
Post-Ischemic Treatment of Recombinant Human Secretory Leukocyte Protease Inhibitor (rhSLPI) Reduced Myocardial Ischemia/Reperfusion Injury


 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Animal and Ethical Approval
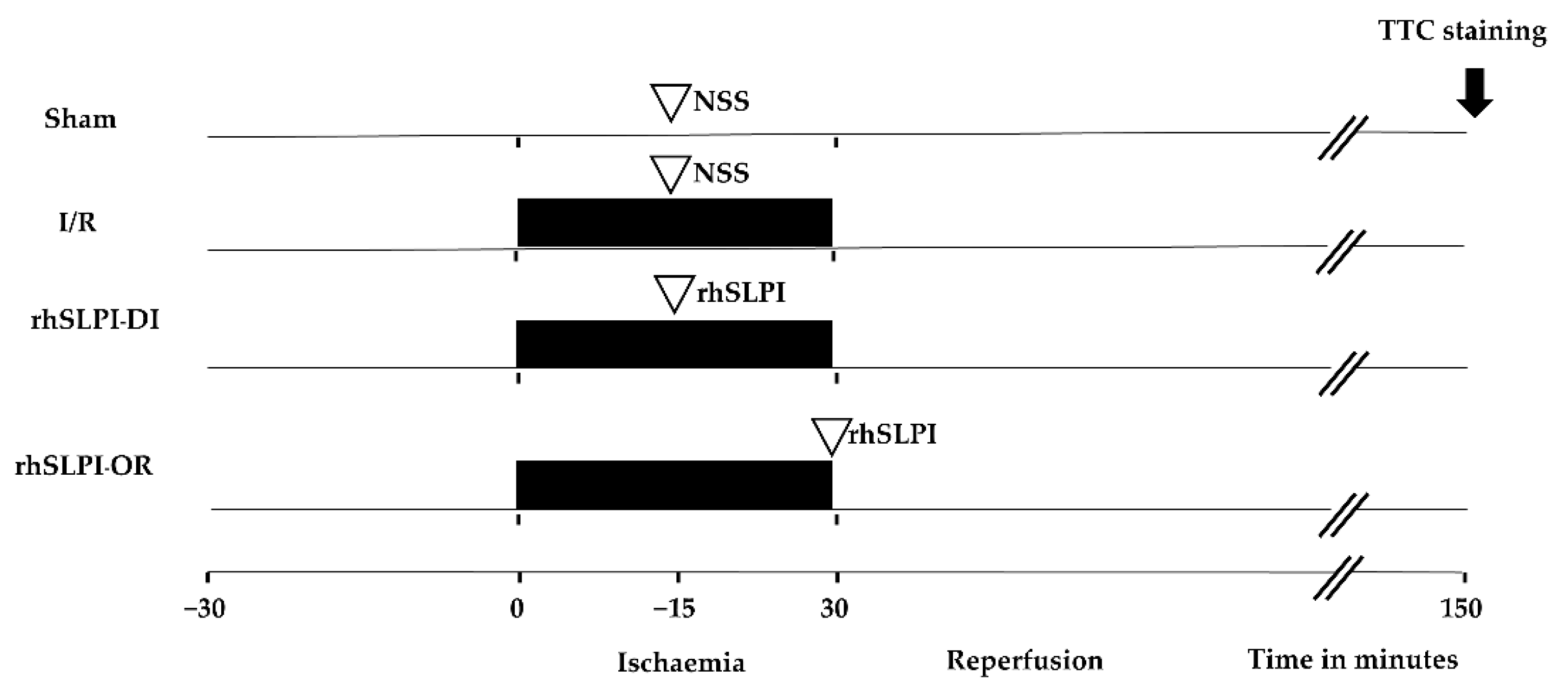
2.2. Experimental Groups
2.3. Surgical Preparation of Myocardial I/R Model in Rats
2.4. Evaluation of Heart Weight, Infarct Size and Area at Risk
2.5. Determination of Serum Creatine Kinase (MB Isoenzyme) and Lactate Dehydrogenase (LDH) Activity
2.6. Heart Tissue Homogenization and Protein Collection
2.7. Determination of Protein Concentration by Bradford Assay
2.8. Determination of Inflammatory Cytokines Level by Enzyme-Linked Immunosorbent Assay (ELISA)
2.9. Spectrophotometric Determination of Protein Carbonyl (PC) Content Level
2.10. Spectrophotometric Determination of Ischemia-Modified Albumin (IMA) Level
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Results
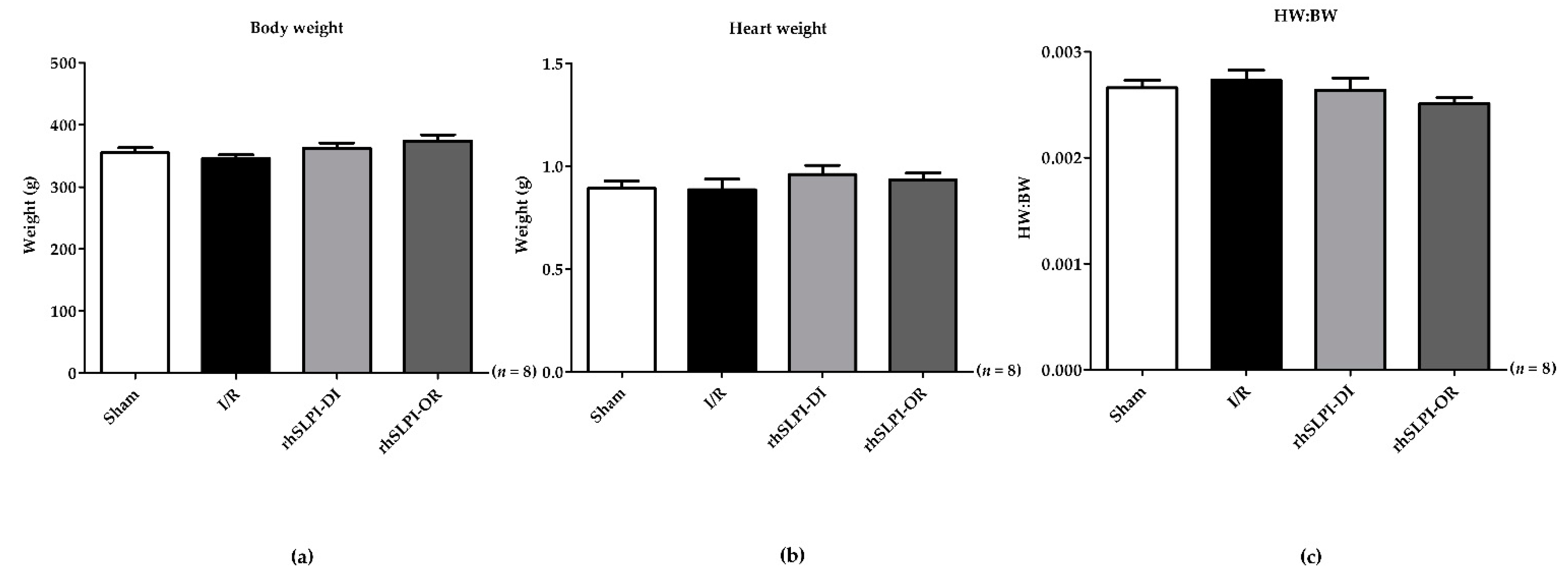
3.1. The Animal Body Weight and Heart Weight of Experimental Animals
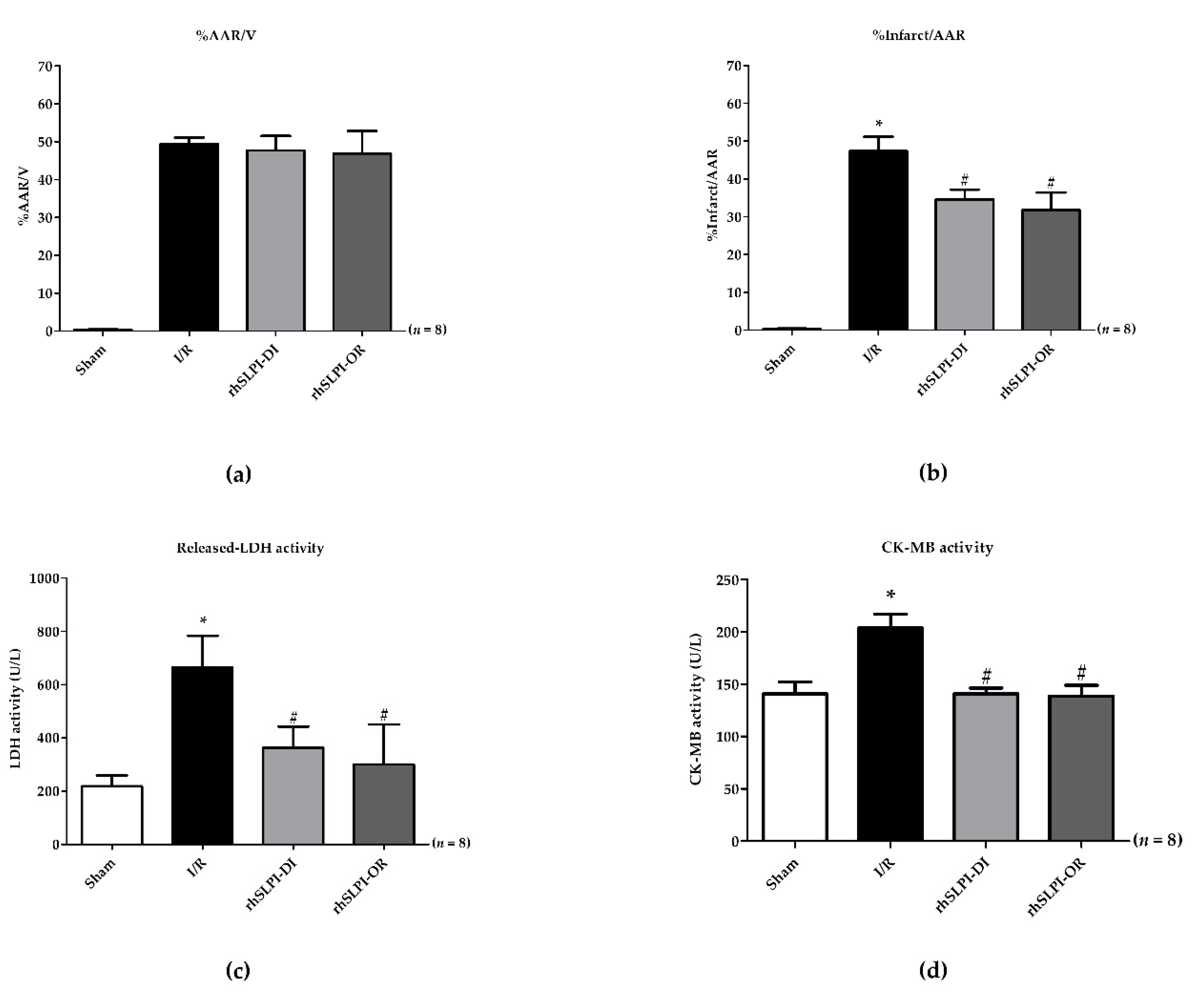
3.2. Effect of Post-Ischemic rhSLPI Treatment on Cardiac Biomarkers
3.3. Effect of rhSLPI Treatment on LVP in I/R Rat
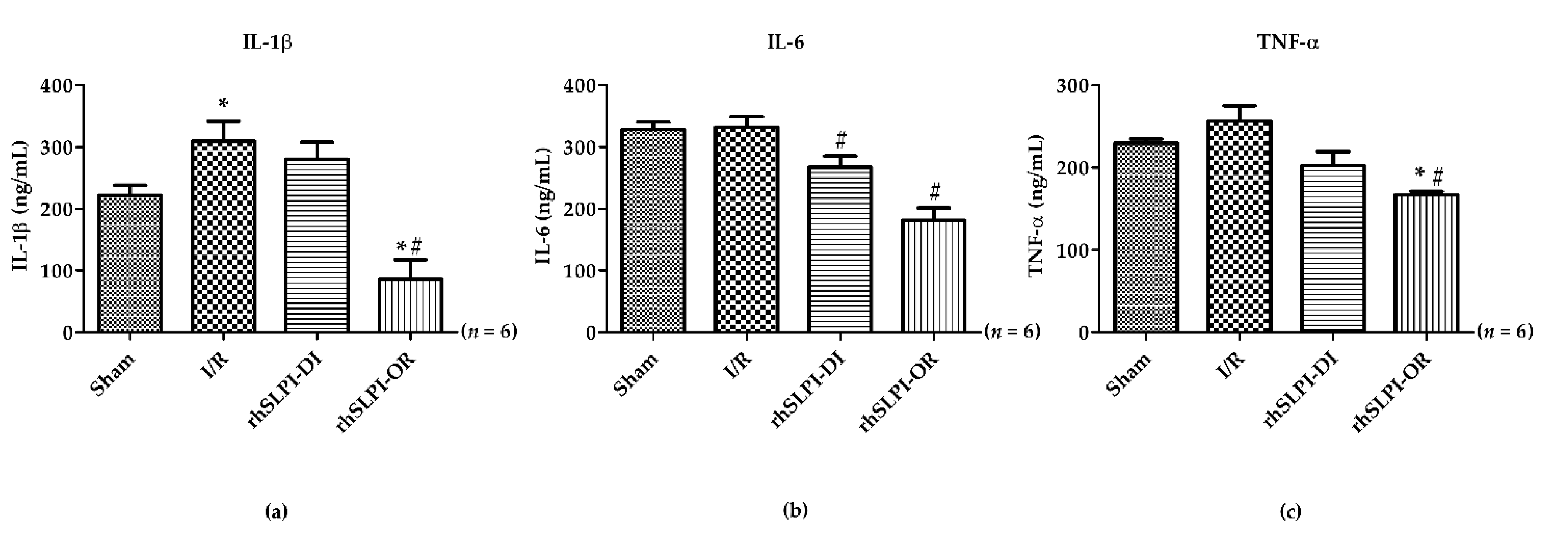
3.4. Effect of Post-Ischemic rhSLPI Treatment on Inflammatory Cytokines Level
3.5. Effect of Post-Ischemic rhSLPI Treatment on Oxidatively Modified Cardiac Proteins Level
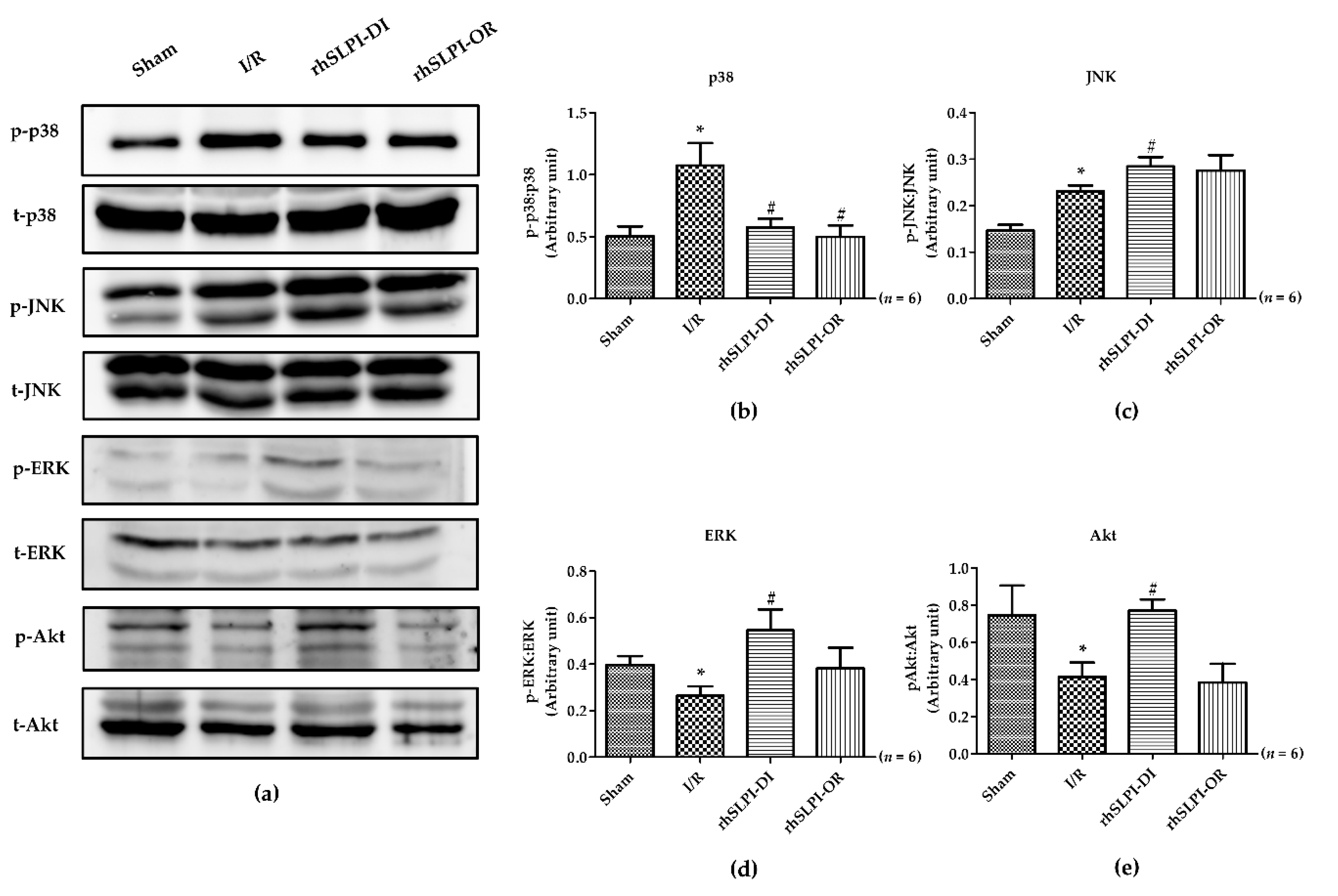
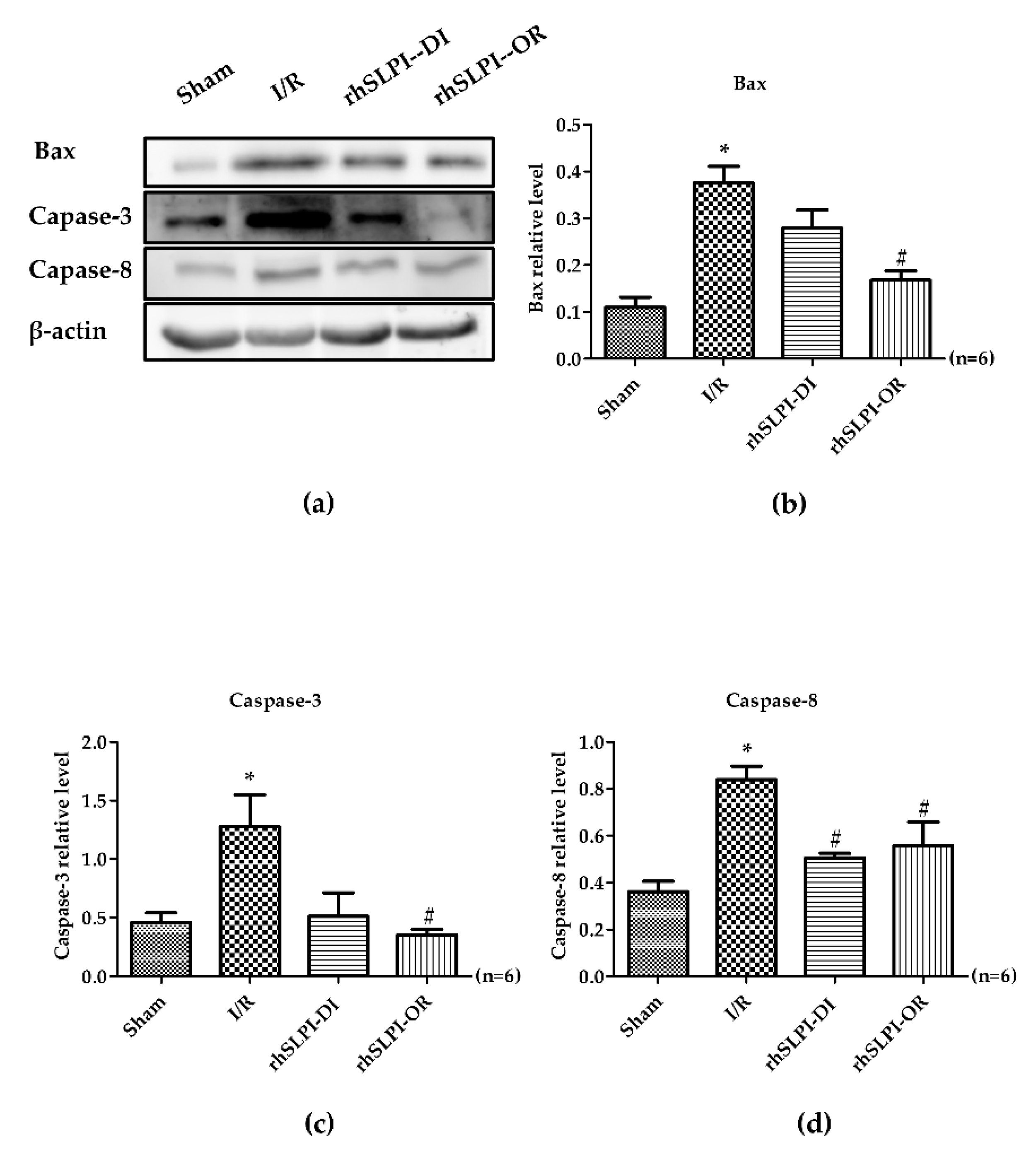
3.6. Effect of Post-Ischemic rhSLPI Treatment on Signal Transduction and Apoptosis Regulatory Molecules
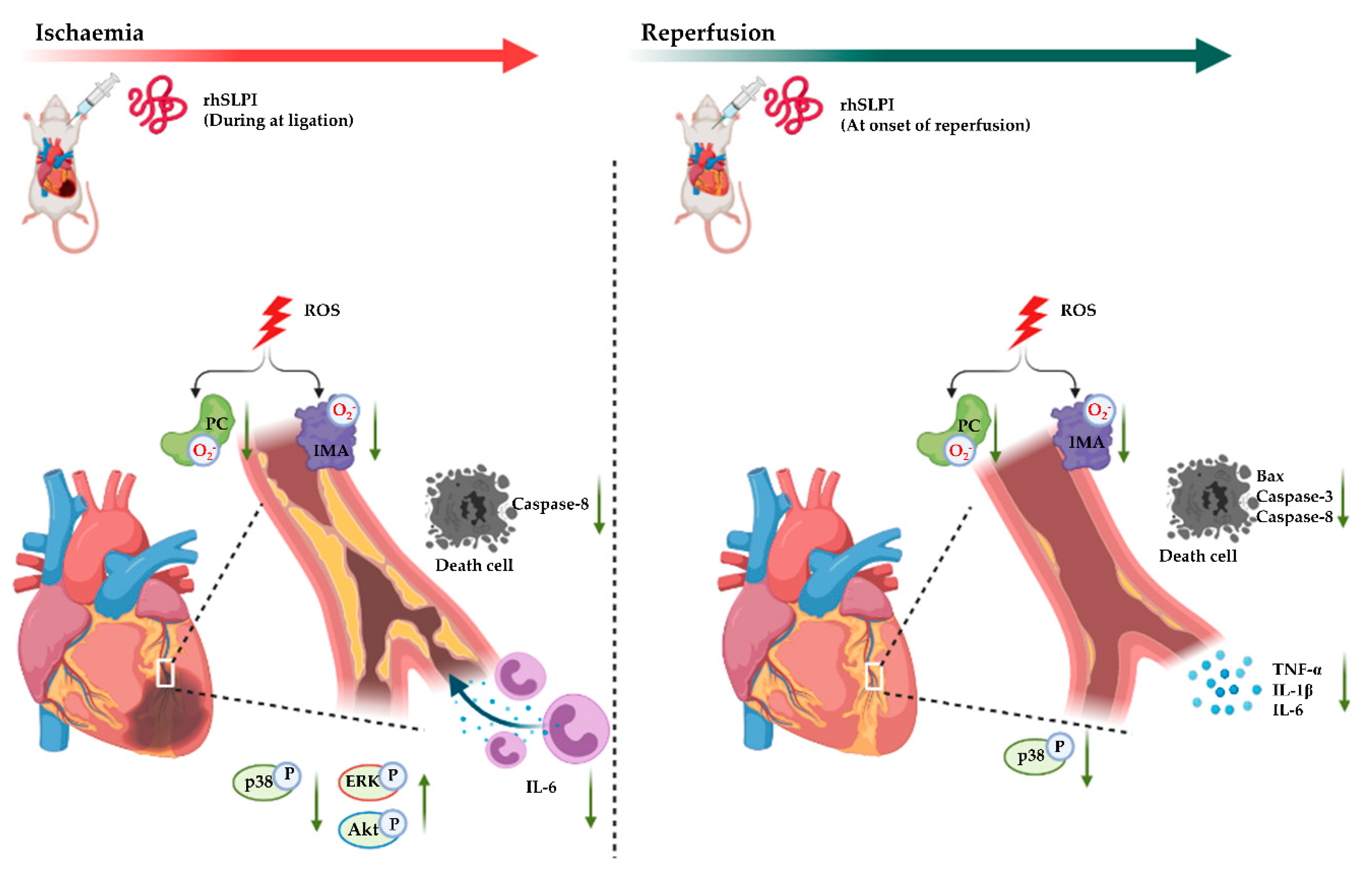
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [PubMed]
- Kongpol, K.; Yodsheewan, R.; Nernpermpisooth, N.; Kumphune, S. Recombinant human secretory leukocyte protease inhibitor ameliorated vessel preservation in experimentally isolated rat arteries. J. Appl. Pharm. Sci. 2020, 10. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002, 53, 31–47. [Google Scholar] [CrossRef]
- Zahler, S.; Massoudy, P.; Hartl, H.; Hähnel, C.; Meisner, H.; Becker, B.F. Acute cardiac inflammatory responses to postischemic reperfusion during cardiopulmonary bypass. Cardiovasc. Res. 1999, 41, 722–730. [Google Scholar] [CrossRef]
- Vinten-Johansen, J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 2004, 61, 481–497. [Google Scholar] [CrossRef]
- Müller, A.L.; Dhalla, N.S. Role of various proteases in cardiac remodeling and progression of heart failure. Heart Fail. Rev. 2012, 17, 395–409. [Google Scholar] [CrossRef]
- Stetler, G.; Brewer, M.T.; Thompson, R.C. Isolation and sequence of a human gene encoding a potent inhibitor of leukocyte proteases. Nucleic Acids Res. 1986, 14, 7883–7896. [Google Scholar] [CrossRef]
- Lentsch, A.B.; Jordan, J.A.; Czermak, B.J.; Diehl, K.M.; Younkin, E.M.; Sarma, V.; Ward, P.A. Inhibition of NF-kappaB activation and augmentation of IkappaBbeta by secretory leukocyte protease inhibitor during lung inflammation. Am. J. Pathol. 1999, 154, 239–247. [Google Scholar] [CrossRef]
- Doumas, S.; Kolokotronis, A.; Stefanopoulos, P. Anti-inflammatory and antimicrobial roles of secretory leukocyte protease inhibitor. Infect. Immun. 2005, 73, 1271–1274. [Google Scholar] [CrossRef]
- Wahl, S.M.; McNeely, T.B.; Janoff, E.N.; Shugars, D.; Worley, P.; Tucker, C.; Orenstein, J.M. Secretory leukocyte protease inhibitor (SLPI) in mucosal fluids inhibits HIV-I. Oral Dis. 1997, 3, S64–S69. [Google Scholar] [CrossRef] [PubMed]
- Gillissen, A.; Birrer, P.; McElvaney, N.G.; Buhl, R.; Vogelmeier, C.; Hoyt, R.F., Jr.; Hubbard, R.C.; Crystal, R.G. Recombinant secretory leukoprotease inhibitor augments glutathione levels in lung epithelial lining fluid. J. Appl. Physiol. 1993, 75, 825–832. [Google Scholar] [CrossRef] [PubMed]
- McGarry, N.; Greene, C.M.; McElvaney, N.G.; Weldon, S.; Taggart, C.C. The Ability of Secretory Leukocyte Protease Inhibitor to Inhibit Apoptosis in Monocytes Is Independent of Its Antiprotease Activity. J. Immunol. Res. 2015, 507315. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, S.; Hautz, T.; Wahl, S.M.; Brandacher, G.; Sucher, R.; Steinmassl, O.; Steinmassl, P.; Wright, C.D.; Obrist, P.; Werner, E.R.; et al. The effect of secretory leukocyte protease inhibitor (SLPI) on ischemia/reperfusion injury in cardiac transplantation. Am. J. Transpl. 2008, 8, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Prompunt, E.; Nernpermpisooth, N.; Sanit, J.; Kumphune, S. Overexpression and pre-treatment of recombinant human Secretory Leukocyte Protease Inhibitor (rhSLPI) reduces an in vitro ischemia/reperfusion injury in rat cardiac myoblast (H9c2) cell. Biomol. Concepts 2018, 9, 17–32. [Google Scholar] [CrossRef]
- Prompunt, E.; Sanit, J.; Barrère-Lemaire, S.; Nargeot, J.; Noordali, H.; Madhani, M.; Kumphune, S. The cardioprotective effects of secretory leukocyte protease inhibitor against myocardial ischemia/reperfusion injury. Exp. Med. 2018, 15, 5231–5242. [Google Scholar] [CrossRef] [PubMed]
- Nernpermpisooth, N.; Prompunt, E.; Kumphune, S. An in vitro endothelial cell protective effect of secretory leukocyte protease inhibitor against simulated ischaemia/reperfusion injury. Exp. Med. 2017, 14, 5793–5800. [Google Scholar] [CrossRef]
- Petchdee, S.; Laosripaiboon, W.; Jarussophon, N.; Kumphune, S. Cardio-Protective Effects of Germinated Brown Rice Extract Against Myocardial Ischemia Reperfusion Injury. High. Blood Press. Cardiovasc. Prev. 2020, 27, 251–258. [Google Scholar] [CrossRef]
- Kongpol, K.; Nernpermpisooth, N.; Prompunt, E.; Kumphune, S. Endothelial-Cell-Derived Human Secretory Leukocyte Protease Inhibitor (SLPI) Protects Cardiomyocytes against Ischemia/Reperfusion Injury. Biomolecules 2019, 9, 678. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Qin, G.; Jiang, W.; Zhao, Y.; Xu, Y.; Lv, X. 6-Gingerol Protects Heart by Suppressing Myocardial Ischemia/Reperfusion Induced Inflammation via the PI3K/Akt-Dependent Mechanism in Rats. Evid Based Complement. Altern. Med. 2018, 6209679. [Google Scholar] [CrossRef]
- Pantke, U.; Volk, T.; Schmutzler, M.; Kox, W.J.; Sitte, N.; Grune, T. Oxidized proteins as a marker of oxidative stress during coronary heart surgery. Free Radic. Biol. Med. 1999, 27, 1080–1086. [Google Scholar] [CrossRef]
- Maneewong, K.; Mekrungruangwong, T.; Luangaram, S.; Thongsri, T.; Kumphune, S. Combinatorial Determination of Ischemia Modified Albumin and Protein Carbonyl in the Diagnosis of NonST-Elevation Myocardial Infarction. Indian J. Clin. Biochem. 2011, 26, 389–395. [Google Scholar] [CrossRef]
- Oran, I.; Oran, B. Ischemia-Modified Albumin as a Marker of Acute Coronary Syndrome: The Case for Revising the Concept of “N-Terminal Modification” to “Fatty Acid Occupation” of Albumin. Dis. Markers 2017, 2017, 5692583. [Google Scholar] [CrossRef]
- Sanit, J.; Prompunt, E.; Adulyaritthikul, P.; Nokkaew, N.; Mongkolpathumrat, P.; Kongpol, K.; Kijtawornrat, A.; Petchdee, S.; Barrère-Lemaire, S.; Kumphune, S. Combination of metformin and p38 MAPK inhibitor, SB203580, reduced myocardial ischemia/reperfusion injury in non-obese type 2 diabetic Goto-Kakizaki rats. Exp. Med. 2019, 18, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Schisler, J.C.; Portbury, A.L.; Patterson, C. Build it up-Tear it down: Protein quality control in the cardiac sarcomere. Cardiovasc. Res. 2009, 81, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E.S.; Martinez-Vicente, M. Protein degradation, aggregation, and misfolding. Mov. Disord. 2010, 25, S49–S54. [Google Scholar] [CrossRef]
- Muller, A.L.; Hryshko, L.V.; Dhalla, N.S. Extracellular and intracellular proteases in cardiac dysfunction due to ischemia-reperfusion injury. Int. J. Cardiol. 2013, 164, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Chohan, P.K.; Dhalla, N.S.; Netticadan, T. The sarcoplasmic reticulum proteins are targets for calpain action in the ischemic-reperfused heart. J. Mol. Cell Cardiol. 2004, 37, 101–110. [Google Scholar] [CrossRef]
- Singh, R.B.; Dandekar, S.P.; Elimban, V.; Gupta, S.K.; Dhalla, N.S. Role of proteases in the pathophysiology of cardiac disease. Mol. Cell Biochem. 2004, 263, 241–256. [Google Scholar] [CrossRef]
- Powell, S.R.; Wang, P.; Katzeff, H.; Shringarpure, R.; Teoh, C.; Khaliulin, I.; Das, D.K.; Davies, K.J.; Schwalb, H. Oxidized and ubiquitinated proteins may predict recovery of postischemic cardiac function: Essential role of the proteasome. Antioxid Redox Signal. 2005, 7, 538–546. [Google Scholar] [CrossRef]
- Chohan, P.K.; Singh, R.B.; Dhalla, N.S.; Netticadan, T. L-arginine administration recovers sarcoplasmic reticulum function in ischemic reperfused hearts by preventing calpain activation. Cardiovasc. Res. 2006, 69, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Takai, S.; Yamada, M.; Sakaguchi, M.; Kamoshita, K.; Ishida, K.; Sukenaga, Y.; Miyazaki, M. Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc. Res. 2003, 60, 413–420. [Google Scholar] [CrossRef][Green Version]
- Kanemitsu, H.; Takai, S.; Tsuneyoshi, H.; Nishina, T.; Yoshikawa, K.; Miyazaki, M.; Ikeda, T.; Komeda, M. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens. Res. 2006, 29, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, S.; Bianchi, C.; Takai, S.; Chu, L.M.; Sellke, F.W. Chymase inhibition reduces infarction and matrix metalloproteinase-9 activation and attenuates inflammation and fibrosis after acute myocardial ischemia/reperfusion. J. Pharm. Exp. 2011, 339, 143–151. [Google Scholar] [CrossRef]
- Zheng, J.; Wei, C.C.; Hase, N.; Shi, K.; Killingsworth, C.R.; Litovsky, S.H.; Powell, P.C.; Kobayashi, T.; Ferrario, C.M.; Rab, A.; et al. Chymase mediates injury and mitochondrial damage in cardiomyocytes during acute ischemia/reperfusion in the dog. PLoS ONE 2014, 9, e94732. [Google Scholar] [CrossRef]
- Hooshdaran, B.; Kolpakov, M.A.; Guo, X.; Miller, S.A.; Wang, T.; Tilley, D.G.; Rafiq, K.; Sabri, A. Dual inhibition of cathepsin G and chymase reduces myocyte death and improves cardiac remodeling after myocardial ischemia reperfusion injury. Basic Res. Cardiol. 2017, 112, 62. [Google Scholar] [CrossRef]
- Arooj, M.; Sakkiah, S.; Cao, G.P.; Kim, S.; Arulalapperumal, V.; Lee, K.W. Finding off-targets, biological pathways, and target diseases for chymase inhibitors via structure-based systems biology approach. Proteins 2015, 83, 1209–1224. [Google Scholar] [CrossRef] [PubMed]
- Subramaniyam, D.; Hollander, C.; Westin, U.; Erjefalt, J.; Stevens, T.; Janciauskiene, S. Secretory leukocyte protease inhibitor inhibits neutrophil apoptosis. Respirology 2011, 16, 300–307. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Ferrari, R.; Guardigli, G.; Mele, D.; Percoco, G.F.; Ceconi, C.; Curello, S. Oxidative stress during myocardial ischaemia and heart failure. Curr. Pharm. Des. 2004, 10, 1699–1711. [Google Scholar] [CrossRef]
- Paiyabhroma, N.; Nernpermpisooth, N.; Kumphune, S. The Recombinant Human Secretory Leukocyte Protease Inhibitor (SLPI) protects cardiac fibroblasts injury against an in vitro ischemia/reperfusion injury ARTICLE INFO ABSTRACT. J. Appl. Pharm. Sci. 2018, 8, 156–162. [Google Scholar] [CrossRef]
- Caimi, G.; Canino, B.; Incalcaterra, E.; Ferrera, E.; Montana, M.; Lo Presti, R. Behaviour of protein carbonyl groups in juvenile myocardial infarction. Clin. Hemorheol. Microcirc. 2013, 53, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Dennis, K.E.; Hill, S.; Rose, K.L.; Sampson, U.K.; Hill, M.F. Augmented cardiac formation of oxidatively-induced carbonylated proteins accompanies the increased functional severity of post-myocardial infarction heart failure in the setting of type 1 diabetes mellitus. Cardiovasc. Pathol. 2013, 22, 473–480. [Google Scholar] [CrossRef]
- Reddy, C.B.; Cyriac, C.; Desle, H.B. Role of “Ischemia Modified Albumin” (IMA) in acute coronary syndromes. Indian Heart J. 2014, 66, 656–662. [Google Scholar] [CrossRef]
- Jin, F.Y.; Nathan, C.; Radzioch, D.; Ding, A. Secretory leukocyte protease inhibitor: A macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell 1997, 88, 417–426. [Google Scholar] [CrossRef]
- Nugteren, S.; Samsom, J.N. Secretory Leukocyte Protease Inhibitor (SLPI) in mucosal tissues: Protects against inflammation, but promotes cancer. Cytokine Growth Factor Rev. 2021. [Google Scholar] [CrossRef]
- Kumphune, S.; Chattipakorn, S.; Chattipakorn, N. Role of p38 inhibition in cardiac ischemia/reperfusion injury. Eur J. Clin. Pharm. 2012, 68, 513–524. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Group | |||
|---|---|---|---|---|
| Sham (n = 8) | I/R (n = 8) | rhSLPI-DI (n = 8) | rhSLPI-OR (n = 8) | |
| EDP (mmHg) | 4.87 ± 0.53 | 4.23 ± 0.16 | 4.43 ± 0.53 | 4.75 ± 0.29 |
| ESP (mmHg) | 113.8 ± 6.34 | 104.80 ± 9.31 | 119.2 ± 15.30 | 118.9 ± 17.25 |
| dp/dtmax (mmHg/s) | 5233 ± 359.2 | 5071 ± 391.1 | 6113 ± 575.4 | 5470 ± 287.1 |
| CtrI | 100.5 ± 2.02 | 106.9 ± 5.59 | 99.73 ± 7.59 | 100.4 ± 6.0 |
| dp/dtmin (mmHg/s) | −4989 ± 383.2 | −4526 ± 487.1 | −5627 ± 831.8 | −5116 ± 362.6 |
| Tau/e (ms) | 10.18 ± 0.21 | 11.03 ± 0.43 | 9.67 ± 0.83 | 9.34 ± 0.73 |
| devP (mmHg) | 109.4 ± 5.40 | 108.1 ± 9.85 | 115.8 ± 16.75 | 109.8 ± 12.96 |
| HR (bpm) | 431.5 ± 18.09 | 426.9 ± 20.09 | 416.4 ± 10.04 | 437.8 ± 13.66 |
| Parameters | Group | |||
|---|---|---|---|---|
| Sham (n = 8) | I/R (n = 8) | rhSLPI DI (n = 8) | rhSLPI-OR (n = 8) | |
| EDP (mmHg) | 5.36 ± 0.40 | 8.63 ± 1.55 * | 3.65 ± 0.46 # | 7.29 ± 0.18 |
| ESP (mmHg) | 111.8 ± 10.1 | 81.81 ± 9.69 * | 131.2 ± 9.08 # | 101.8 ± 17.0 |
| dp/dtmax (mmHg/s) | 5072 ± 725.1 | 3547 ± 415.7 * | 5983 ± 373.3 # | 4520 ± 541.0 |
| CtrI | 97.63 ± 5.21 | 101.5 ± 6.60 | 100.6 ± 2.55 | 80.88 ± 21.69 |
| dp/dtmin (mmHg/s) | −4185 ± 206.6 | −3139 ± 93.69 * | −5646 ± 701.1 # | −4565 ± 590.5 |
| Tau/e (ms) | 10.11 ± 0.46 | 12.38 ± 1.05 * | 10.04 ± 0.37 # | 8.744 ± 0.22 |
| devP (mmHg) | 106.9 ± 9.78 | 83.47 ± 7.72 * | 126.7 ± 12.82 # | 105.5 ± 8.85 |
| HR (bpm) | 429.3 ± 19.13 | 405.3 ± 24.52 | 390.1 ± 15.31 | 421.2 ± 12.75 |
| Parameters | Group | |||
|---|---|---|---|---|
| Sham (n = 8) | I/R (n = 8) | rhSLPI-DI (n = 8) | rhSLPI-OR (n = 8) | |
| EDP (mmHg) | 5.85 ± 0.60 | 7.63 ± 0.08 * | 2.871 ± 0.19 # | 4.084 ± 0.60 # |
| ESP (mmHg) | 105.6 ± 6.65 | 82.74 ± 6.63 * | 111.1 ± 10.01 # | 94.47 ± 8.80 # |
| dp/dtmax (mmHg/s) | 4509 ± 527.4 | 2836 ± 373.4 * | 4739 ± 466.8 # | 4458 ± 376 # |
| CtrI | 91.27 ± 5.79 | 94.41 ± 9.65 | 90.42 ± 2.79 | 72.85 ± 20.30 |
| dp/dtmin (mmHg/s) | −4890 ± 569.3 | −2418 ± 669.7 * | −4127 ± 244.6 # | −3990 ± 264.3 # |
| Tau/e (ms) | 11.24 ± 0.53 | 14.83 ± 3.58 | 9.73 ± 0.54 | 8.754 ± 1.37 |
| devP (mmHg) | 104.4 ± 7.40 | 80.96 ± 7.25 | 96.28 ± 4.08 | 95.51 ± 6.61 |
| HR (Bpm) | 408.5 ± 20.81 | 373.9 ± 42.74 | 402.7 ± 6.47 | 410.3 ± 10.92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mongkolpathumrat, P.; Kijtawornrat, A.; Prompunt, E.; Panya, A.; Chattipakorn, N.; Barrère-Lemaire, S.; Kumphune, S. Post-Ischemic Treatment of Recombinant Human Secretory Leukocyte Protease Inhibitor (rhSLPI) Reduced Myocardial Ischemia/Reperfusion Injury. Biomedicines 2021, 9, 422. https://doi.org/10.3390/biomedicines9040422
Mongkolpathumrat P, Kijtawornrat A, Prompunt E, Panya A, Chattipakorn N, Barrère-Lemaire S, Kumphune S. Post-Ischemic Treatment of Recombinant Human Secretory Leukocyte Protease Inhibitor (rhSLPI) Reduced Myocardial Ischemia/Reperfusion Injury. Biomedicines. 2021; 9(4):422. https://doi.org/10.3390/biomedicines9040422
Chicago/Turabian StyleMongkolpathumrat, Podsawee, Anusak Kijtawornrat, Eakkapote Prompunt, Aussara Panya, Nipon Chattipakorn, Stephanie Barrère-Lemaire, and Sarawut Kumphune. 2021. "Post-Ischemic Treatment of Recombinant Human Secretory Leukocyte Protease Inhibitor (rhSLPI) Reduced Myocardial Ischemia/Reperfusion Injury" Biomedicines 9, no. 4: 422. https://doi.org/10.3390/biomedicines9040422
APA StyleMongkolpathumrat, P., Kijtawornrat, A., Prompunt, E., Panya, A., Chattipakorn, N., Barrère-Lemaire, S., & Kumphune, S. (2021). Post-Ischemic Treatment of Recombinant Human Secretory Leukocyte Protease Inhibitor (rhSLPI) Reduced Myocardial Ischemia/Reperfusion Injury. Biomedicines, 9(4), 422. https://doi.org/10.3390/biomedicines9040422