrossRef]
- Nagar, S.; Walther, S.; Blanchard, R.L. Sulfotransferase (SULT) 1A1 Polymorphic Variants *1, *2, and *3 Are Associated with Altered Enzymatic Activity, Cellular Phenotype, and Protein Degradation. Mol. Pharmacol. 2006, 69, 2084–2092. [Google Scholar] [CrossRef]
- Raftogianis, R.B.; Wood, T.C.; Otterness, D.M.; Van Loon, J.A.; Weinshilboum, R.M. Phenol Sulfotransferase Pharmacogenetics in Humans: Association of CommonSULT1A1Alleles with TS PST Phenotype. Biochem. Biophys. Res. Commun. 1997, 239, 298–304. [Google Scholar] [CrossRef]
- Fuji, T.; Fujita, S.; Kawai, Y.; Nakamura, M.; Kimura, T.; Fukuzawa, M.; Abe, K.; Tachibana, S. Efficacy and safety of edoxaban versus enoxaparin for the prevention of venous thromboembolism following total hip arthroplasty: STARS J-V. Thromb. J. 2015, 13, 1–9. [Google Scholar] [CrossRef]
- Fuji, T.; Wang, C.-J.; Fujita, S.; Kawai, Y.; Kimura, T.; Tachibana, S. Safety and Efficacy of Edoxaban, an Oral Factor Xa Inhibitor, for Thromboprophylaxis After Total Hip Arthroplasty in Japan and Taiwan. J. Arthroplast. 2014, 29, 2439–2446. [Google Scholar] [CrossRef]
- Fuji, T.; Fujita, S.; Kawai, Y.; Nakamura, M.; Kimura, T.; Kiuchi, Y.; Abe, K.; Tachibana, S. Safety and ef-ficacy of edoxaban in pa-tients undergoing hip fracture surgery. Thromb Res. 2014, 133, 1016–1022. [Google Scholar] [CrossRef]
- Samama, M.M.; Mendell, J.; Guinet, C.; Le Flem, L.; Kunitada, S. In vitro study of the anticoagulant effects of edoxaban and its effect on thrombin generation in comparison to fondaparinux. Thromb. Res. 2012, 129, e77–e82. [Google Scholar] [CrossRef] [PubMed]
- Ogata, K.; Mendell-Harary, J.; Tachibana, M.; Masumoto, H.; Oguma, T.; Kojima, M.; Kunitada, S. Clinical Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Novel Factor Xa Inhibitor Edoxaban in Healthy Volunteers. J. Clin. Pharmacol. 2010, 50, 743–753. [Google Scholar] [CrossRef]
- Matsushima, N.; Lee, F.; Sato, T.; Weiss, D.; Mendell, J. Bioavailability and Safety of the Factor Xa Inhibitor Edoxaban and the Effects of Quinidine in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2013, 2, 358–366. [Google Scholar] [CrossRef]
- Parasrampuria, D.A.; Kanamaru, T.; Connor, A.; Wilding, I.; Ogata, K.; Shimoto, Y.; Kunitada, S. Evaluation of regional gastrointestinal absorption of edoxaban using the enterion capsule. J. Clin. Pharmacol. 2015, 55, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Parasrampuria, D.A.; Truitt, K.E. Pharmacokinetics and Pharmacodynamics of Edoxaban, a Non-Vitamin K Antagonist Oral Anticoagulant that Inhibits Clotting Factor Xa. Clin. Pharmacokinet. 2016, 55, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Bathala, M.S.; Masumoto, H.; Oguma, T.; He, L.; Lowrie, C.; Mendell, J. Pharmacokinetics, Biotransformation, and Mass Balance of Edoxaban, a Selective, Direct Factor Xa Inhibitor, in Humans. Drug Metab. Dispos. 2012, 40, 2250–2255. [Google Scholar] [CrossRef]
- Daiichi Sankyo, Inc. Savaysa (Edoxabantosylate): FDA Cardiovascular and Renal Drugs Adviso-ry Committee Briefing Document. NDA 206316. Available online: http://www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/ucm420703.htm (accessed on 20 December 2020).
- Jönsson, S.; Simonsson, U.S.H.; Miller, R.; Karlsson, M.O. Population pharmacokinetics of edoxaban and its main metabolite in a dedicated renal impairment study. J. Clin. Pharmacol. 2015, 55, 1268–1279. [Google Scholar] [CrossRef] [PubMed]
- Mikkaichi, T.; Yoshigae, Y.; Masumoto, H.; Imaoka, T.; Rozehnal, V.; Fischer, T.; Okudaira, N.; Izumi, T. Edoxaban Transport via P-Glycoprotein Is a Key Factor for the Drug’s Disposition. Drug Metab. Dispos. 2014, 42, 520–528. [Google Scholar] [CrossRef]
- FDA Center for Drug Evaluation Research. FDA Draft Guidance for Industry: Drug Interaction Studies—Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. Available online: http://www.fda.gov/downloads/Drugs/Guidances/ucm292362.pdf (accessed on 20 December 2020).
- Flockhart, D.A. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine. Available online: https://static.medicine.iupui.edu/divisions/clinpharm/content/p450_Table_Oct_11_2009.pdf (accessed on 20 December 2020).
- Parasrampuria, D.A.; Mendell, J.; Shi, M.; Matsushima, N.; Zahir, H.; Truitt, K. Edoxaban drug–drug interactions with ketoconazole, erythromycin, and cyclosporine. Br. J. Clin. Pharmacol. 2016, 82, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.; Zahir, H.; Matsushima, N.; Noveck, R.; Lee, F.; Chen, S.; Zhang, G.; Shi, M. Drug-Drug Interaction Studies of Cardiovascular Drugs Involving P-Glycoprotein, an Efflux Transporter, on the Pharmacokinetics of Edoxaban, an Oral Factor Xa Inhibitor. Am. J. Cardiovasc. Drugs 2013, 13, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Cordarone®. (Amiodarone HCl) Tablets: Full Prescribing Information. Wyeth Pharmaceuticals Inc., a Subsidiary of Pfizer Inc.; Philadelphia. Available online: http://labeling.pfizer.com/showlabeling.aspx?id=93 (accessed on 20 December 2020).
- Mendell, J.; Chen, S.; He, L.; Desai, M.; Parasramupria, D.A. The Effect of Rifampin on the Pharmacokinetics of Edoxaban in Healthy Adults. Clin. Drug Investig. 2015, 35, 447–453. [Google Scholar] [CrossRef]
- Mendell, J.; Lee, F.; Chen, S.; Worland, V.; Shi, M.; Samama, M.M. The Effects of the Antiplatelet Agents, Aspirin and Naproxen, on Pharmacokinetics and Pharmacodynamics of the Anticoagulant Edoxaban, a Direct Factor Xa Inhibitor. J. Cardiovasc. Pharmacol. 2013, 62, 212–221. [Google Scholar] [CrossRef]
- Matsushima, N.; Halim, A.-B.; He, L.; Zhang, G.; Lee, F.; Worland, V.; Mendell, J.; Zahir, H. Edoxaban administration following enoxaparin: A pharmacodynamic, pharmacokinetic, and tolerability assessment in human subjects. Thromb. Haemost. 2012, 108, 166–175. [Google Scholar] [CrossRef]
- Vandell, A.G.; Lee, J.; Shi, M.; Rubets, I.; Brown, K.S.; Walker, J.R. An integrated pharmacokinetic/pharmacogenomic analysis of ABCB1 and SLCO1B1 polymorphisms on edoxaban exposure. Pharm. J. 2016, 18, 153–159. [Google Scholar] [CrossRef]
- Bounameaux, H.; Camm, A.J. Edoxaban: An Update on the New Oral Direct Factor Xa Inhibitor. Drugs 2014, 74, 1209–1231, Erratum in: Drugs 2014, 74, 1455. [Google Scholar] [CrossRef] [PubMed]
- Stangier, J. Clinical Pharmacokinetics and Pharmacodynamics of the Oral Direct Thrombin??Inhibitor Dabigatran Etexilate. Clin. Pharmacokinet. 2008, 47, 285–295. [Google Scholar] [CrossRef]
- Umamaheswaran, G.; Kumar, D.K.; Adithan, C. Distribution of genetic polymorphisms of genes en-coding drug metabolizing enzymes & drug transporters—A review with Indian perspective. Indian J. Med. Res. 2014, 139, 27–65. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Direct Oral Anticoagulants (DOACs) | Drugs That Increase DOACs’ Concentration in the Serum | Drugs That Decrease DOACs’ Concentration in the Serum |
|---|---|---|
| Apixaban | Olanzapine Tamoxifen Irinotecan Docetaxel Vincristine Meflohin Artemether Lumefantrine Tacrolimus Cyclosporine Chlorphenamine Terfenadine Astemizole Clopidgrel Nifedipine Amlodipine Felodipine Verapamil Indinavir Nelfinavir Ritonavir Saquinavir Atorvastatin Cerivastatin Clarothromycin Testosterone Estradiol Progesterone | Pifampicin |
| Dabigatran etexilate | Ketoconazole Chinidine | Rifampicin |
| Rivaroxaban | n/a | Rifampicin Phenytoine Carbamazepine Phenobarbital |
| Edoxaban | Amiodarone Ketoconazole Chinidine Erythromycin Cyclosporine | n/a |
| Drugs | Enzymes Involved in the Metabolism of DOACs | Candidate Genes Involved in the Metabolism of DOACs |
|---|---|---|
| Apixaban | Isoenzyme 3A4/3A5 of the hepatic cytochrome 450 Sulfotransferase 1 | CYP3A4/CYP2A5 SULT1A1 |
| Dabigatran etexilate | Isoenzymes 1A2, 2C8, 2C9, 2C19 and 2J2 of the hepatic cytochrome 450 | CYP1A2, CYP2C8, CYP2C9, CYPC19, CYP2J2 |
| Rivaroxaban | Sulfotransferase 1 | CES1 |
| Edoxaban | Isoenzymes 3A4/3A5 and 2J2 of the hepatic cytochrome 450 | CYP3A4/CYP3A5, CYP2J2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shnayder, N.A.; Petrova, M.M.; Shesternya, P.A.; Savinova, A.V.; Bochanova, E.N.; Zimnitskaya, O.V.; Pozhilenkova, E.A.; Nasyrova, R.F. Using Pharmacogenetics of Direct Oral Anticoagulants to Predict Changes in Their Pharmacokinetics and the Risk of Adverse Drug Reactions. Biomedicines 2021, 9, 451. https://doi.org/10.3390/biomedicines9050451
Shnayder NA, Petrova MM, Shesternya PA, Savinova AV, Bochanova EN, Zimnitskaya OV, Pozhilenkova EA, Nasyrova RF. Using Pharmacogenetics of Direct Oral Anticoagulants to Predict Changes in Their Pharmacokinetics and the Risk of Adverse Drug Reactions. Biomedicines. 2021; 9(5):451. https://doi.org/10.3390/biomedicines9050451
Chicago/Turabian StyleShnayder, Natalia A., Marina M. Petrova, Pavel A. Shesternya, Alina V. Savinova, Elena N. Bochanova, Olga V. Zimnitskaya, Elena A. Pozhilenkova, and Regina F. Nasyrova. 2021. "Using Pharmacogenetics of Direct Oral Anticoagulants to Predict Changes in Their Pharmacokinetics and the Risk of Adverse Drug Reactions" Biomedicines 9, no. 5: 451. https://doi.org/10.3390/biomedicines9050451
APA StyleShnayder, N. A., Petrova, M. M., Shesternya, P. A., Savinova, A. V., Bochanova, E. N., Zimnitskaya, O. V., Pozhilenkova, E. A., & Nasyrova, R. F. (2021). Using Pharmacogenetics of Direct Oral Anticoagulants to Predict Changes in Their Pharmacokinetics and the Risk of Adverse Drug Reactions. Biomedicines, 9(5), 451. https://doi.org/10.3390/biomedicines9050451