Urinary Cyclophilin A as Marker of Tubular Cell Death and Kidney Injury

 ,
,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Western Blot
2.3. Cell Viability Assay
2.4. NSS and KT Patients
2.5. Urine Samples Collection and ELISA
2.6. Statistics
3. Results
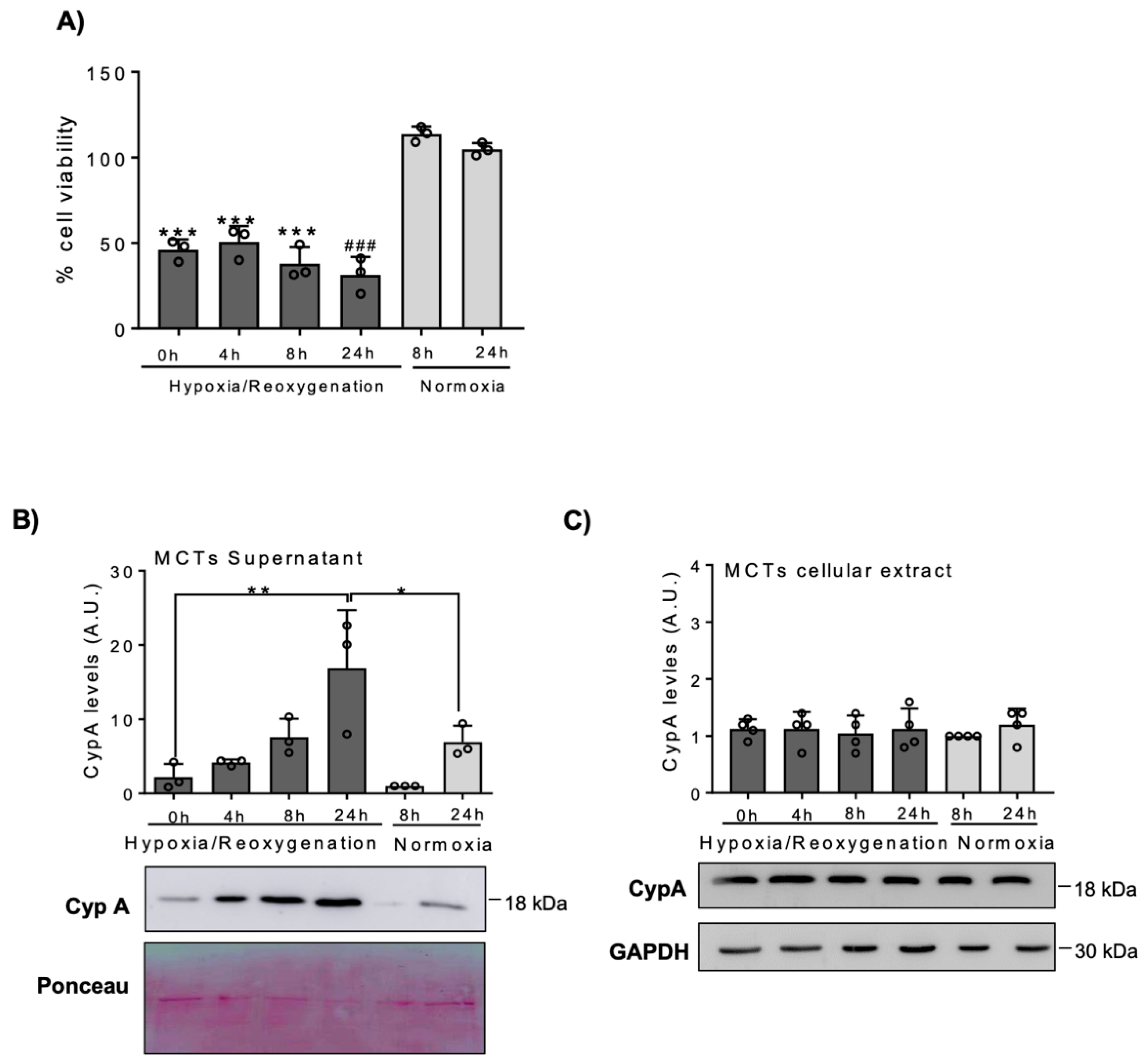
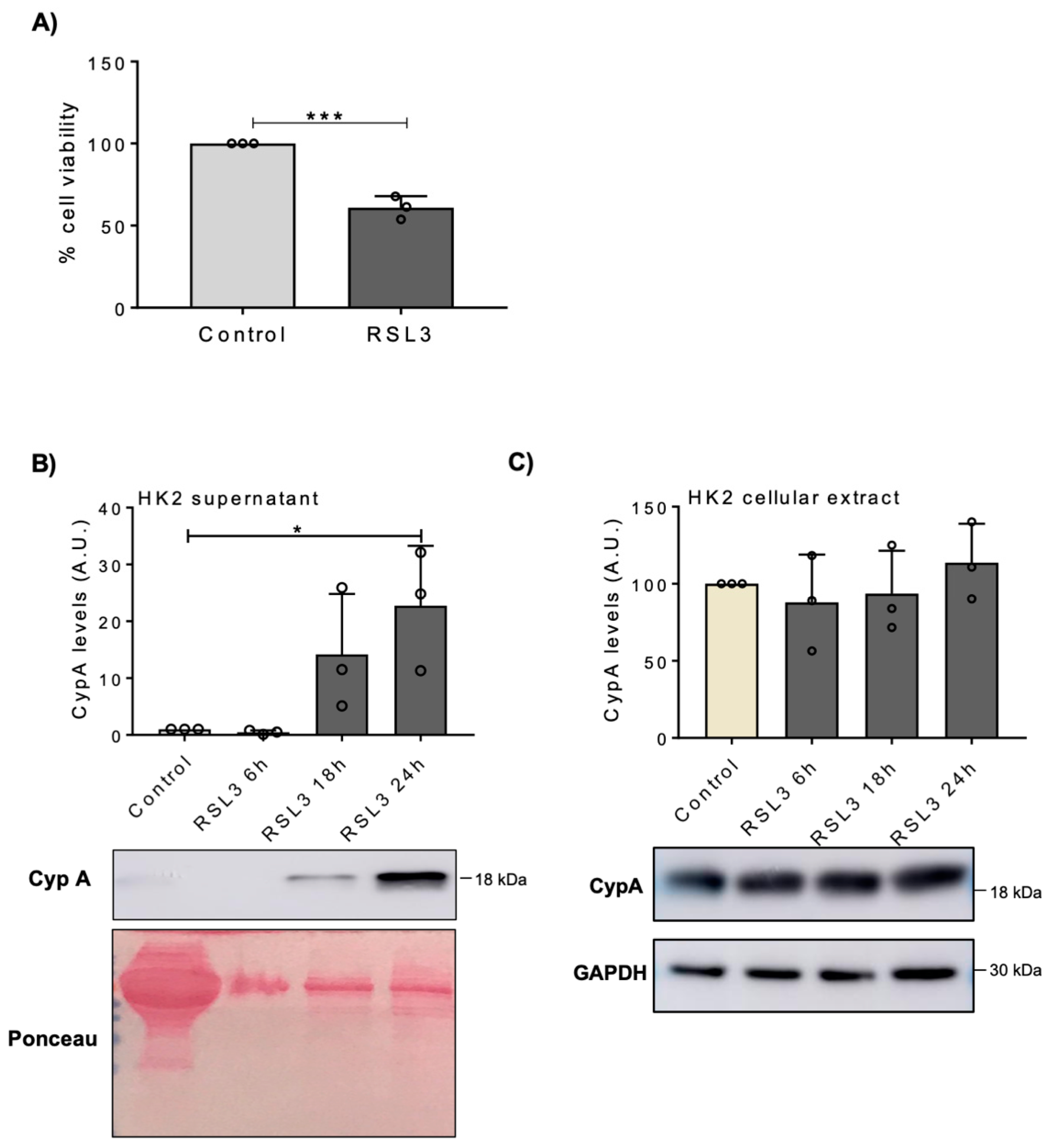
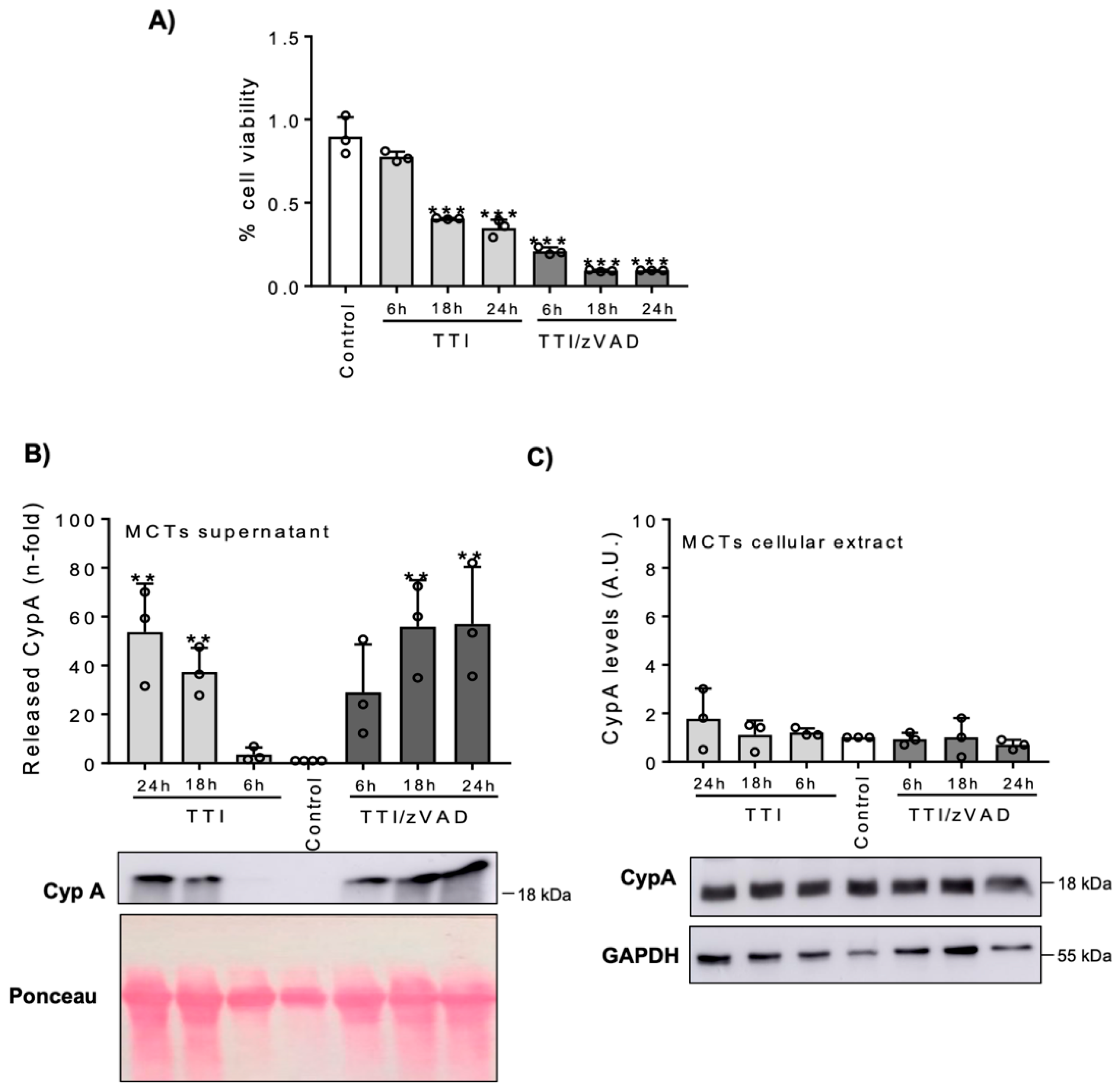
3.1. Lethal Stimuli Promote CypA Release from Cultured Kidney Tubular Cells
3.2. Clinical Study: Patient Characteristics
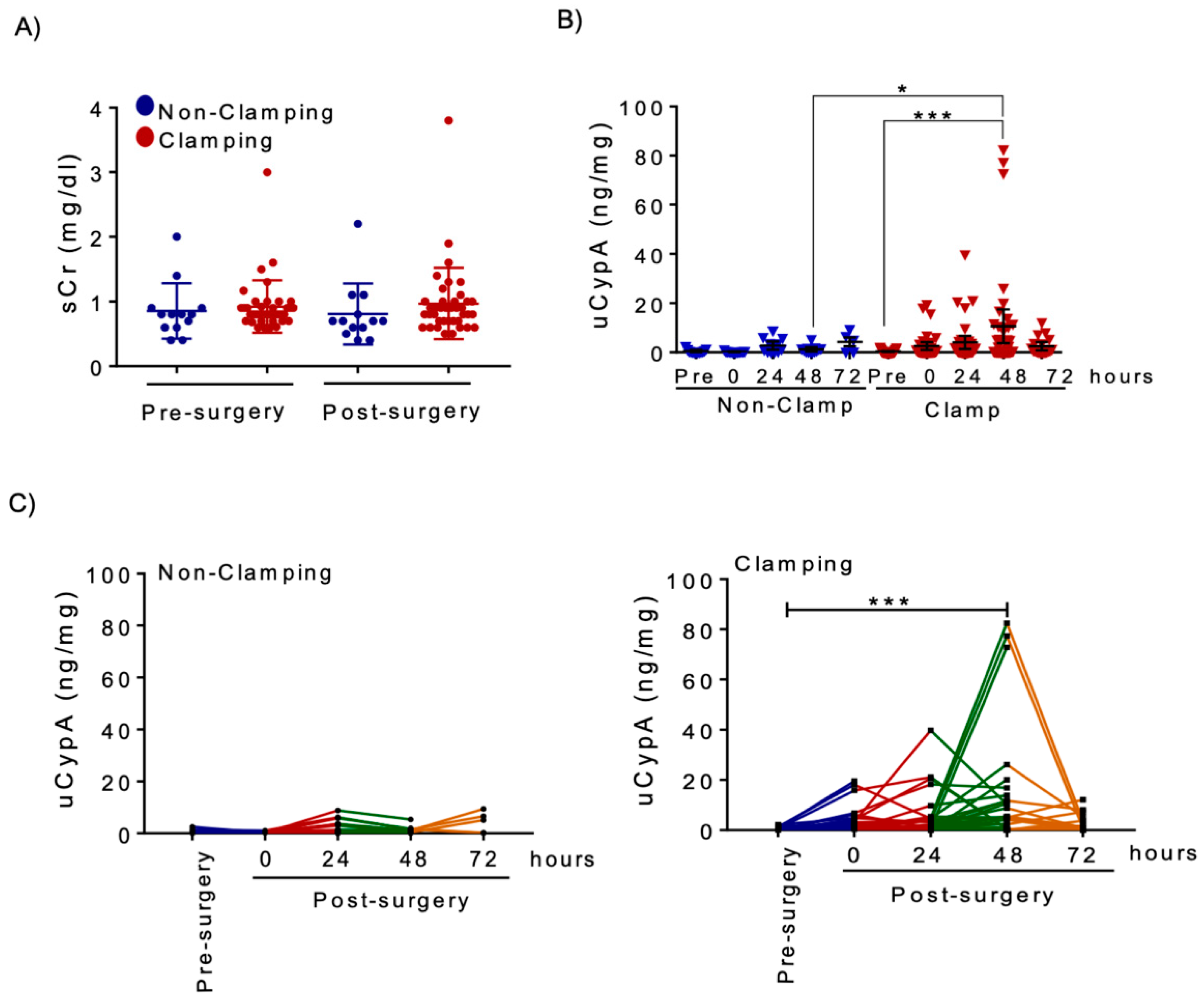
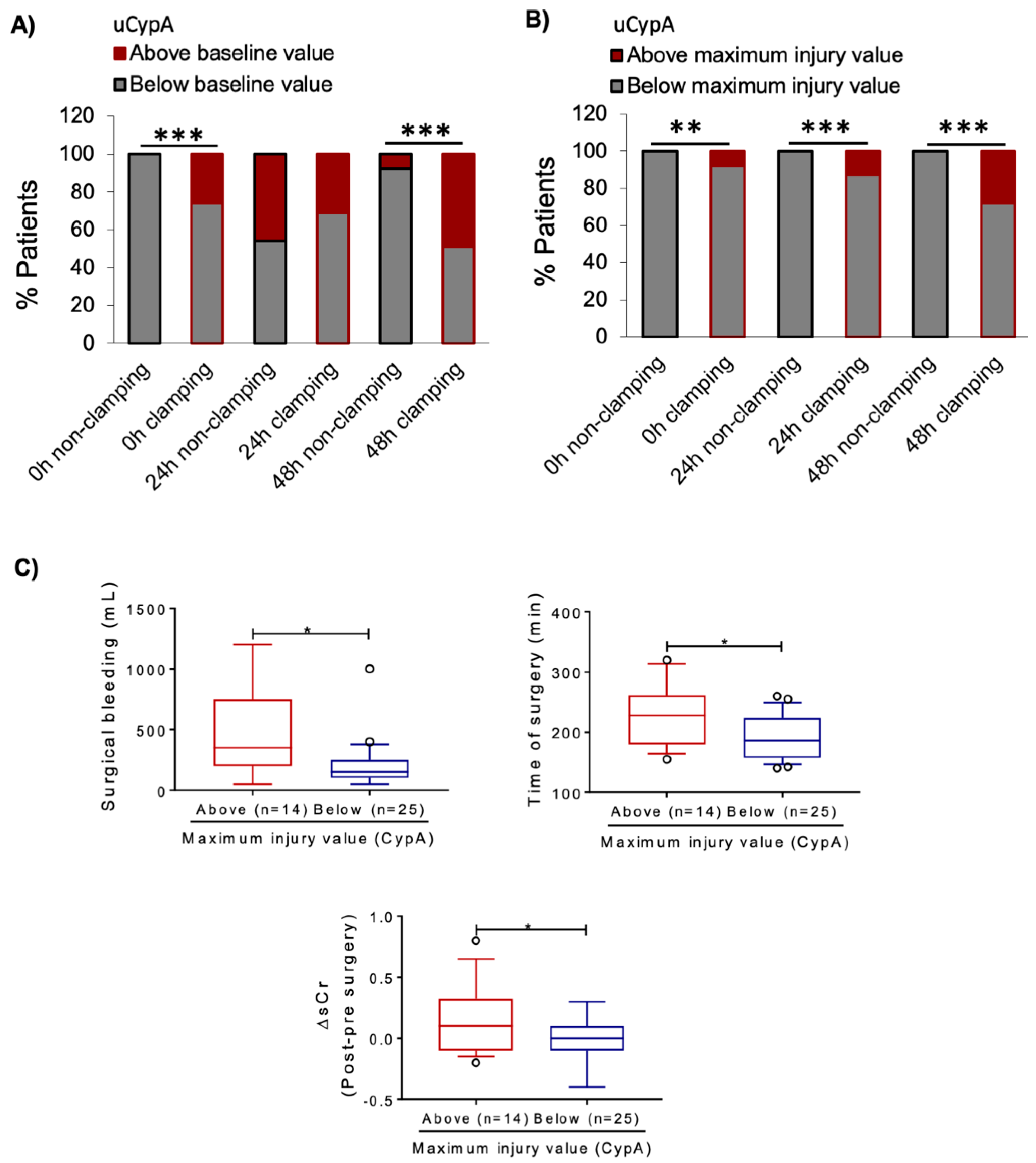
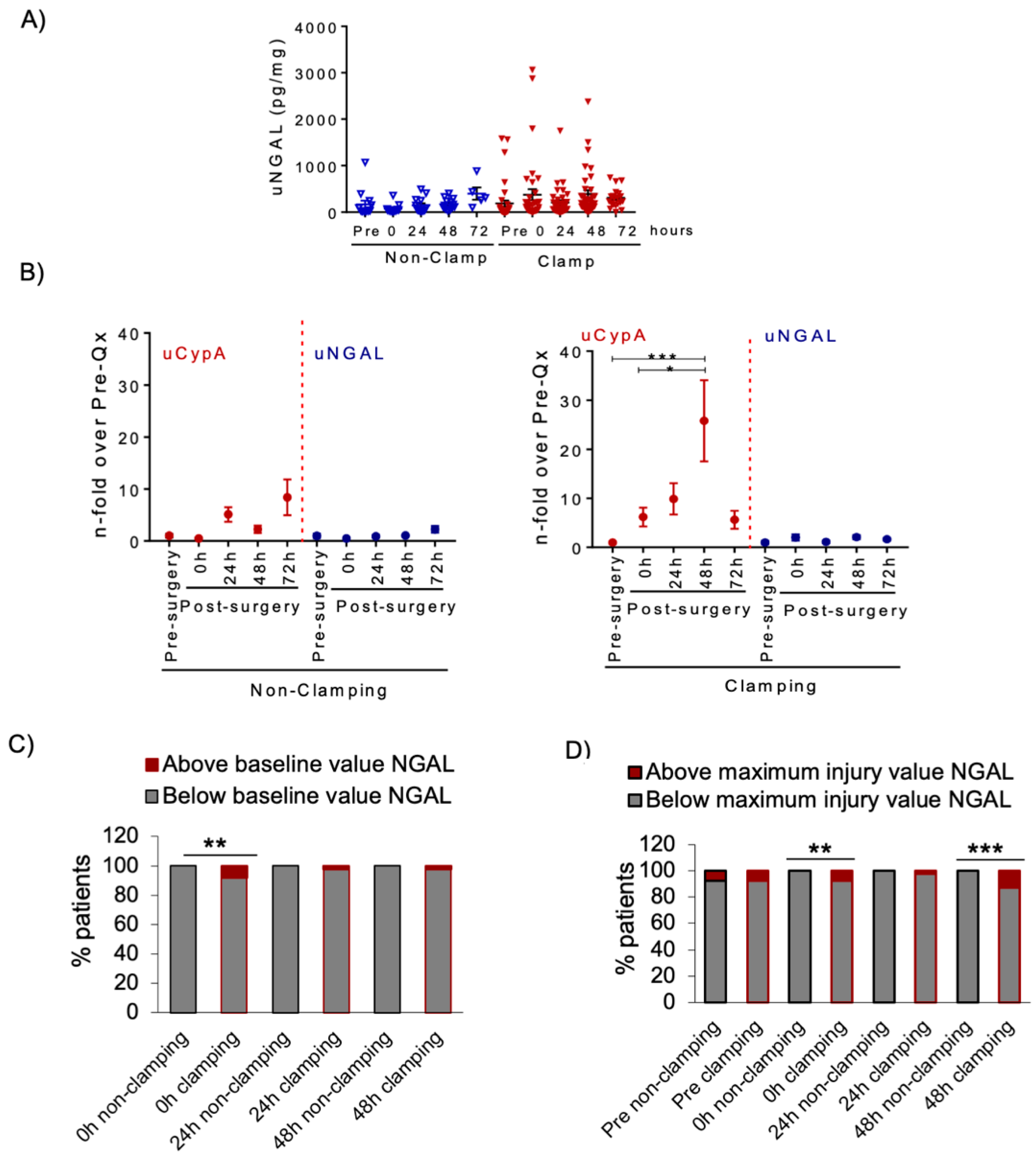
3.3. Renal Artery Clamping during NSS Is Associated with Increased uCypA
3.4. Urine NGAL in NSS Patients
3.5. Urine CypA Levels Following Living Donor KT
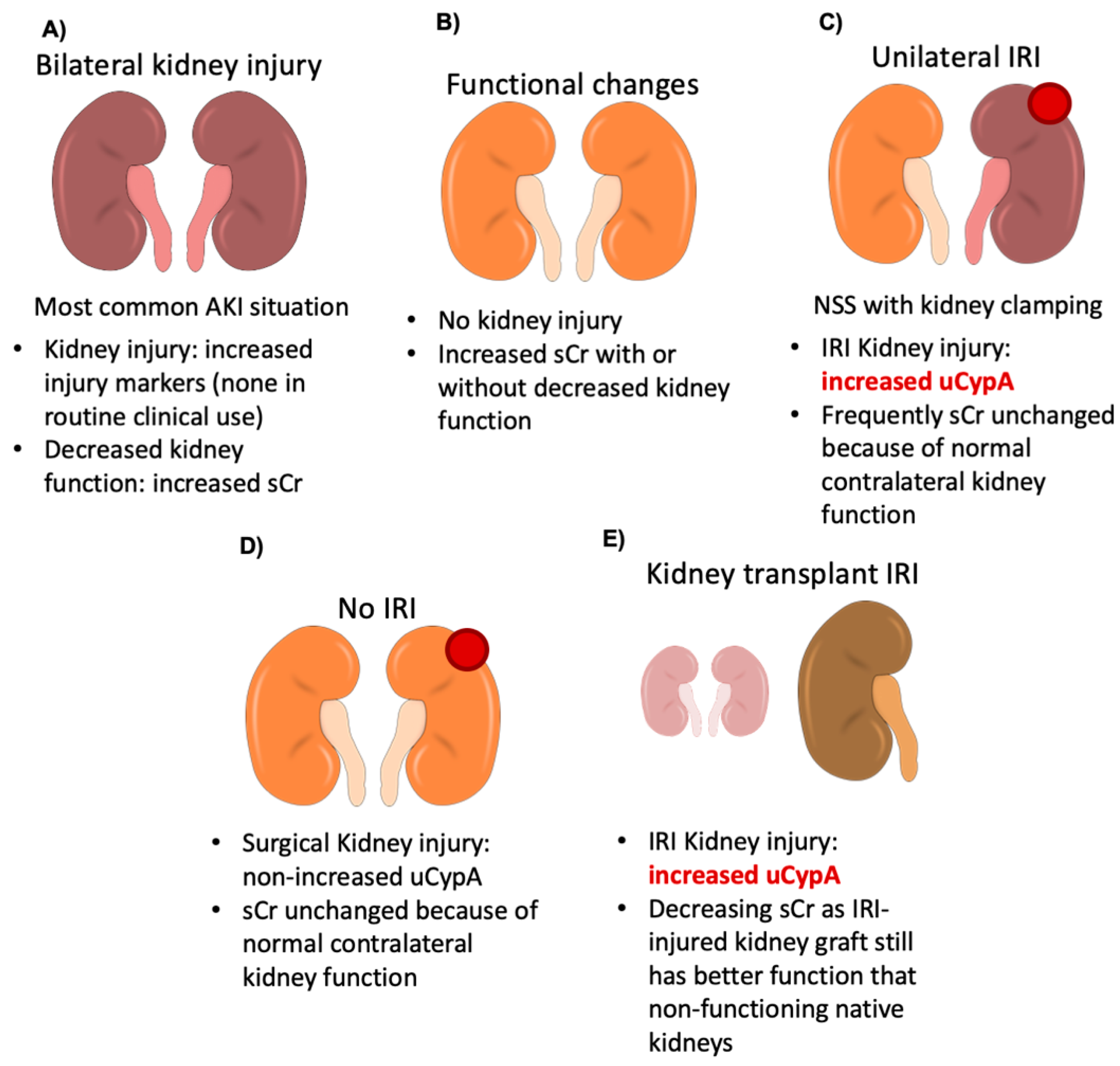
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khwaja, A. KDIGO Clinical Practice Guidelines for Acute Kidney Injury. Nephron 2012, 120, c179–c184. [Google Scholar] [CrossRef] [PubMed]
- Pefanis, A.; Ierino, F.L.; Murphy, J.M.; Cowan, P.J. Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int. 2019, 96, 291–301. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Poveda, J.; Fontecha-Barriuso, M.; Ruiz-Andres, O.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting of regulated necrosis in kidney disease. Nefrología 2018, 38, 125–135. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef] [PubMed]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Carrasco, S.; Sanchez-Niño, M.D.; Von Mässenhausen, A.; Linkermann, A.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; et al. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc. Natl. Acad. Sci. USA 2018, 115, 4182–4187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonelli, A.; Allinovi, M.; Cocci, A.; Russo, G.I.; Schiavina, R.; Rocco, B.; Giovannalberto, P.; Celia, A.; Galfano, A.; Varca, V.; et al. The Predictive Role of Biomarkers for the Detection of Acute Kidney Injury After Partial or Radical Nephrectomy: A Systematic Review of the Literature. Eur. Urol. Focus 2020, 6, 344–353. [Google Scholar] [CrossRef]
- Hong, F.; Lee, J.; Piao, Y.J.; Jae, Y.K.; Kim, Y.-J.; Oh, C.; Seo, J.-S.; Yun, Y.S.; Yang, C.W.; Ha, J.; et al. Transgenic mice overexpressing cyclophilin A are resistant to cyclosporin A-induced nephrotoxicity via peptidyl-prolyl cis–trans isomerase activity. Biochem. Biophys. Res. Commun. 2004, 316, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Roy, S.; Singh, P.; Singla-Pareek, S.L.; Pareek, A. Cyclophilins: Proteins in search of function. Plant Signal. Behav. 2013, 8, e22734. [Google Scholar] [CrossRef] [Green Version]
- Colgan, J.; Asmal, M.; Neagu, M.; Yu, B.; Schneidkraut, J.; Lee, Y.; Sokolskaja, E.; Andreotti, A.; Luban, J. Cyclophilin A Regulates TCR Signal Strength in CD4+ T Cells via a Proline-Directed Conformational Switch in Itk. Immun. 2004, 21, 189–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, P.; Pompilio, G.; Capogrossi, M.C. Cyclophilin A: A key player for human disease. Cell Death Dis. 2013, 4, e888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dornan, J.; Taylor, P.; Walkinshaw, M.D. Structures of immunophilins and their ligand complexes. Curr. Top. Med. Chem. 2003, 3, 1392–1409. [Google Scholar] [CrossRef] [PubMed]
- Dear, J.W.; Simpson, K.J.; Nicolai, M.P.J.; Catterson, J.H.; Street, J.; Huizinga, T.; Craig, D.G.; Dhaliwal, K.; Webb, S.; Bateman, D.N.; et al. Cyclophilin A Is a Damage-Associated Molecular Pattern Molecule That Mediates Acetaminophen-Induced Liver Injury. J. Immunol. 2011, 187, 3347–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofferson, D.E.; Yuan, J. Cyclophilin A release as a biomarker of necrotic cell death. Cell Death Differ. 2010, 17, 1942–1943. [Google Scholar] [CrossRef] [PubMed]
- Ryffel, B.; Woerly, G.; Greiner, B.; Haendler, B.; Mihatsch, M.J.; Foxwell, B.M. Distribution of the cyclosporine binding protein cyclophilin in human tissues. Immunol. 1991, 72, 399–404. [Google Scholar]
- Demeule, M.; Laplante, A.; Sepehr-Araé, A.; Murphy, G.M.; Wenger, R.M.; Béliveau, R. Association of cyclophilin A with renal brush border membranes: Redistribution by cyclosporine A. Kidney Int. 2000, 57, 1590–1598. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.-F.; Su, C.-W.; Wu, M.-J.; Chen, C.-H.; Fu, C.-P.; Liu, C.-S.; Hsieh, M. Urinary Cyclophilin A as a New Marker for Diabetic Nephropathy. Med. 2015, 94, e1802. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Ziyadeh, F.N.; Neilson, E.G. Expression of apoptosis-regulatory genes in renal proximal tubular epithelial cells exposed to high ambient glucose and in diabetic kidneys. J. Investig. Med. 1997, 45, 50–56. [Google Scholar]
- Lee, C.-C.; Chang, C.-H.; Cheng, Y.-L.; Kuo, G.; Chen, S.-W.; Li, Y.-J.; Chen, Y.-T.; Tian, Y.-C. Diagnostic Performance of Cyclophilin A in Cardiac Surgery-Associated Acute Kidney Injury. J. Clin. Med. 2019, 9, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poveda, J.; Sanchez-Niño, M.D.; Glorieux, G.; Sanz, A.B.; Egido, J.; Vanholder, R.; Ortiz, A. p-Cresyl sulphate has pro-inflammatory and cytotoxic actions on human proximal tubular epithelial cells. Nephrol. Dial. Transplant. 2014, 29, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haverty, T.P.; Kelly, C.J.; Hines, W.H.; Amenta, P.S.; Watanabe, M.; Harper, R.A.; Kefalides, N.A.; Neilson, E.G. Characterization of a renal tubular epithelial cell line which secretes the autologous target antigen of autoimmune experimental interstitial nephritis. J. Cell Biol. 1988, 107, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Justo, P.; Sanz, A.; Sanchez-Niño, M.D.; Winkles, J.; Lorz, C.; Egido, J.; Ortiz, A.; Arduan, A.O.; Sanchez-Ni, M.D. Cytokine cooperation in renal tubular cell injury: The role of TWEAK. Kidney Int. 2006, 70, 1750–1758. [Google Scholar] [CrossRef] [Green Version]
- Conde, E.; Alegre, L.; Blanco-Sanchez, I.; Sáenz-Morales, D.; Aguado-Fraile, E.; Ponte, B.; Ramos, E.; Saiz, A.; Jiménez, C.; Ordoñez, A.; et al. Hypoxia Inducible Factor 1-Alpha (HIF-1 Alpha) Is Induced during Reperfusion after Renal Ischemia and Is Critical for Proximal Tubule Cell Survival. PLoS ONE 2012, 7, e33258. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martín-Sanchez, D.; Martinez-Moreno, J.M.; Cardenas-Villacres, D.; Carrasco, S.; Sanchez-Niño, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Molecular pathways driving omeprazole nephrotoxicity. Redox Biol. 2020, 32, 101464. [Google Scholar] [CrossRef]
- Kutikov, A.; Uzzo, R.G. The RENAL Nephrometry Score: A Comprehensive Standardized System for Quantitating Renal Tumor Size, Location and Depth. J. Urol. 2009, 182, 844–853. [Google Scholar] [CrossRef]
- Ficarra, V.; Novara, G.; Secco, S.; Macchi, V.; Porzionato, A.; De Caro, R.; Artibani, W. Preoperative Aspects and Dimensions Used for an Anatomical (PADUA) Classification of Renal Tumours in Patients who are Candidates for Nephron-Sparing Surgery. Eur. Urol. 2009, 56, 786–793. [Google Scholar] [CrossRef]
- Simmons, M.N.; Ching, C.B.; Samplaski, M.K.; Park, C.H.; Gill, I.S. Kidney Tumor Location Measurement Using the C Index Method. J. Urol. 2010, 183, 1708–1713. [Google Scholar] [CrossRef] [PubMed]
- Simmons, M.N. Morphometric characterization of kidney tumors. Curr. Opin. Urol. 2011, 21, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Hakky, T.S.; Baumgarten, A.S.; Allen, B.; Lin, H.-Y.; Ercole, C.E.; Sexton, W.J.; Spiess, P.E. Zonal NePhRO Scoring System: A Superior Renal Tumor Complexity Classification Model. Clin. Genitourin. Cancer 2014, 12, e13–e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaliviero, M.; Poon, B.Y.; Karlo, C.A.; Guglielmetti, G.B.; Di Paolo, P.L.; Corradi, R.B.; Martin-Malburet, A.G.; Campos-Juanatey, F.; Escudero-Fontano, E.; Sjoberg, D.D.; et al. An Arterial Based Complexity (ABC) Scoring System to Assess the Morbidity Profile of Partial Nephrectomy. Eur. Urol. 2016, 69, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaszewski, J.J.; Smaldone, M.C.; Cung, B.; Li, T.; Mehrazin, R.; Kutikov, A.; Canter, D.J.; Viterbo, R.; Chen, D.Y.; Greenberg, R.E.; et al. Internal validation of the renal pelvic score: A novel marker of renal pelvic anatomy that predicts urine leak after partial nephrectomy. Urol. 2014, 84, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Davidiuk, A.J.; Parker, A.S.; Thomas, C.S.; Leibovich, B.C.; Castle, E.P.; Heckman, M.G.; Custer, K.; Thiel, D.D. Mayo Adhesive Probability Score: An Accurate Image-based Scoring System to Predict Adherent Perinephric Fat in Partial Nephrectomy. Eur. Urol. 2014, 66, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Rodriguez, E.; Fernandez-Prado, R.; Martin-Cleary, C.; Pizarro-Sánchez, M.S.; Sanchez-Niño, M.D.; Sanz, A.B.; Fernandez-Fernandez, B.; Ortiz, A. Kidney Injury Marker 1 and Neutrophil Gelatinase-Associated Lipocalin in Chronic Kidney Disease. Nephron 2016, 136, 263–267. [Google Scholar] [CrossRef]
- Srisawat, N.; Kellum, J.A. The Role of Biomarkers in Acute Kidney Injury. Crit. Care Clin. 2020, 36, 125–140. [Google Scholar] [CrossRef]
- Moledina, D.G.; Parikh, C.R. Phenotyping of Acute Kidney Injury: Beyond Serum Creatinine. Semin. Nephrol. 2018, 38, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Paragas, N.; Qiu, A.; Zhang, Q.; Samstein, B.; Deng, S.-X.; Schmidt-Ott, K.M.; Viltard, M.; Yu, W.; Forster, C.S.; Gong, G.; et al. The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat. Med. 2011, 17, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Kashani, K.; Cheungpasitporn, W.; Ronco, C. Biomarkers of acute kidney injury: The pathway from discovery to clinical adoption. Clin. Chem. Lab. Med. 2017, 55, 1074–1089. [Google Scholar] [CrossRef]
- Albert, C.; Zapf, A.; Haase, M.; Röver, C.; Pickering, J.W.; Albert, A.; Bellomo, R.; Breidthardt, T.; Camou, F.; Chen, Z.; et al. Neutrophil Gelatinase-Associated Lipocalin Measured on Clinical Laboratory Platforms for the Prediction of Acute Kidney Injury and the Associated Need for Dialysis Therapy: A Systematic Review and Meta-analysis. Am. J. Kidney Dis. 2020, 76, 826–841.e1. [Google Scholar] [CrossRef]
- Suzuki, J.; Jin, Z.-G.; Meoli, D.F.; Matoba, T.; Berk, B.C. Cyclophilin A Is Secreted by a Vesicular Pathway in Vascular Smooth Muscle Cells. Circ. Res. 2006, 98, 811–817. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Lin, M.; Zhang, H.; Li, J.; Wu, M.; Lv, H.; Wang, J.; Xie, S. The Release of Cyclophilin A from Rapamycin-Stimulated Vascular Smooth Muscle Cells Mediated by Myosin II Activation: Involvement of Apoptosis but Not Autophagy. J. Vasc. Res. 2020, 57, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.; Lin, C.; Chen, T.; Hung, T.; Lin, H.; Chiu, H.; Chiang, C.; Lin, F.; Fu, E. Fibroblast-enhanced cyclophilin A releasing from U937 cell upregulates MMP-2 in gingival fibroblast. J. Periodontal Res. 2020, 55, 705–712. [Google Scholar] [CrossRef]
- Ramachandran, S.; Venugopal, A.; Charles, S.; Chandran, N.P.; Mullassari, A.; Pillai, M.R.; Kartha, C.C. Proteomic profiling of high glucose primed monocytes identifies cyclophilin A as a potential secretory marker of inflammation in type 2 diabetes. Proteom. 2012, 12, 2808–2821. [Google Scholar] [CrossRef] [PubMed]
- Seko, Y.; Fujimura, T.; Taka, H.; Mineki, R.; Murayama, K.; Nagai, R. Hypoxia followed by reoxygenation induces secretion of cyclophilin A from cultured rat cardiac myocytes. Biochem. Biophys. Res. Commun. 2004, 317, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.H.; Song, J.W.; Shin, E.-J.; Kwak, Y.-L.; Choi, N.; Shim, J.-K. Ethyl pyruvate is renoprotective against ischemia-reperfusion injury under hyperglycemia. J. Thorac. Cardiovasc. Surg. 2018, 155, 1650–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ortega, M.R.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid–Induced AKI. J. Am. Soc. Nephrol. 2016, 28, 218–229. [Google Scholar] [CrossRef]
- Dear, J.W.; Leelahavanichkul, A.; Aponte, A.; Hu, X.; Constant, S.L.; Hewitt, S.M.; Yuen, P.S.; Star, R.A. Liver proteomics for therapeutic drug discovery: Inhibition of the cyclophilin receptor CD147 attenuates sepsis-induced acute renal failure*. Crit. Care Med. 2007, 35, 2319–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Niño, M.-D.; Benito-Martin, A.; Ortiz, A.; Arduan, A.O. New paradigms in cell death in human diabetic nephropathy. Kidney Int. 2010, 78, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NSS without Arterial Clamp (n = 13) | NSS with Arterial Clamp (n = 39) | p Value | |
|---|---|---|---|
| Age (years, mean ± SD) | 65.50 ± 8.97 | 60.30 ± 11.50 | 0.155 |
| Female sex | 5 (38.4%) | 10 (25%) | 0.278 |
| Right kidney tumor | 4 (30.7%) | 19 (48.7%) | 0.235 |
| Tumor size * (mm, mean ± SD) | 22.5 ± 8.93 | 31.3 ± 8.93 | 0.006 |
| Nephrometric scores | |||
| RENAL (median (IQR)) [25] | 6 (1.00) | 6 (2.00) | 0.521 |
| PADUA (median (IQR)) [26] | 7 (0.00) | 8 (2.00) | 0.071 |
| C.INDEX (mean ± SD) [27] | 3.25 ± 1.47 | 2.52 ± 0.92 | 0.141 |
| DAP (median (IQR)) [28] | 5 (2.00) | 6 (2.00) | 0.021 |
| NePhRO (median (IQR)) [29] | 6 (2.00) | 8 (2.00) | 0.011 |
| ABC category [30] | |||
| 1 | 5 (38.4%) | 8 (20.5%) | 0.124 |
| 2 | 7 (53.8%) | 23 (58.9%) | |
| 3H | 1 (7.6%) | 2 (5.1%) | |
| 3S | 0 | 6 (15,3%) | |
| RPS [31] <50% | 3 (23%) | 7 (17.9%) | 1.000 |
| Mayo score (median (IQR) [32] | 3 (2.00) | 3 (3.00) | 0.468 |
| Ischemia time WIT (min, mean ± SD) | 0 | 23.83 ± 8.38 | |
| KDIGO criteria for AKI | 1 (7.69%) | 4 (10.25%) | 0.613 |
| Kidney Donors (n = 6) | Kidney Recipients (n = 6) | |
|---|---|---|
| Age (years) | 58.83 ± 5.91 | 54.00 ± 20.43 |
| Female sex | 4 (66.6%) | 3 (50%) |
| Renal ischemia | ||
| CIT (min) | 83.50 ± 7.60 | |
| WIT (min) | 3.17 ± 1.27 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabello, R.; Fontecha-Barriuso, M.; Martin-Sanchez, D.; Lopez-Diaz, A.M.; Carrasco, S.; Mahillo, I.; Gonzalez-Enguita, C.; Sanchez-Niño, M.D.; Ortiz, A.; Sanz, A.B. Urinary Cyclophilin A as Marker of Tubular Cell Death and Kidney Injury. Biomedicines 2021, 9, 217. https://doi.org/10.3390/biomedicines9020217
Cabello R, Fontecha-Barriuso M, Martin-Sanchez D, Lopez-Diaz AM, Carrasco S, Mahillo I, Gonzalez-Enguita C, Sanchez-Niño MD, Ortiz A, Sanz AB. Urinary Cyclophilin A as Marker of Tubular Cell Death and Kidney Injury. Biomedicines. 2021; 9(2):217. https://doi.org/10.3390/biomedicines9020217
Chicago/Turabian StyleCabello, Ramio, Miguel Fontecha-Barriuso, Diego Martin-Sanchez, Ana M. Lopez-Diaz, Susana Carrasco, Ignacio Mahillo, Carmen Gonzalez-Enguita, Maria D. Sanchez-Niño, Alberto Ortiz, and Ana B. Sanz. 2021. "Urinary Cyclophilin A as Marker of Tubular Cell Death and Kidney Injury" Biomedicines 9, no. 2: 217. https://doi.org/10.3390/biomedicines9020217
APA StyleCabello, R., Fontecha-Barriuso, M., Martin-Sanchez, D., Lopez-Diaz, A. M., Carrasco, S., Mahillo, I., Gonzalez-Enguita, C., Sanchez-Niño, M. D., Ortiz, A., & Sanz, A. B. (2021). Urinary Cyclophilin A as Marker of Tubular Cell Death and Kidney Injury. Biomedicines, 9(2), 217. https://doi.org/10.3390/biomedicines9020217