
Macrophage Plasticity and Function in the Lung Tumour Microenvironment Revealed in 3D Heterotypic Spheroid and Explant Models

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. CD14+ Cell Isolation and Monocyte-Derived Macrophage Culture
2.3. Multi-Component Spheroid Culture
2.4. Brightfield Microscopy
2.5. Primary Tissue Processing and Explant Culture
2.6. Flow Cytometry
2.7. T Cell Functional Assay
2.8. LEGENDplexTM Bead-Based Immunoassay
2.9. Statistical Analyses
3. Results
3.1. NSCLC Spheroid-Conditioned Polarisation of Mφs In Vitro: Morphology and Two-Marker Analysis Based on CD64 and CD206 Expression
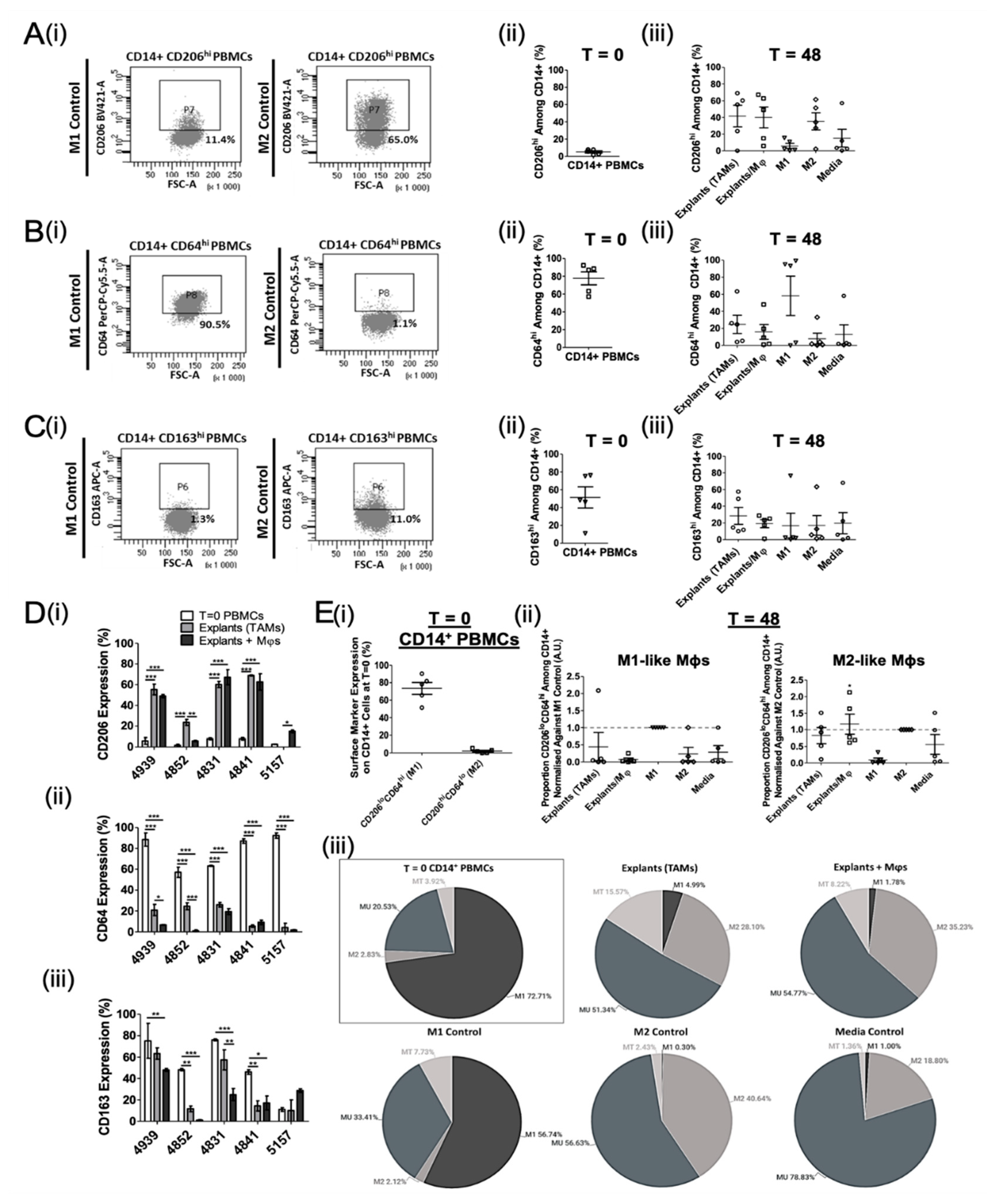
3.2. Ex Vivo Study of Mφ Polarisation Induced by Lung Tumour Explants, Using the Two-Marker Approach
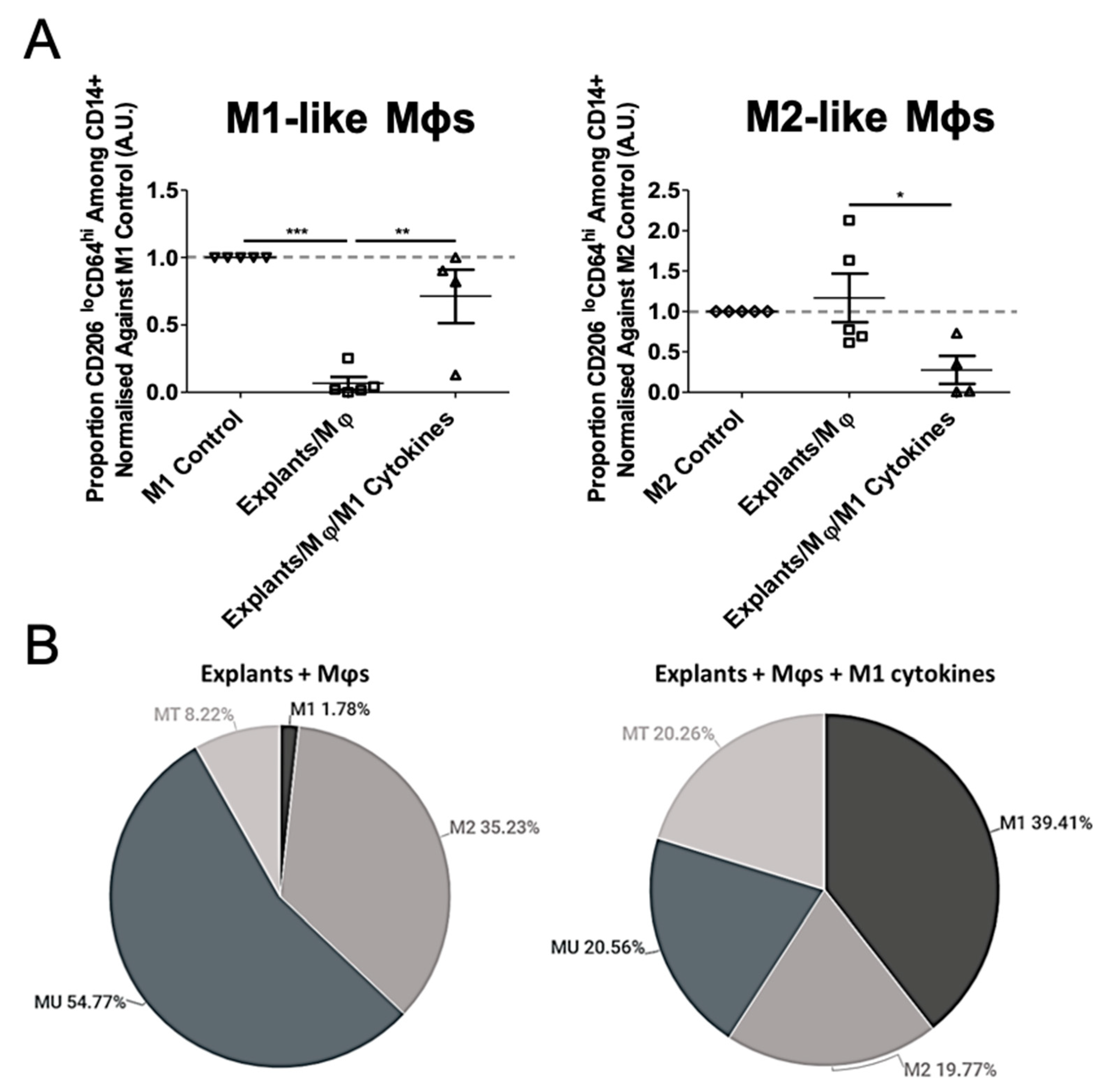
3.3. Mφ Plasticity in the TME Model
3.4. Tumour Explant, but Not Spheroid, Co-Cultures Suppress CD8+ T Cell Responses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGFB | AG02603 fibroblasts |
| CAFs | Cancer-associated fibroblasts |
| EBV | Epstein-Barr virus |
| FBS | Fetal bovine serum |
| HCMV | Human cytomegalovirus |
| IHC | Immunohistochemistry |
| iNOS | Nitric oxide synthase |
| LPS | Lipopolysaccharide |
| Mφ | Monocyte-derived macrophages |
| MDSCs | Myeloid-derived suppressor cells |
| MFI | Mean fluorescence intensity |
| MT | Transitional myeloid cells |
| MU | Unpolarised myeloid cells |
| NSCLC | Non-small cell lung cancer |
| PBMCs | Peripheral blood mononuclear cells |
| PBS | Phosphate-buffered saline |
| RT | Room temperature |
| TAM | Tumour-associated macrophage |
| TGFβ1 | Transforming growth factor β1 |
| TME | Tumour microenvironment |
| TT | Tetanus toxoid |
| VP | Viral peptide |
References
- Gordon, S. The macrophage: Past, present and future. Eur. J. Immunol. 2007, 37 (Suppl. 1), S9–S17. [Google Scholar] [CrossRef]
- Genin, M.; Clement, F.; Fattaccioli, A.; Raes, M.; Michiels, C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015, 15, 1–14. [Google Scholar] [CrossRef]
- Takahashi, H.; Sakakura, K.; Kudo, T.; Toyoda, M.; Kaira, K.; Oyama, T.; Chikamatsu, K. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 2017, 8, 8633–8647. [Google Scholar] [CrossRef]
- Genard, G.; Lucas, S.; Michiels, C. Reprogramming of Tumor-Associated Macrophages with Anticancer Therapies: Radiotherapy versus Chemo- and Immunotherapies. Front. Immunol 2017, 8, 1–19. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Yuan, A.; Hsiao, Y.J.; Chen, H.Y.; Chen, H.W.; Ho, C.C.; Chen, Y.Y.; Liu, Y.C.; Hong, T.H.; Yu, S.L.; Chen, J.J.; et al. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef]
- Zhang, B.; Yao, G.; Zhang, Y.; Gao, J.; Yang, B.; Rao, Z. M2-Polarized tumor-associated macrophages are associated with poor prognoses resulting from accelerated lymphangiogenesis in lung adenocarcinoma. Clinics (Sao Paulo) 2011, 66, 1879–1886. [Google Scholar] [CrossRef]
- Chen, J.J.; Lin, Y.C.; Yao, P.L.; Yuan, A.; Chen, H.Y.; Shun, C.T.; Tsai, M.F.; Chen, C.H.; Yang, P.C. Tumor-associated macrophages: The double-edged sword in cancer progression. J. Clin. Oncol. 2005, 23, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Yusen, W.; Xia, W.; Shengjun, Y.; Shaohui, Z.; Hongzhen, Z. The expression and significance of tumor associated macrophages and CXCR4 in non-small cell lung cancer. J. Buon 2018, 23, 398–402. [Google Scholar] [PubMed]
- Sumitomo, R.; Hirai, T.; Fujita, M.; Murakami, H.; Otake, Y.; Huang, C. M2 Tumor-Associated Macrophages Promote Tumor Progression in Non-Small-Cell Lung Cancer. Exp. Ther. Med. 2019, 18, 4490–4498. [Google Scholar] [CrossRef]
- Johnson, D.B.; Rioth, M.J.; Horn, L. Immune Checkpoint Inhibitors in NSCLC. Curr. Treat. Options Oncol 2014, 15, 658–669. [Google Scholar] [CrossRef] [PubMed]
- Karekla, E.; Liao, W.J.; Sharp, B.; Pugh, J.; Reid, H.; Quesne, J.L.; Moore, D.; Pritchard, C.; MacFarlane, M.; Pringle, J.H. Ex Vivo Explant Cultures of Non-Small Cell Lung Carcinoma Enable Evaluation of Primary Tumor Responses to Anticancer Therapy. Cancer Res. 2017, 77, 2029–2039. [Google Scholar] [CrossRef]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chester, C.; Melero, I.; Kohrt, H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol 2016, 27, 1190–1198. [Google Scholar] [CrossRef]
- Rebelo, S.P.; Pinto, C.; Martins, T.R.; Harrer, N.; Estrada, M.F.; Loza-Alvarez, P.; Cabeçadas, J.; Alves, P.M.; Gualda, E.J.; Sommergruber, W.; et al. 3D-3-culture: A tool to unveil macrophage plasticity in the tumour microenvironment. Biomaterials 2018, 163, 185–197. [Google Scholar] [CrossRef]
- Kuen, J.; Darowski, D.; Kluge, T.; Majety, M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS ONE 2017, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tevis, K.M.; Cecchi, R.J.; Colson, Y.L.; Grinstaff, M.W. Mimicking the Tumor Microenvironment to Regulate Macrophage Phenotype and Assessing Chemotherapeutic Efficacy in Embedded Cancer Cell/Macrophage Spheroid Models. Acta Biomater 2017, 50, 271–279. [Google Scholar] [CrossRef]
- Raghavan, S.; Mehta, P.; Xie, Y.; Lei, Y.L.; Mehta, G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J. Immunother Cancer 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Schmid, J.O.; Dong, M.; Haubeiss, S.; Friedel, G.; Bode, S.; Grabner, A.; Ott, G.; Murdter, T.E.; Oren, M.; Aulitzky, W.E.; et al. Cancer cells cue the p53 response of cancer-associated fibroblasts to cisplatin. Cancer Res. 2012, 72, 5824–5832. [Google Scholar] [CrossRef]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chandorkar, P.; Posch, W.; Zaderer, V.; Blatzer, M.; Steger, M.; Ammann, C.G.; Binder, U.; Hermann, M.; Hortnagl, P.; Lass-Florl, C.; et al. Fast-track development of an in vitro 3D lung/immune cell model to study Aspergillus infections. Sci Rep. 2017, 7, 11644. [Google Scholar] [CrossRef]
- Lang, D.S.; Droemann, D.; Schultz, H.; Branscheid, D.; Martin, C.; Ressmeyer, A.R.; Zabel, P.; Vollmer, E.; Goldmann, T. A novel human ex vivo model for the analysis of molecular events during lung cancer chemotherapy. Respir Res. 2007, 8, 43. [Google Scholar] [CrossRef]
- Parry-Jones, A.; Spary, L. The Wales Cancer Bank (WCB). Open J. Bioresour. 2018, 5, 1–5. [Google Scholar] [CrossRef]
- Maritan, S.; Lian, E.; Mulligan, L. An Efficient and Flexible Cell Aggregation Method for 3D Spheroid Production. J. Vis. Exp. JoVE 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ryu, N.; Lee, S.; Park, H. Spheroid Culture System Methods and Applications for Mesenchymal Stem Cells. Cells 2019, 8, 1620. [Google Scholar] [CrossRef]
- Coleman, S.; Clayton, A.; Mason, M.D.; Jasani, B.; Adams, M.; Tabi, Z. Recovery of CD8+ T-cell function during systemic chemotherapy in advanced ovarian cancer. Cancer Res. 2005, 65, 7000–7006. [Google Scholar] [CrossRef]
- Currier, J.; Kuta, E.; Turk, E.; Earhart, L.; Loomis-Price, L.; Janetzki, S.; Ferrari, G.; Birx, D.; Cox, J. A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays. J. Immunol. Methods 2002, 260, 157–172. [Google Scholar] [CrossRef]
- Clappaert, E.J.; Murgaski, A.; Van Damme, H.; Kiss, M.; Laoui, D. Diamonds in the Rough: Harnessing Tumor-Associated Myeloid Cells for Cancer Therapy. Front. Immunol 2018, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Porta, C.; Morlacchi, S.; Banfi, S.; Strauss, L.; Rimoldi, M.; Totaro, M.G.; Riboldi, E. Origin and Functions of Tumor-Associated Myeloid Cells (TAMCs). Cancer Microenviron. 2012, 5, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Barron, L. Macrophages: Master Regulators of Inflammation and Fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D.; Cihakova, D. Alternatively activated macrophages in infection and autoimmunity. J. Autoimmun. 2009, 33, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Spary, L.K.; Salimu, J.; Webber, J.P.; Clayton, A.; Mason, M.D.; Tabi, Z. Tumor stroma-derived factors skew monocyte to dendritic cell differentiation toward a suppressive CD14(+) PD-L1(+) phenotype in prostate cancer. Oncoimmunology 2014, 3, e955331. [Google Scholar] [CrossRef]
- Balestrini, J.L.; Chaudhry, S.; Sarrazy, V.; Koehler, A.; Hinz, B. The mechanical memory of lung myofibroblasts. Integr. Biol. (Camb) 2012, 4, 410–421. [Google Scholar] [CrossRef]
- Hinz, B. The myofibroblast: Paradigm for a mechanically active cell. J. Biomech. 2010, 43, 146–155. [Google Scholar] [CrossRef]
- Webber, J.; Steadman, R.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010, 70, 9621–9630. [Google Scholar] [CrossRef]
- Webber, J.P.; Spary, L.K.; Sanders, A.J.; Chowdhury, R.; Jiang, W.G.; Steadman, R.; Wymant, J.; Jones, A.T.; Kynaston, H.; Mason, M.D.; et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015, 34, 290–302. [Google Scholar] [CrossRef]
- Menzies, F.M.; Henriquez, F.L.; Alexander, J.; Roberts, C.W. Sequential expression of macrophage anti-microbial/inflammatory and wound healing markers following innate, alternative and classical activation. Clin. Exp. Immunol. 2010, 160, 369–379. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Mills, C.D.; Thomas, A.C.; Lenz, L.L.; Munder, M. Macrophage: SHIP of Immunity. Front. Immunol. 2014, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef]
- Buechler, C.; Ritter, M.; Orso, E.; Langmann, T.; Klucken, J.; Schmitz, G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J. Leukoc Biol. 2000, 67, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Almatroodi, S.A.; McDonald, C.F.; Darby, I.A.; Pouniotis, D.S. Characterization of M1/M2 Tumour-Associated Macrophages (TAMs) and Th1/Th2 Cytokine Profiles in Patients with NSCLC. Cancer Microenviron. 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jackute, J.; Zemaitis, M.; Pranys, D.; Sitkauskiene, B.; Miliauskas, S.; Vaitkiene, S.; Sakalauskas, R. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol. 2018, 19, 3. [Google Scholar] [CrossRef]
- Maniecki, M.B.; Etzerodt, A.; Moestrup, S.K.; Moller, H.J.; Graversen, J.H. Comparative assessment of the recognition of domain-specific CD163 monoclonal antibodies in human monocytes explains wide discrepancy in reported levels of cellular surface CD163 expression. Immunobiology 2011, 216, 882–890. [Google Scholar] [CrossRef]
- Tarique, A.A.; Logan, J.; Thomas, E.; Holt, P.G.; Sly, P.D.; Fantino, E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am. J. Respir Cell Mol. Biol. 2015, 53, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Ambarus, C.A.; Krausz, S.; van Eijk, M.; Hamann, J.; Radstake, T.R.; Reedquist, K.A.; Tak, P.P.; Baeten, D.L. Systematic validation of specific phenotypic markers for in vitro polarized human macrophages. J. Immunol. Methods 2012, 375, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Segura, C.P.; Taddei, M.L.; Lanciotti, M.; Serni, S.; Morandi, A.; Chiarugi, P.; Giannoni, E. Zoledronic acid impairs stromal reactivity by inhibiting M2-macrophages polarization and prostate cancer-associated fibroblasts. Oncotarget 2017, 8, 118–132. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Fuertes, M.; Spaapen, R.; Zheng, Y.; Kline, J. Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Curr. Opin. Immunol. 2011, 23, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Peng, M.; Huang, B.; Zhang, H.; Wang, H.; Xue, Z.; Zhang, L.; Da, Y.; Yang, D.; Yao, Z.; et al. Immune microenvironment profiles of tumor immune equilibrium and immune escape states of mouse sarcoma. Cancer Lett. 2013, 340, 124–133. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic | All Patients n = 11 |
|---|---|
| Age at operation, years | |
| Median (Min, Max) | 72.0 (58.0, 78.0) |
| Mean | 71.3 |
| SD | 6.3 |
| Sex | |
| Male | 6 (54.5%) |
| Female | 5 (45.5%) |
| Histology | |
| Adenocarcinoma | 3 (27.3%) |
| Squamous cell carcinoma | 8 (72.7%) |
| T stage | |
| T2a | 4 (36.4%) |
| T2b | 2 (18.1%) |
| T3 | 5 (45.5%) |
| N stage | |
| N0 | 6 (54.5%) |
| N1 | 4 (36.4%) |
| N2 | 1 (9.1%) |
| M stage | |
| MX | 7 (63.6%) |
| M0 | 3 (27.3%) |
| M1b | 1 (9.1%) |
| Cytokine/Chemokine Concentration (pg/mL) | ||||
|---|---|---|---|---|
| IFNγ | ||||
| 4898 | 4836 | 2780 | 5178 | |
| T cells Only | <0.53 | 212.47 | 113.23 | 3.32 |
| + Explants | 0.62 | 105.40 | 150.66 | 1.55 |
| + Explants/Mφ | 1.13 | 18.65 | 209.30 | 78.56 |
| TNFα | ||||
| 4898 | 4836 | 2780 | 5178 | |
| T cells Only | <1.20 | 1.21 | 1.71 | 3.52 |
| + Explants | <1.20 | <1.20 | 1.95 | <1.20 |
| + Explants/Mφ | <1.20 | <1.20 | 1.58 | 1.58 |
| IL-10 | ||||
| 4898 | 4836 | 2780 | 5178 | |
| T cells Only | <1.09 | 1.675 | 1.18 | 1.77 |
| + Explants | 2.95 | 3.44 | 3.96 | 2.74 |
| + Explants/Mφ | 1.73 | 17.03 | 8.12 | 8.31 |
| Arginase | ||||
| 4898 | 4836 | 2780 | 5178 | |
| T cells Only | 3663.90 | 3923.90 | 2346.75 | 3975.41 |
| + Explants | 3052.86 | 3285.92 | 2346.46 | 2819.79 |
| + Explants/Mφ | 5712.62 | 4302.11 | 4212.98 | 5670.56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evans, L.; Milward, K.; Attanoos, R.; Clayton, A.; Errington, R.; Tabi, Z. Macrophage Plasticity and Function in the Lung Tumour Microenvironment Revealed in 3D Heterotypic Spheroid and Explant Models. Biomedicines 2021, 9, 302. https://doi.org/10.3390/biomedicines9030302
Evans L, Milward K, Attanoos R, Clayton A, Errington R, Tabi Z. Macrophage Plasticity and Function in the Lung Tumour Microenvironment Revealed in 3D Heterotypic Spheroid and Explant Models. Biomedicines. 2021; 9(3):302. https://doi.org/10.3390/biomedicines9030302
Chicago/Turabian StyleEvans, Lauren, Kate Milward, Richard Attanoos, Aled Clayton, Rachel Errington, and Zsuzsanna Tabi. 2021. "Macrophage Plasticity and Function in the Lung Tumour Microenvironment Revealed in 3D Heterotypic Spheroid and Explant Models" Biomedicines 9, no. 3: 302. https://doi.org/10.3390/biomedicines9030302
APA StyleEvans, L., Milward, K., Attanoos, R., Clayton, A., Errington, R., & Tabi, Z. (2021). Macrophage Plasticity and Function in the Lung Tumour Microenvironment Revealed in 3D Heterotypic Spheroid and Explant Models. Biomedicines, 9(3), 302. https://doi.org/10.3390/biomedicines9030302