
Exploiting the Potential of Drosophila Models in Lysosomal Storage Disorders: Pathological Mechanisms and Drug Discovery

 ,
, 
Abstract
1. Introduction
2. Cellular Pathways Involved in LSDs
2.1. The Lysosome and Its Spatial Distribution in the Cytosol
2.1.1. Lysosome Structure and Formation
2.1.2. Lysosome Positioning
2.2. Functions of the Lysosome and Its Central Role in Cell Homeostasis
2.2.1. Lysosome as a Regulatory Hub
2.2.2. Endocytosis
2.2.3. Autophagy
2.2.4. Autophagic Lysosomal Reformation (ALR)
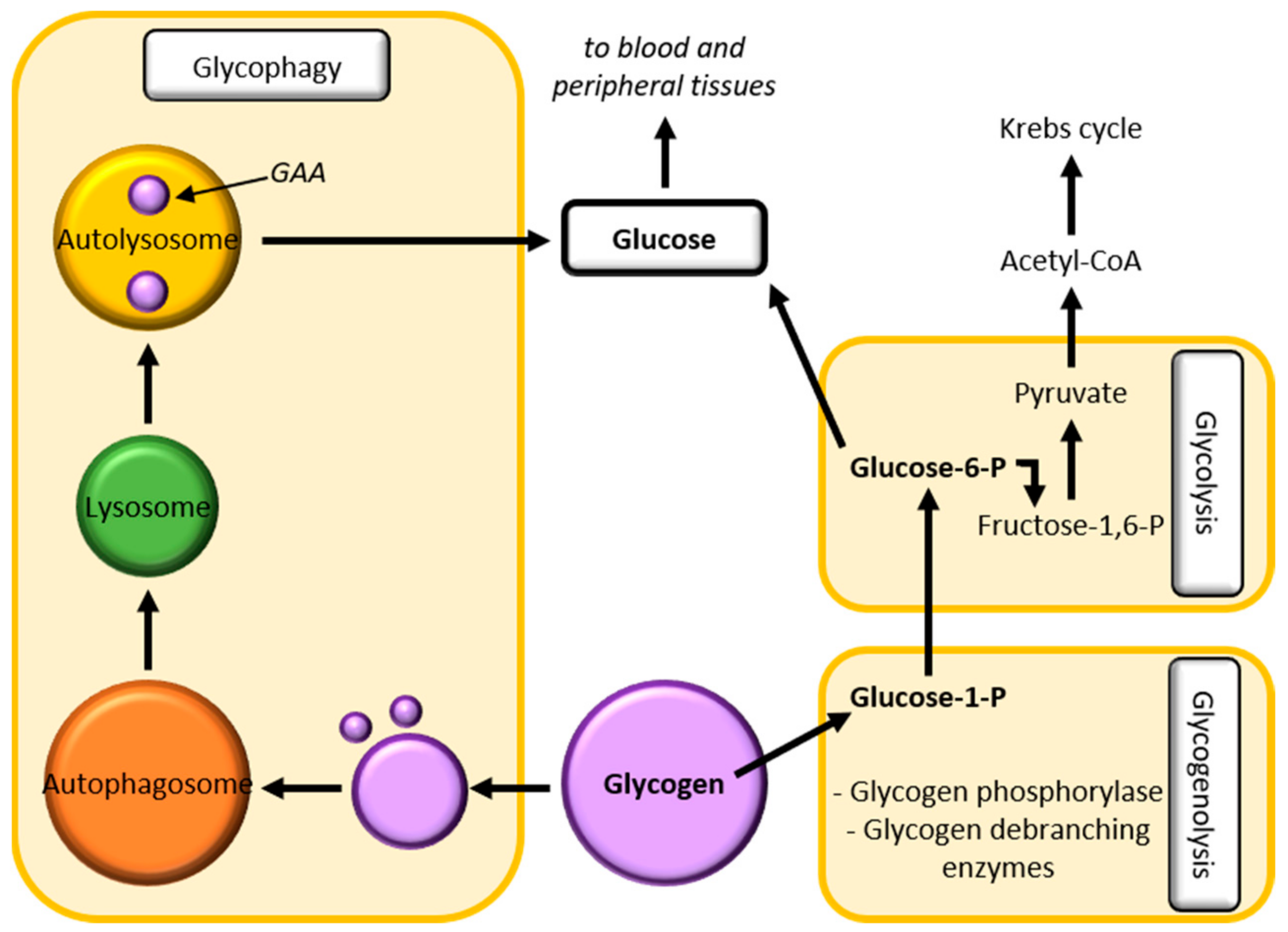
2.2.5. Glycogen Homeostasis
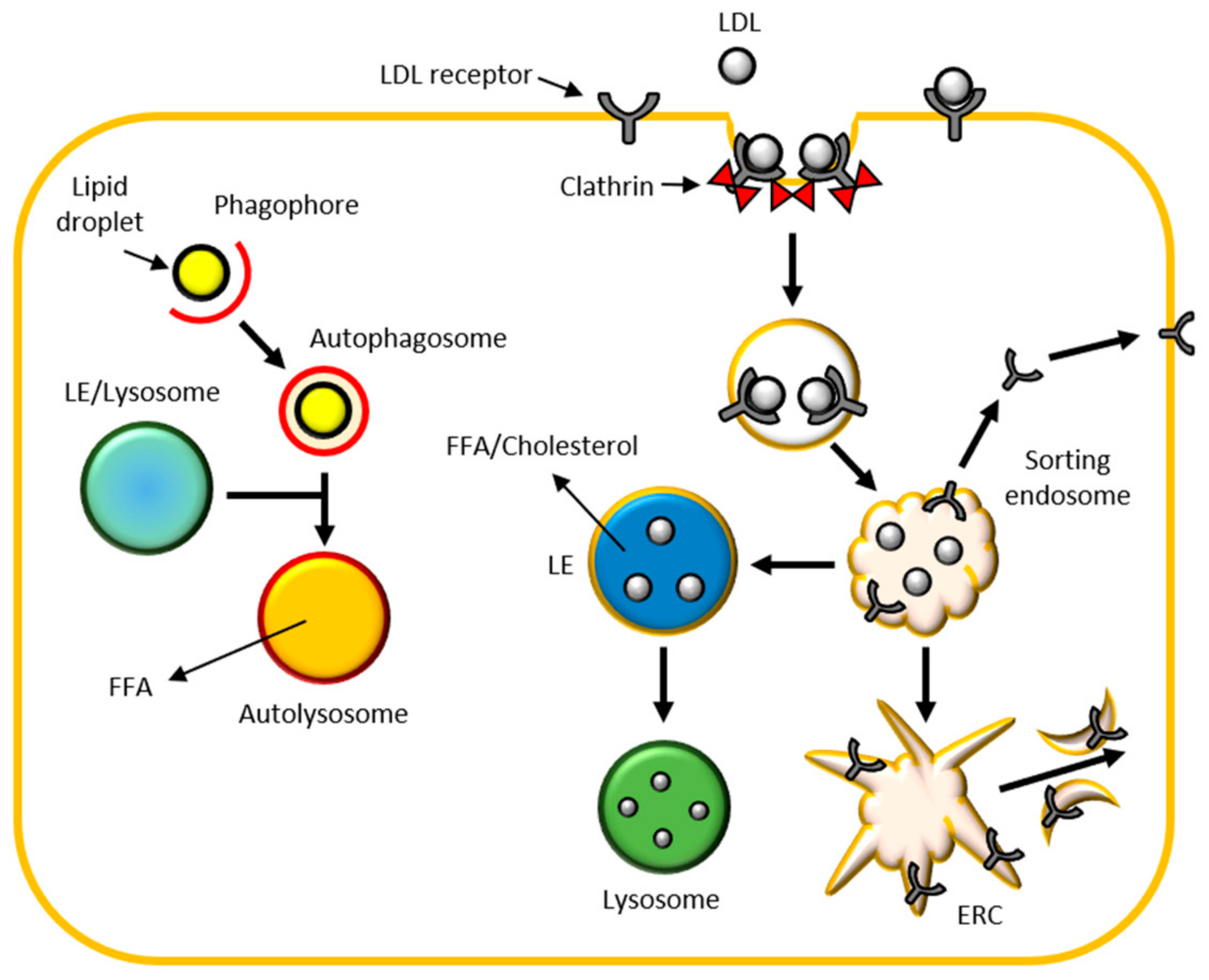
2.2.6. Lipid Metabolism
2.3. Lysosomal Diseases and the Major Metabolic Pathways Altered in in These Pathologies
3. Modelling LSDs in Drosophila
3.1. Approaches to Model Diseases in Drosophila
3.1.1. Transposable Elements
3.1.2. The CRISPR/Cas9 Approach
3.1.3. The GAL4/UAS System
3.2. Tools to Study LSDs’ Pathways in Drosophila
3.2.1. Reporter Lines
3.2.2. Vital Dyes and Antibodies
3.2.3. Other Tools
3.3. Next-Generation Analysis and Metabolic Studies
4. Current Drosophila Models of LSDs: An Unexplored Potential
4.1. Mucolipidosis Type IV (MLIV) Drosophila Model
4.2. Batten Disease/Neuronal Ceroid Lipofuscinose (NCL) Drosophila Models
4.2.1. Palmitoyl Protein Thioesterase I (CLN1) Drosophila Model
4.2.2. CLN3 Drosophila Model
4.2.3. CLN4 Drosophila Model
4.2.4. Cathepsin D (CLN10) Drosophila Model
4.3. Mucopolysaccharidosis Drosophila Models
4.3.1. Mucopolysaccharidosis Type II (MPS II) Drosophila Model
4.3.2. Mucopolysaccharidosis Type IIIA (MPS IIIA) Drosophila Model
4.3.3. α-N-acetylglucosaminidase (NAGLU) Homologous Identified in Drosophila
4.3.4. Mucopolysaccharidosis Type VII (MPS VII) Drosophila Model
4.4. Sphingolipidoses Drosophila Models
4.4.1. Niemann–Pick Type C Disease (NPC) Drosophila Model
4.4.2. Gaucher Disease (GD) Drosophila Model
4.4.3. Metachromatic Leukodystrophy (MLD) Drosophila Model
4.4.4. Fabry Disease Drosophila Model
4.4.5. Saposin Deficient Sphingolipidoses Drosophila Model
4.5. LSDs-Like Drosophila Models
Spinster/Benchwarmer Drosophila Model
5. Drosophila as a Tool for Drug Testing and Screening
5.1. Drug Delivery in Drosophila
5.2. Drosophila BBB as a Tool for CNS Treatments Discovery
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Bellettato, C.M.; Hubert, L.; Scarpa, M.; Wangler, M.F. Inborn errors of metabolism involving complex molecules: Lysosomal and peroxisomal storage diseases. Pediatr. Clin. N. Am. 2018, 65, 353–373. [Google Scholar] [CrossRef]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Hers, H.G. Inborn lysosomal diseases. Gastroenterology 1965, 48, 625–633. [Google Scholar] [CrossRef]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders–challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef]
- Kingma, S.D.K.; Bodamer, O.A.; Wijburg, F.A. Epidemiology and diagnosis of lysosomal storage disorders; Challenges of screening. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 145–157. [Google Scholar] [CrossRef]
- Bulfield, G. Inherited metabolic disease in laboratory animals: A review. J. Inherit. Metab. Dis. 1980, 3, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Hindle, S.; Hebbar, S.; Sweeney, S.T. Invertebrate models of lysosomal storage disease: What have we learned so far? Invertebr. Neurosci. 2011, 11, 59–71. [Google Scholar] [CrossRef]
- Pastores, G.M.; Torres, P.A.; Zeng, B.-J. Animal models for lysosomal storage disorders. Biochem. 2013, 78, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Favret, J.M.; Weinstock, N.I.; Feltri, M.L.; Shin, D. Pre-clinical mouse models of neurodegenerative lysosomal storage diseases. Front. Mol. Biosci. 2020, 7, 57. [Google Scholar] [CrossRef]
- Zhang, T.; Peterson, R.T. Modeling lysosomal storage diseases in the zebrafish. Front. Mol. Biosci. 2020, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Sym, M.; Basson, M.; Johnson, C. A model for niemann-pick type C disease in the nematode Caenorhabditis elegans. Curr. Biol. 2000, 10, 527–530. [Google Scholar] [CrossRef]
- Nakano, Y.; Fujitani, K.; Kurihara, J.; Ragan, J.; Usui-Aoki, K.; Shimoda, L.; Lukacsovich, T.; Suzuki, K.; Sezaki, M.; Sano, Y.; et al. Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in drosophila melanogaster. Mol. Cell. Biol. 2001, 21, 3775–3788. [Google Scholar] [CrossRef]
- Julian, L.M.; Stanford, W.L. Organelle cooperation in stem cell fate: Lysosomes as emerging regulators of cell identity. Front. Cell Dev. Biol. 2020, 8, 591. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef]
- Saftig, P.; Schröder, B.; Blanz, J. Lysosomal membrane proteins: Life between acid and neutral conditions: Figure 1. Biochem. Soc. Trans. 2010, 38, 1420–1423. [Google Scholar] [CrossRef]
- Appelqvist, H.; Wa, P. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2018, 5, 214–226. [Google Scholar] [CrossRef]
- Reggiori, F.; Klumperman, J. Lysosome biogenesis and autophagy. In Lysosomes: Biology, Diseases, and Therapeutics; Maxfield, F.R., Willard, J.M., Lu, S., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 7–31. [Google Scholar]
- Matteoni, R.; Kreis, T.E. Translocation and clustering of endosomes and lysosomes depends on microtubules. J. Cell Biol. 1987, 105, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Pu, J.; Guardia, C.M.; Keren-Kaplan, T.; Bonifacino, J.S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 2016, 129, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Ba, Q.; Raghavan, G.; Kiselyov, K.; Yang, G. Whole-cell scale dynamic organization of lysosomes revealed by spatial statistical analysis. Cell Rep. 2018, 23, 3591–3606. [Google Scholar] [CrossRef]
- Perera, R.M.; Zoncu, R. The lysosome as a regulatory hub. Annu. Rev. Cell Dev. Biol. 2016, 32, 223–253. [Google Scholar]
- Bajaj, L.; Lotfi, P.; Pal, R.; di Ronza, A.; Sharma, J.; Sardiello, M. Lysosome biogenesis in health and disease. J. Neurochem. 2018, 148, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Zeigerer, A.; Gilleron, J.; Bogorad, R.L.; Marsico, G.; Nonaka, H.; Seifert, S.; Epstein-Barash, H.; Kuchimanchi, S.; Peng, C.G.; Ruda, V.M.; et al. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature 2012, 485, 465–470. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Rink, J.; Ghigo, E.; Kalaidzidis, Y.; Zerial, M. Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005, 122, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Lőrincz, P.; Mauvezin, C.; Juhász, G. Exploring autophagy in Drosophila. Cells 2017, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Jacomin, A.C.; Fauvarque, M.O.; Taillebourg, E. A functional endosomal pathway is necessary for lysosome biogenesis in Drosophila. BMC Cell Biol. 2016, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluijs, P.; Hull, M.; Webster, P.; Mâle, P.; Goud, B.; Mellman, I. The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell 1992, 70, 729–740. [Google Scholar] [CrossRef]
- Campa, C.C.; Hirsch, E. Rab11 and phosphoinositides: A synergy of signal transducers in the control of vesicular trafficking. Adv. Biol. Regul. 2017, 63, 132–139. [Google Scholar] [CrossRef]
- Goldenring, J.R. Recycling endosomes. Curr. Opin. Cell Biol. 2015, 35, 117–122. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, M.; Li, X.; Liu, J.; Li, H. Origin of the autophagosome membrane in mammals. BioMed Res. Int. 2018, 2018, 1012789. [Google Scholar] [CrossRef]
- Gaudecker, V. On variation in some cell organelles during formation of reserve substances in fatty bodies of Drosophila larvae. Z. Zellforsch. Mikrosk. Anat. 1963, 61, 56–95. [Google Scholar] [CrossRef]
- Mulakkal, N.C.; Nagy, P.; Takats, S.; Tusco, R.; Juhász, G.; Nezis, I.P. Autophagy in Drosophila: from historical studies to current knowledge. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Lund, V.K.; Madsen, K.L.; Kjaerulff, O. Drosophila Rab2 controls endosome-lysosome fusion and LAMP delivery to late endosomes. Autophagy 2018, 14, 1520–1542. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, L. Development of research into autophagic lysosome reformation. Mol. Cells 2018, 41, 45–49. [Google Scholar]
- Rong, Y.; McPhee, C.K.; Deng, S.; Huang, L.; Chen, L.; Liu, M.; Tracy, K.; Baehrecke, E.H.; Yu, L.; Lenardo, M.J. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 7826–7831. [Google Scholar] [CrossRef] [PubMed]
- Kilimann, M.W.; Oldfors, A. Glycogen pathways in disease: New developments in a classical field of medical genetics. J. Inherit. Metab. Dis. 2015, 38, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tang, M.; Liu, M.; Chen, L. Glycophagy: An emerging target in pathology. Clin. Chim. Acta 2018, 484, 298–303. [Google Scholar] [CrossRef]
- Zirin, J.; Nieuwenhuis, J.; Perrimon, N. Role of Autophagy in Glycogen Breakdown and Its Relevance to Chloroquine Myopathy. PLoS Biol. 2013, 11, e1001708. [Google Scholar] [CrossRef]
- Kobayashi, T.; Beuchat, M.H.; Lindsay, M.; Frias, S.; Palmiter, R.D.; Sakuraba, H.; Parton, R.G.; Gruenberg, J. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1999, 1, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Thelen, A.M.; Zoncu, R. Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol. 2017, 27, 833–850. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Huang, X. Lipid metabolism in Drosophila: Development and disease. Acta Biochim. Biophys. Sin. 2013, 45, 44–50. [Google Scholar] [CrossRef]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar] [PubMed]
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef]
- Allocca, M.; Zola, S.; Bellosta, P. The fruit fly, Drosophila melanogaster: Modeling of human diseases (part II). In Drosophila Melanogaster Model for Recent Advances in Genetics and Therapeutics; Perveen, F.K., Ed.; IntechOpen: London, UK, 2018. [Google Scholar]
- Chintapalli, V.R.; Wang, J.; Dow, J.A.T. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 2007, 39, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nichols, C.D. Human disease models in drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol. Rev. 2011, 63, 411–436. [Google Scholar] [CrossRef] [PubMed]
- Moulton, M.J.; Letsou, A. Modeling congenital disease and inborn errors of development in Drosophila melanogaster. Dis. Model. Mech. 2016, 9, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Ryder, E.; Russell, S. Transposable elements as tools for genomics and genetics in Drosophila. Briefings Funct. Genomics Proteomics 2003, 2, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, A.; Knust, E. The Use of P-Element Transposons to Generate Transgenic Flies; Humana Press: Totowa, NJ, USA, 2008; pp. 61–77. [Google Scholar]
- Port, F.; Chen, H.-M.; Lee, T.; Bullock, S.L. Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc. Natl. Acad. Sci. USA 2014, 111, E2967–E2976. [Google Scholar] [CrossRef]
- Port, F.; Muschalik, N.; Bullock, S.L. Systematic evaluation of Drosophila CRISPR tools reveals safe and robust alternatives to autonomous gene drives in basic research. G3 Genes Genomes Genet. 2015, 5, 1493–1502. [Google Scholar] [CrossRef]
- Hales, K.G.; Korey, C.A.; Larracuente, A.M.; Roberts, D.M. Genetics on the fly: A primer on the Drosophila model system. Genetics 2015, 201, 815–842. [Google Scholar] [CrossRef]
- Gratz, S.J.; Rubinstein, C.D.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. CRISPR-Cas9 genome editing in Drosophila. Curr. Protoc. Mol. Biol. 2015, 111, 31.2.1–31.2.20. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar]
- St Johnston, D. The art and design of genetic screens: Drosophila melanogaster. Nat. Rev. Genet. 2002, 3, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.S.; Bang, S.M.; Toh, A. Lipids and lipid signaling in Drosophila models of neurodegenerative diseases. Omega-3 Faty Acids Brain Neurol. Health 2014, 327–336. [Google Scholar] [CrossRef]
- Riedel, F.; Gillingham, A.K.; Rosa-Ferreira, C.; Galindo, A.; Munro, S. An antibody toolkit for the study of membrane traffic in Drosophila melanogaster. Biol. Open 2016, 5, 987–992. [Google Scholar] [CrossRef]
- An, P.N.T.; Fukusaki, E. Metabolomics: State-of-the-art technologies and applications on Drosophila melanogaster. Adv. Exp. Med. Biol. 2018, 1076, 257–276. [Google Scholar] [PubMed]
- Bargal, R.; Avidan, N.; Ben-Asher, E.; Olender, Z.; Zeigler, M.; Frumkin, A.; Raas-Rothschild, A.; Glusman, G.; Lancet, D.; Bach, G. Identification of the gene causing mucolipidosis type IV. Nat. Genet. 2000, 26, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Slaugenhaupt, S.A.; Acierno, J.S.; Helbling, L.A.; Bove, C.; Goldin, E.; Bach, G.; Schiffmann, R.; Gusella, J.F. Mapping of the mucolipidosis type IV gene to chromosome 19p and definition of founder haplotypes. Am. J. Hum. Genet. 1999, 65, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Amir, N.; Ziotogora, J.; Bach, G. Mucolipidosis type IV: Clinical spectrum and natural history. Pediatrics 1987, 79, 953–959. [Google Scholar]
- Venkatachalam, K.; Long, A.A.; Elsaesser, R.; Nikolaeva, D.; Montell, C. Motor deficit in a Drosophila model of mucolipidosis Type IV due to defective clearance of apoptotic cells. Cell 2008, 135, 838–851. [Google Scholar] [CrossRef]
- Wong, C.-O.; Li, R.; Montell, C.; Venkatachalam, K. Drosophila TRPML is required for TORC1 activation. Curr. Biol. 2012, 22, 1616–1621. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.-O.; Palmieri, M.; Li, J.; Akhmedov, D.; Chao, Y.; Broadhead, G.T.; Zhu, M.X.; Berdeaux, R.; Collins, C.A.; Sardiello, M.; et al. Diminished MTORC1-Dependent JNK activation underlies the neurodevelopmental defects associated with lysosomal dysfunction. Cell Rep. 2015, 12, 2009–2020. [Google Scholar] [CrossRef]
- Onyenwoke, R.U.; Sexton, J.Z.; Yan, F.; Díaz, M.C.H.; Forsberg, L.J.; Major, M.B.; Brenman, J.E. The mucolipidosis IV Ca2+ channel TRPML1 (MCOLN1) is regulated by the TOR kinase. Biochem. J. 2015, 470, 331–342. [Google Scholar] [CrossRef]
- Haltia, M.; Goebel, H.H. The neuronal ceroid-lipofuscinoses: A historical introduction. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Kohlschütter, A.; Schulz, A. Towards understanding the neuronal ceroid lipofuscinoses. Brain Dev. 2009, 31, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.L.; Hickey, A.J.; Chotkowski, H.L.; Chu-LaGraff, Q. Characterization of Drosophila palmitoyl-protein thioesterase 1. Gene 2003, 312, 271–279. [Google Scholar] [CrossRef]
- Hickey, A.J.; Chotkowski, H.L.; Singh, N.; Ault, J.G.; Korey, C.A.; MacDonald, M.E.; Glaser, R.L. Palmitoyl-protein thioesterase 1 deficiency in Drosophila melanogaster causes accumulation of abnormal storage material and reduced life span. Genetics 2006, 172, 2379–2390. [Google Scholar] [CrossRef] [PubMed]
- Chu-LaGraff, Q.; Blanchette, C.; O’Hern, P.; Denefrio, C. The Batten disease Palmitoyl Protein Thioesterase 1 gene regulates neural specification and axon connectivity during Drosophila embryonic development. PLoS ONE 2010, 5, e14402. [Google Scholar] [CrossRef]
- Munroe, P.B.; Mitchison, H.M.; O’rawe, A.M.; Anderson, J.W.; Boustany, R.-M.; Lerner, T.J.; Taschner, P.E.M.; De Vos, N.; Breuning, M.H.; Gardiner, R.M.; et al. Spectrum of Mutations in the Batten Disease Gene, CLN3. Am. J. Hum. Genet. 1997, 61, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Tuxworth, R.I.; Vivancos, V.; O’Hare, M.B.; Tear, G. Interactions between the juvenile Batten disease gene, CLN3, and the Notch and JNK signalling pathways. Hum. Mol. Genet. 2009, 18, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Tuxworth, R.I.; Chen, H.; Vivancos, V.; Carvajal, N.; Huang, X.; Tear, G. The Batten disease gene CLN3 is required for the response to oxidative stress. Hum. Mol. Genet. 2011, 20, 2037–2047. [Google Scholar] [CrossRef]
- Henderson, M.X.; Wirak, G.S.; Zhang, Y.-q.; Dai, F.; Ginsberg, S.D.; Dolzhanskaya, N.; Staropoli, J.F.; Nijssen, P.C.G.; Lam, T.K.T.; Roth, A.F.; et al. Neuronal ceroid lipofuscinosis with DNAJC5/CSPα mutation has PPT1 pathology and exhibit aberrant protein palmitoylation. Acta Neuropathol. 2016, 131, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Nosková, L.; Stránecký, V.; Hartmannová, H.; Přistoupilová, A.; Barešová, V.; Ivánek, R.; Hlková, H.; Jahnová, H.; Van Der Zee, J.; Staropoli, J.F.; et al. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 2011, 89, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Imler, E.; Pyon, J.S.; Kindelay, S.; Torvund, M.; Zhang, Y.Q.; Chandra, S.S.; Zinsmaier, K.E. A drosophila model of neuronal ceroid lipofuscinosis CLN4 reveals a hypermorphic gain of function mechanism. eLife 2019, 8, e46607. [Google Scholar] [CrossRef] [PubMed]
- Myllykangas, L.; Tyynela, J.; Page-McCaw, A.; Rubin, G.M.; Haltia, M.J.; Feany, M.B. Cathepsin D-deficient Drosophila recapitulate the key features of neuronal ceroid lipofuscinoses. Neurobiol. Dis. 2005, 19, 194–199. [Google Scholar] [CrossRef]
- Kuronen, M.; Talvitie, M.; Lehesjoki, A.E.; Myllykangas, L. Genetic modifiers of degeneration in the cathepsin D deficient Drosophila model for neuronal ceroid lipofuscinosis. Neurobiol. Dis. 2009, 36, 488–493. [Google Scholar] [CrossRef]
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of mucopolysaccharidoses, an update. Int. J. Mol. Sci. 2020, 21, 2515. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef]
- Rathmann, M.; Bunge, S.; Beck, M.; Kresse, H.; Tylki-Szymanska, A.; Gal, A. Mucopolysaccharidosis type II (Hunter syndrome): Mutation “hot spots” in the iduronate-2-sulfatase gene. Am. J. Hum. Genet. 1996, 59, 1202–1209. [Google Scholar]
- D’Avanzo, F.; Rigon, L.; Zanetti, A.; Tomanin, R. Mucopolysaccharidosis type II: One hundred years of research, diagnosis, and treatment. Int. J. Mol. Sci. 2020, 21, 1258. [Google Scholar] [CrossRef]
- Young, I.D.; Harper, P.S.; Newcombe, R.G.; Archer, I.M. A clinical and genetic study of Hunter’s syndrome. 2. Differences between the mild and severe forms. J. Med. Genet. 1982, 19, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.S. Comparative studies of vertebrate iduronate 2-sulfatase (IDS) genes and proteins: Evolution of A mammalian X-linked gene. 3 Biotech 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rigon, L.; Kucharowski, N.; Eckardt, F.; Bauer, R. Modeling mucopolysaccharidosis type II in the fruit fly by using the RNA interference approach. Life 2020, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Fedele, A. Sanfilippo syndrome: Causes, consequences, and treatments. Appl. Clin. Genet. 2015, 8, 269. [Google Scholar] [CrossRef]
- Neufeld, E.; Muenzer, J. The mucopolysaccharidoses. In The Online Metabolic and Molecular Bases of Inherited Disease; McGraw-Hill Medical: New York, NY, USA, 2001. [Google Scholar]
- Webber, D.L.; Choo, A.; Hewson, L.J.; Trim, P.J.; Snel, M.F.; Hopwood, J.J.; Richards, R.I.; Hemsley, K.M.; O’Keefe, L.V. Neuronal-specific impairment of heparan sulfate degradation in Drosophila reveals pathogenic mechanisms for Mucopolysaccharidosis type IIIA. Exp. Neurol. 2018, 303, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Aronovich, E.L.; Johnston, J.M.; Wang, P.; Giger, U.; Whitley, C.B. Molecular basis of mucopolysaccharidosis type IIIB in Emu (Dromaius novaehollandiae): An avian model of sanfilippo syndrome type B<. Genomics 2001, 74, 299–305. [Google Scholar] [PubMed]
- Bar, S.; Prasad, M.; Datta, R. Neuromuscular degeneration and locomotor deficit in a Drosophila model of mucopolysaccharidosis VII is attenuated by treatment with resveratrol. Dis. Model. Mech. 2018, 11, dmm036954. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.I.; Waheed, A.; Grubb, J.H.; Klei, H.E.; Korolev, S.; Sly, W.S. High Resolution crystal structure of human β-glucuronidase reveals structural basis of lysosome targeting. PLoS ONE 2013, 8, e79687. [Google Scholar] [CrossRef] [PubMed]
- Grubb, J.H.; Vogler, C.; Levy, B.; Galvin, N.; Tan, Y.; Sly, W.S. Chemically modified glucuronidase crosses blood-brain barrier and clears neuronal storage in murine mucopolysaccharidosis VII. Proc. Natl. Acad. Sci. USA 2008, 105, 2616–2621. [Google Scholar] [CrossRef]
- Islam, M.R.; Tomatsu, S.; Shah, G.N.; Grubb, J.H.; Jain, S.; Sly, W.S. Active site residues of human beta-glucuronidase. Evidence for Glu(540) as the nucleophile and Glu(451) as the acid-base residue. J. Biol. Chem. 1999, 274, 23451–23455. [Google Scholar] [CrossRef] [PubMed]
- Sellin, J.; Schulze, H.; Paradis, M.; Gosejacob, D.; Papan, C.; Shevchenko, A.; Psathaki, O.E.; Paululat, A.; Thielisch, M.; Sandhoff, K.; et al. Characterization of Drosophila Saposin-related mutants as a model for lysosomal sphingolipid storage diseases. Dis. Model. Mech. 2017, 10, 737–750. [Google Scholar] [CrossRef]
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef]
- Sturley, S.L.; Patterson, M.C.; Balch, W.; Liscum, L. The pathophysiology and mechanisms of NP-C disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2004, 1685, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Maxfield, F.R. Lipid and cholesterol trafficking in NPC. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2004, 1685, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Liscum, L.; Sturley, S.L. Intracellular trafficking of Niemann–Pick C proteins 1 and 2: Obligate components of subcellular lipid transport. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2004, 1685, 22–27. [Google Scholar] [CrossRef]
- Huang, X.; Suyama, K.; Buchanan, J.; Zhu, A.J.; Scott, M.P. A Drosophila model of the Niemann-Pick type C lysosome storage disease: dnpc1a is required for molting and sterol homeostasis. Development 2005, 132, 5115–5124. [Google Scholar] [CrossRef]
- Huang, X.; Warren, J.T.; Buchanan, J.; Gilbert, L.I.; Scott, M.P. Drosophila Niemann-Pick type C-2 genes control sterol homeostasis and steroid biosynthesis: A model of human neurodegenerative disease. Development 2007, 134, 3733–3742. [Google Scholar] [CrossRef] [PubMed]
- Fluegel, M.L.; Parker, T.J.; Pallanck, L.J. Mutations of a Drosophila NPC1 gene confer sterol and ecdysone metabolic defects. Genetics 2006, 172, 185–196. [Google Scholar] [CrossRef]
- Phillips, S.E.; Woodruff, E.A.; Liang, P.; Patten, M.; Broadie, K. Neuronal loss of Drosophila NPC1a causes cholesterol aggregation and age-progressive neurodegeneration. J. Neurosci. 2008, 28, 6569–6582. [Google Scholar] [CrossRef]
- Dandana, A.; Ben Khelifa, S.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher disease: Clinical, biological and therapeutic aspects. Pathobiology 2016, 83, 13–23. [Google Scholar] [CrossRef]
- Cox, T.M. Gaucher disease: Clinical profile and therapeutic developments. Biologics 2010, 4, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.W.; Herzyk, P.; Dow, J.A.T.; Leader, D.P. FlyAtlas: Database of gene expression in the tissues of Drosophila melanogaster. Nucleic Acids Res. 2013, 41, D744–D750. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Suzuki, T.; Ito, K.; Takahara, T.; Goto-Inoue, N.; Setou, M.; Sakata, K.; Ishida, N. Minos-insertion mutant of the Drosophila GBA gene homologue showed abnormal phenotypes of climbing ability, sleep and life span with accumulation of hydroxy-glucocerebroside. Gene 2017, 614, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.Y.; Trinh, K.; Thomas, R.E.; Yu, S.; Germanos, A.A.; Whitley, B.N.; Sardi, S.P.; Montine, T.J.; Pallanck, L.J. Glucocerebrosidase Deficiency in Drosophila Results in α-Synuclein-Independent Protein Aggregation and Neurodegeneration. PLoS Genet. 2016, 12, e1005944. [Google Scholar] [CrossRef]
- Maor, G.; Rencus-Lazar, S.; Filocamo, M.; Steller, H.; Segal, D.; Horowitz, M. Unfolded protein response in Gaucher disease: From human to Drosophila. Orphanet J. Rare Dis. 2013, 8, 140. [Google Scholar] [CrossRef]
- Cabasso, O.; Paul, S.; Dorot, O.; Maor, G.; Krivoruk, O.; Pasmanik-Chor, M.; Mirzaian, M.; Ferraz, M.; Aerts, J.; Horowitz, M. Drosophila melanogaster mutated in its GBA1b ortholog recapitulates neuronopathic Gaucher disease. J. Clin. Med. 2019, 8, 1420. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.K.; Schejter, E.; Bialik, S.; Shkedy, A.; Levin-Salomon, V.; Levin-Zaidman, S.; Kimchi, A. Death by over-eating: The Gaucher disease associated gene GBA1, identified in a screen for mediators of autophagic cell death, is necessary for developmental cell death in Drosophila midgut. Cell Cycle 2017, 16, 2003–2010. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Grönke, S.; Castillo-Quan, J.I.; Woodling, N.S.; Li, L.; Sirka, E.; Gegg, M.; Mills, K.; Hardy, J.; Bjedov, I.; et al. A Drosophila model of neuronopathic Gaucher Disease demonstrates lysosomal-autophagic defects and altered mTOR signalling and is functionally rescued by rapamycin. J. Neurosci. 2016, 36, 11654–11670. [Google Scholar] [CrossRef]
- Suzuki, T.; Shimoda, M.; Ito, K.; Hanai, S.; Aizawa, H.; Kato, T.; Kawasaki, K.; Yamaguchi, T.; Ryoo, H.D.; Goto-Inoue, N.; et al. Expression of human Gaucher disease gene GBA generates neurodevelopmental defects and ER stress in Drosophila eye. PLoS ONE 2013, 8, e69147. [Google Scholar] [CrossRef] [PubMed]
- Maor, G.; Cabasso, O.; Krivoruk, O.; Rodriguez, J.; Steller, H.; Segal, D.; Horowitz, M. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar] [PubMed]
- Maor, G.; Rapaport, D.; Horowitz, M. The effect of mutant GBA1 on accumulation and aggregation of a synuclein. Hum. Mol. Genet. 2019, 28, 1768–1781. [Google Scholar] [CrossRef]
- Matthes, F.; Stroobants, S.; Gerlach, D.; Wohlenberg, C.; Wessig, C.; Fogh, J.; Gieselmann, V.; Eckhardt, M.; D’Hooge, R.; Matzner, U. Efficacy of enzyme replacement therapy in an aggravated mouse model of metachromatic leukodystrophy declines with age. Hum. Mol. Genet. 2012, 21, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Van Rappard, D.F.; Boelens, J.J.; Wolf, N.I. Metachromatic leukodystrophy: Disease spectrum and approaches for treatment. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 261–273. [Google Scholar] [CrossRef]
- Lee, J.S.; Kanai, K.; Suzuki, M.; Kim, W.S.; Yoo, H.S.; Fu, Y.; Kim, D.-K.; Jung, B.C.; Choi, M.; Oh, K.W.; et al. Arylsulfatase A, a genetic modifier of Parkinson’s disease, is an α-synuclein chaperone. Brain 2019, 142, 2845–2859. [Google Scholar] [CrossRef]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; Van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef]
- Schiffmann, R.; Moore, D.F. Neurological Manifestations of Fabry Disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; ISBN 1-903539-03-X. [Google Scholar]
- McCafferty, E.H.; Scott, L.J. Migalastat: A Review in Fabry Disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Kalliokoski, R.J.; Kalliokoski, K.K.; Penttinen, M.; Kantola, I.; Leino, A.; Viikari, J.S.; Simell, O.; Nuutila, P.; Raitakari, O.T. Structural and functional changes in peripheral vasculature of Fabry patients. J. Inherit. Metab. Dis. 2006, 29, 660–666. [Google Scholar] [CrossRef]
- Barbey, F.; Brakch, N.; Linhart, A.; Jeanrenaud, X.; Palecek, T.; Bultas, J.; Burnier, M.; Hayoz, D. Increased carotid intima-media thickness in the absence of atherosclerotic plaques in an adult population with Fabry disease. Acta Paediatr. 2007, 95, 63–68. [Google Scholar] [CrossRef]
- Braunstein, H.; Papazian, M.; Maor, G.; Lukas, J.; Rolfs, A.; Horowitz, M. Misfolding of lysosomal α-galactosidase a in a fly model and its alleviation by the pharmacological chaperone migalastat. Int. J. Mol. Sci. 2020, 21, 7397. [Google Scholar] [CrossRef]
- Burkhardt, J.K.; Hüttler, S.; Klein, A.; Möbius, W.; Habermann, A.; Griffiths, G.; Sandhoff, K. Accumulation of sphingolipids in SAP-precursor (prosaposin)-deficient fibroblasts occurs as intralysosomal membrane structures and can be completely reversed by treatment with human SAP-precursor. Eur. J. Cell Biol. 1997, 73, 10–18. [Google Scholar] [PubMed]
- Hindle, S.J.; Hebbar, S.; Schwudke, D.; Elliott, C.J.H.; Sweeney, S.T. A saposin deficiency model in Drosophila: Lysosomal storage, progressive neurodegeneration and sensory physiological decline. Neurobiol. Dis. 2017, 98, 77–87. [Google Scholar] [CrossRef]
- Huizing, M.; Helip-Wooley, A.; Westbroek, W.; Gunay-Aygun, M.; Gahl, W.A. Disorders of lysosome-related organelle biogenesis: Clinical and molecular genetics. Annu. Rev. Genomics Hum. Genet. 2008, 9, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, S.; Khandelwal, A.; Jayashree, R.; Hindle, S.J.; Chiang, Y.N.; Yew, J.Y.; Sweeney, S.T.; Schwudke, D. Lipid metabolic perturbation is an early-onset phenotype in adult spinster mutants: A Drosophila model for lysosomal storage disorders. Mol. Biol. Cell 2017, 28, 3728–3740. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.T.; Davis, G.W. Unrestricted Synaptic Growth in spinster—a late endosomal protein implicated in TGF-β-Mediated synaptic growth regulation. Neuron 2002, 36, 403–416. [Google Scholar] [CrossRef]
- Dermaut, B.; Norga, K.K.; Kania, A.; Verstreken, P.; Pan, H.; Zhou, Y.; Callaerts, P.; Bellen, H.J. Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J. Cell Biol. 2005, 170, 127–139. [Google Scholar] [CrossRef]
- Fernández-Hernández, I.; Scheenaard, E.; Pollarolo, G.; Gonzalez, C. The translational relevance of Drosophila in drug discovery. EMBO Rep. 2016, 17, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, L.F.; Schlosser, T.; Manning, S.A.; Parisot, J.P.; Street, I.P.; Richardson, H.E.; Humbert, P.O.; Brumby, A.M. An in vivo large-scale chemical screening platform using Drosophila for anti-cancer drug discovery. Dis. Model. Mech. 2013, 6, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Limmer, S.; Weiler, A.; Volkenhoff, A.; Babatz, F.; Klämbt, C. The Drosophila blood-brain barrier: Development and function of a glial endothelium. Front. Neurosci. 2014, 8, 365. [Google Scholar] [CrossRef]
- Daneman, R.; Barres, B.A. The blood-brain barrier-Lessons from moody flies. Cell 2005, 123, 9–12. [Google Scholar] [CrossRef][Green Version]
- Schulman, V.K.; Folker, E.S.; Baylies, M.K. A method for reversible drug delivery to internal tissues of Drosophila embryos. Fly 2013, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Fantin, M.; Garelli, F.; Napoli, B.; Forgiarini, A.; Gumeni, S.; De Martin, S.; Montopoli, M.; Vantaggiato, C.; Orso, G. Flavonoids regulate lipid droplets biogenesis in Drosophila melanogaster. Nat. Prod. Commun. 2019, 14, 1934578X1985243. [Google Scholar] [CrossRef]
- Napoli, B.; Gumeni, S.; Forgiarini, A.; Fantin, M.; De Filippis, C.; Panzeri, E.; Vantaggiato, C.; Orso, G. Naringenin ameliorates Drosophila ReepA hereditary spastic paraplegia-linked phenotypes. Front. Neurosci. 2019, 13, 1202. [Google Scholar] [CrossRef]
- Forgiarini, A.; Wang, Z.; D’Amore, C.; Jay-Smith, M.; Li, F.F.; Hopkins, B.; Brimble, M.A.; Pagetta, A.; Bersani, S.; De Martin, S.; et al. Live applications of norbormide-based fluorescent probes in Drosophila melanogaster. PLoS ONE 2019, 14, e0211169. [Google Scholar] [CrossRef]
- Zabihihesari, A.; Hilliker, A.J.; Rezai, P. Localized microinjection of intact Drosophila melanogaster larva to investigate the effect of serotonin on heart rate. Lab Chip 2020, 20, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.D.; Ronesi, J.; Pratt, W.; Sanders-Bush, E. Hallucinogens and Drosophila: Linking serotonin receptor activation to behavior. Neuroscience 2002, 115, 979–984. [Google Scholar] [CrossRef]
- Turin, L.; Skoulakis, E.M.C.; Horsfield, A.P. Electron spin changes during general anesthesia in Drosophila. Proc. Natl. Acad. Sci. USA 2014, 111, E3524–E3533. [Google Scholar] [CrossRef]
- Zalucki, O.H.; Menon, H.; Kottler, B.; Faville, R.; Day, R.; Bademosi, A.T.; Lavidis, N.; Karunanithi, S.; Van Swinderen, B. Syntaxin1A-mediated Resistance and Hypersensitivity to Isoflurane in Drosophila melanogaster. Anesthesiology 2015, 122, 1060–1074. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.S.; DeZazzo, J.; Luk, A.Y.; Tully, T.; Singh, C.M.; Heberlein, U. Ethanol intoxication in Drosophila: Genetic and pharmacological evidence for regulation by the cAMP signaling pathway. Cell 1998, 93, 997–1007. [Google Scholar] [CrossRef]
- McClung, C.; Hirsh, J. Stereotypic behavioral responses to free-base cocaine and the development of behavioral sensitization in drosophila. Curr. Biol. 1998, 8, 109–112. [Google Scholar] [CrossRef]
- Poudel, S.; Kim, Y.; Kwak, J.S.; Jeong, S.; Lee, Y. Gustatory receptor 22e is essential for sensing chloroquine and strychnine in Drosophila melanogaster. Insect Biochem. Mol. Biol. 2017, 88, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Charlu, S.; Wisotsky, Z.; Medina, A.; Dahanukar, A. Acid sensing by sweet and bitter taste neurons in Drosophila melanogaster. Nat. Commun. 2013, 4, 1–10. [Google Scholar] [CrossRef]
- Gasque, G.; Conway, S.; Huang, J.; Rao, Y.; Vosshall, L.B. Small molecule drug screening in Drosophila identifies the 5HT2A receptor as a feeding modulation target. Sci. Rep. 2013, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ja, W.W.; Carvalho, G.B.; Mak, E.M.; De La Rosa, N.N.; Fang, A.Y.; Liong, J.C.; Brummel, T.; Benzer, S. Prandiology of Drosophila and the CAFE assay. Proc. Natl. Acad. Sci. USA 2007, 104, 8253–8256. [Google Scholar] [CrossRef]
- Deshpande, S.A.; Carvalho, G.B.; Amador, A.; Phillips, A.M.; Hoxha, S.; Lizotte, K.J.; Ja, W.W. Quantifying Drosophila food intake: Comparative analysis of current methodology. Nat. Methods 2014, 11, 535–540. [Google Scholar] [CrossRef]
- Shell, B.C.; Schmitt, R.E.; Lee, K.M.; Johnson, J.C.; Chung, B.Y.; Pletcher, S.D.; Grotewiel, M. Measurement of solid food intake in Drosophila via consumption-excretion of a dye tracer. Sci. Rep. 2018, 8, 11536. [Google Scholar] [CrossRef] [PubMed]
- Kuklinski, N.J.; Berglund, E.C.; Ewing, A.G. Micellar capillary electrophoresis-Electrochemical detection of neurochemicals from Drosophila. J. Sep. Sci. 2010, 33, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Levario, T.J.; Zhao, C.; Rouse, T.; Shvartsman, S.Y.; Lu, H. An integrated platform for large-scale data collection and precise perturbation of live Drosophila embryos. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef]
- Ali, S.N.; Dayarathna, T.K.; Ali, A.N.; Osumah, T.; Ahmed, M.; Cooper, T.T.; Power, N.E.; Zhang, D.; Kim, D.; Kim, R.; et al. Drosophila melanogaster as a function-based high-throughput screening model for antinephrolithiasis agents in kidney stone patients. DMM Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Filošević, A.; Al-samarai, S.; Andretić Waldowski, R. High Throughput measurement of locomotor sensitization to volatilized cocaine in Drosophila melanogaster. Front. Mol. Neurosci. 2018, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Giacomotto, J.; Ségalat, L. High-throughput screening and small animal models, where are we? Br. J. Pharmacol. 2010, 160, 204–216. [Google Scholar] [CrossRef]
- Yadav, A.K.; Srikrishna, S.; Gupta, S.C. Cancer drug development using Drosophila as an in vivo Tool: From bedside to bench and back. Trends Pharmacol. Sci. 2016, 37, 789–806. [Google Scholar] [CrossRef]
- Castillo-Quan, J.I.; Tain, L.S.; Kinghorn, K.J.; Li, L.; Grönke, S.; Hinze, Y.; Blackwell, T.K.; Bjedov, I.; Partridge, L. A triple drug combination targeting components of the nutrient-sensing network maximizes longevity. Proc. Natl. Acad. Sci. USA 2019, 116, 20817–20819. [Google Scholar] [CrossRef]
- Segarra, M.; Aburto, M.R.; Acker-Palmer, A. Blood–brain barrier dynamics to maintain brain homeostasis. Trends Neurosci. 2021, 2020. [Google Scholar] [CrossRef]
- Benz, F.; Liebner, S. Structure and Function of the Blood–Brain Barrier (BBB); Springer: Berlin/Heidelberg, Germany, 2020; pp. 1–29. [Google Scholar]
- O’Brown, N.M.; Pfau, S.J.; Gu, C. Bridging barriers: A comparative look at the blood-brain barrier across organisms. Genes Dev. 2018, 32, 466–478. [Google Scholar] [CrossRef]
- Rotstein, B.; Paululat, A. On the morphology of the Drosophila Heart. J. Cardiovasc. Dev. Dis. 2016, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Stork, T.; Engelen, D.; Krudewig, A.; Silies, M.; Bainton, R.J.; Klämbt, C. Organization and function of the blood-brain barrier in Drosophila. J. Neurosci. 2008, 28, 587–597. [Google Scholar] [CrossRef]
- Rouka, E.; Gourgoulianni, N.; Lüpold, S.; Hatzoglou, C.; Gourgoulianis, K.; Blanckenhorn, W.U.; Zarogiannis, S.G. The Drosophila septate junctions beyond barrier function: Review of the literature, prediction of human orthologs of the SJ-related proteins and identification of protein domain families. Acta Physiol. 2021, 231, e13527. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Singh, P.; Ali, V.; Verma, M. Role of membrane-embedded drug efflux ABC transporters in the cancer chemotherapy. Oncol. Rev. 2020, 14, 448. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Buchon, N.; Scott, J.G. Mdr65 decreases toxicity of multiple insecticides in Drosophila melanogaster. Insect Biochem. Mol. Biol. 2017, 89, 11–16. [Google Scholar] [CrossRef] [PubMed]
- DeSalvo, M.K.; Hindle, S.J.; Rusan, Z.M.; Orng, S.; Eddison, M.; Halliwill, K.; Bainton, R.J. The Drosophila surface glia transcriptome: Evolutionary conserved blood-brain barrier processes. Front. Neurosci. 2014, 8, 346. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2011. [Google Scholar] [CrossRef] [PubMed]
- Fünfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Möbius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef]
- Holcroft, C.E.; Jackson, W.D.; Lin, W.-H.; Bassiri, K.; Baines, R.A.; Phelan, P. Innexins Ogre and Inx2 are required in glial cells for normal postembryonic development of the Drosophila central nervous system. J. Cell Sci. 2013, 126, 3823–3834. [Google Scholar] [CrossRef] [PubMed]
- Spéder, P.; Brand, A.H. Gap Junction proteins in the blood-brain barrier control nutrient-dependent reactivation of Drosophila neural stem cells. Dev. Cell 2014, 30, 309–321. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletić-Savatić, M.; Bellen, H.J. The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab. 2017, 26, 719–737.e6. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.M.; Ip, M.; Cheng, E.; Minami, M.; Pellerin, L.; Magistretti, P.; Sapolsky, R.M. Dual-gene, dual-cell type therapy against an excitotoxic insult by bolstering neuroenergetics. J. Neurosci. 2004, 24, 6202–6208. [Google Scholar] [CrossRef] [PubMed]
- Berthet, C.; Lei, H.; Thevenet, J.; Gruetter, R.; Magistretti, P.J.; Hirt, L. Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow Metab. 2009, 29, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Noe, C.R.; Noe-Letschnig, M.; Handschuh, P.; Noe, C.A.; Lanzenberger, R. Dysfunction of the blood-brain barrier—A key step in neurodegeneration and dementia. Front. Aging Neurosci. 2020, 12, 24. [Google Scholar] [CrossRef]
- Shigemoto-Mogami, Y.; Hoshikawa, K.; Sato, K. Activated microglia disrupt the blood-brain barrier and induce chemokines and cytokines in a rat in vitro model. Front. Cell. Neurosci. 2018, 12, 494. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.; Logan, M.A.; Taşdemir, Ö.E.; Freeman, M.R. Ensheathing glia function as phagocytes in the adult Drosophila brain. J. Neurosci. 2009, 29, 4768–4781. [Google Scholar] [CrossRef] [PubMed]
- Hakim-Mishnaevski, K.; Flint-Brodsly, N.; Shklyar, B.; Levy-Adam, F.; Kurant, E. Glial phagocytic receptors promote neuronal loss in adult Drosophila brain. Cell Rep. 2019, 29, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Zhang, S.L.; Sehgal, A. Regulation of the blood–brain barrier by circadian rhythms and sleep. Trends Neurosci. 2019, 42, 500–510. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yue, Z.; Arnold, D.M.; Artiushin, G.; Sehgal, A. A Circadian clock in the blood-brain barrier regulates xenobiotic efflux. Cell 2018, 173, 130–139.e10. [Google Scholar] [CrossRef] [PubMed]
- Sarantseva, S.V.; Bolshakova, O.I.; Timoshenko, S.I.; Kolobov, A.A.; Schwarzman, A.L. Dendrimer D5 is a vector for peptide transport to brain cells. Bull. Exp. Biol. Med. 2011, 150, 429–431. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Human Gene | Protein | Protein Localization | Drosophila Gene | Protein Alignment Data | Model Generation Method | References |
|---|---|---|---|---|---|---|---|
| Neuronal Ceroid-Lipofuscinosis (CLN) or Batten Disease | |||||||
| CLN1 | CLN1/PPT1 | Palmitoyl-protein thioesterase 1 (PPT1) | Cytosol; Golgi apparatus; Lysosomal lumen; Nucleus | CG12108/Ppt1 | 72% similarity, 55% identity | RNAi | [78,79] |
| CLN3 | CLN3 | Transmembrane protein | Endoplasmic reticulum; Early endosome; Late endosome; Golgi apparatus; Golgi membrane; Lysosomal membrane; Mitochondria; Nucleus; Plasma membrane | CG5582/Cln3 | Data not available | Minos transposable element imprecise excision; RNAi | [73,82] |
| CLN4 | CLN4/DNAJC5 | Soluble cysteine string protein α (CSPα) | Cytosol | CG6395/Csp | Data not available | P-element insertion | [85] |
| CLN10 | CLN10/CTSD | Cathepsin D (CTSD) | Lysosomal lumen | CG1548/cathD | 65% similarity, 50% identity | P-element imprecision excision | [86,87] |
| Mucolipidosis (ML) and Mucopolysaccharidoses (MPSs) | |||||||
| MLIV | MCOLN1 | Mucolipin-1 (TRPML1) | Lysosome membrane; Late endosome membrane; Cell membrane; Phagosome membrane | CG8743/Trpml | 40% identity | P-element insertion | [71,72,73] |
| MPS II, Hunter Syndrome | IDS | Iduronate 2-sulfatase (IDS) | Lysosomal lumen | CG12014/Ids | 47% identity | RNAi | [93,94] |
| MPS IIIA, San Filippo Syndrome type A | SGSH | N-sulfoglucosamine sulfohydrolase (SGSH) | Lysosomal lumen | CG14291/Sgsh | 53% identity | RNAi | [97] |
| MPS IIIB, San Filippo Syndrome type B | NAGLU | α-N-acetylglucosaminidase (NAGLU) | Lysosomal lumen | CG13397 | 41% identity | none | [98] |
| MPS VII, Sly Syndrome | GUSB | Glucuronidase beta (GUSB) | Lysosomal lumen | CG2135/βGlu | 40% identity, 60% similarity | Homologous recombination | [99] |
| Sphingolipidosis | |||||||
| Gaucher disease (GD) or glucocerebrosidase deficiency | GBA | Glucosylceramidase beta (GBA) | Lysosomal lumen; Lysosomal membrane | CG31148/GBA1a CG31414/GBA1b | 31% identity, 49% similarity | Minos transposable element insertion; Homologous recombination; Transposon insertion and precise excision; RNAi | [116,118]; [121,122]; [117]; [120] |
| Niemann Pick disease type 1C (NPC1) | NPC1 | NPC intracellular cholesterol transporter 1 (NPC1) | Endoplasmic reticulum; Late endosome membrane; Golgi apparatus; Lysosomal membrane; Nuclear envelope; Plasma membrane | CG5722/Npc1a CG12092/Npc1b | Npc1a: 44% similarity, 63% identity; Npc1b: 55% similarity, 38% identity | RNAi | [109,111,112] |
| Niemann Pick disease type 2C (NPC2) | NPC2 | NPC intracellular cholesterol transporter 2 (NPC2) | Endoplasmic reticulum; Lysosomal lumen | CG7291, CG3153, CG3934, CG12813, CG31410, CG6164, CG11314, CG11315 (Npc2a-h) | Npc2a: 53% similarity, 36% identity | P-element insertion and imprecise excision | [110] |
| Metachromatic leukodystrophy | ARSA | Arylsulfatase A | Endoplasmic reticulum; Lysosomal lumen | CG32191 | Data not-available | PhiC31 integrase system | [127] |
| Fabry disease | GLA | α-Galactosidase | Lysosomal lumen | CG5731 | Data not available | [133] | |
| Saposin deficiency sphingolipidoses | PSAP | Prosaposin (PSAP) | Lysosomal lumen; Lysosomal membrane; Plasma membrane | CG12010 (Saposin-related) | Data not available | P-element insertion and imprecise excision; FLP-FRT based deletion | [103,135] |
| LSD-like | |||||||
| Spinster/Benchwarmer | - | transmembrane protein and putative late-endosomal/lysosomal efflux permease | Late endolysosomal compartment | CG8428 (spin or bnch) | Data not available | P-element insertion and imprecise excision P-element insertion | [138,139]; [13,137] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigon, L.; De Filippis, C.; Napoli, B.; Tomanin, R.; Orso, G. Exploiting the Potential of Drosophila Models in Lysosomal Storage Disorders: Pathological Mechanisms and Drug Discovery. Biomedicines 2021, 9, 268. https://doi.org/10.3390/biomedicines9030268
Rigon L, De Filippis C, Napoli B, Tomanin R, Orso G. Exploiting the Potential of Drosophila Models in Lysosomal Storage Disorders: Pathological Mechanisms and Drug Discovery. Biomedicines. 2021; 9(3):268. https://doi.org/10.3390/biomedicines9030268
Chicago/Turabian StyleRigon, Laura, Concetta De Filippis, Barbara Napoli, Rosella Tomanin, and Genny Orso. 2021. "Exploiting the Potential of Drosophila Models in Lysosomal Storage Disorders: Pathological Mechanisms and Drug Discovery" Biomedicines 9, no. 3: 268. https://doi.org/10.3390/biomedicines9030268
APA StyleRigon, L., De Filippis, C., Napoli, B., Tomanin, R., & Orso, G. (2021). Exploiting the Potential of Drosophila Models in Lysosomal Storage Disorders: Pathological Mechanisms and Drug Discovery. Biomedicines, 9(3), 268. https://doi.org/10.3390/biomedicines9030268