
Zoom in on Antibody Aggregates: A Potential Pitfall in the Search of Rare EV Populations

 , , and
, , and
Abstract
1. Introduction
2. Experimental Section
2.1. Samples
2.2. Labels
2.3. Label Panels
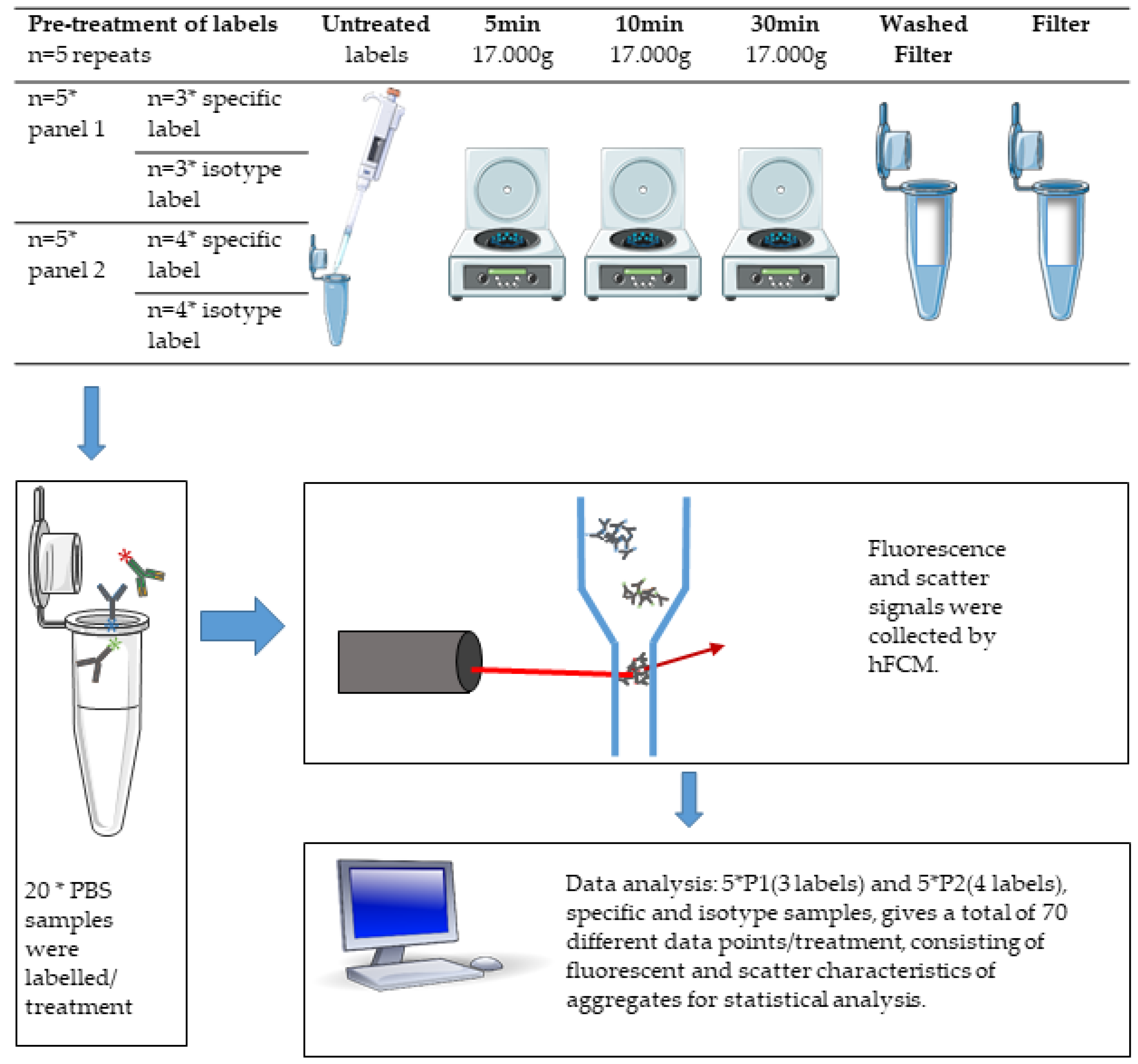
2.4. Pre-Treatment of Labels
2.5. Staining of Samples
2.6. Setup
2.7. Analysis of Label-Aggregates by hFCM
2.8. Statistical Data Analysis
3. Results
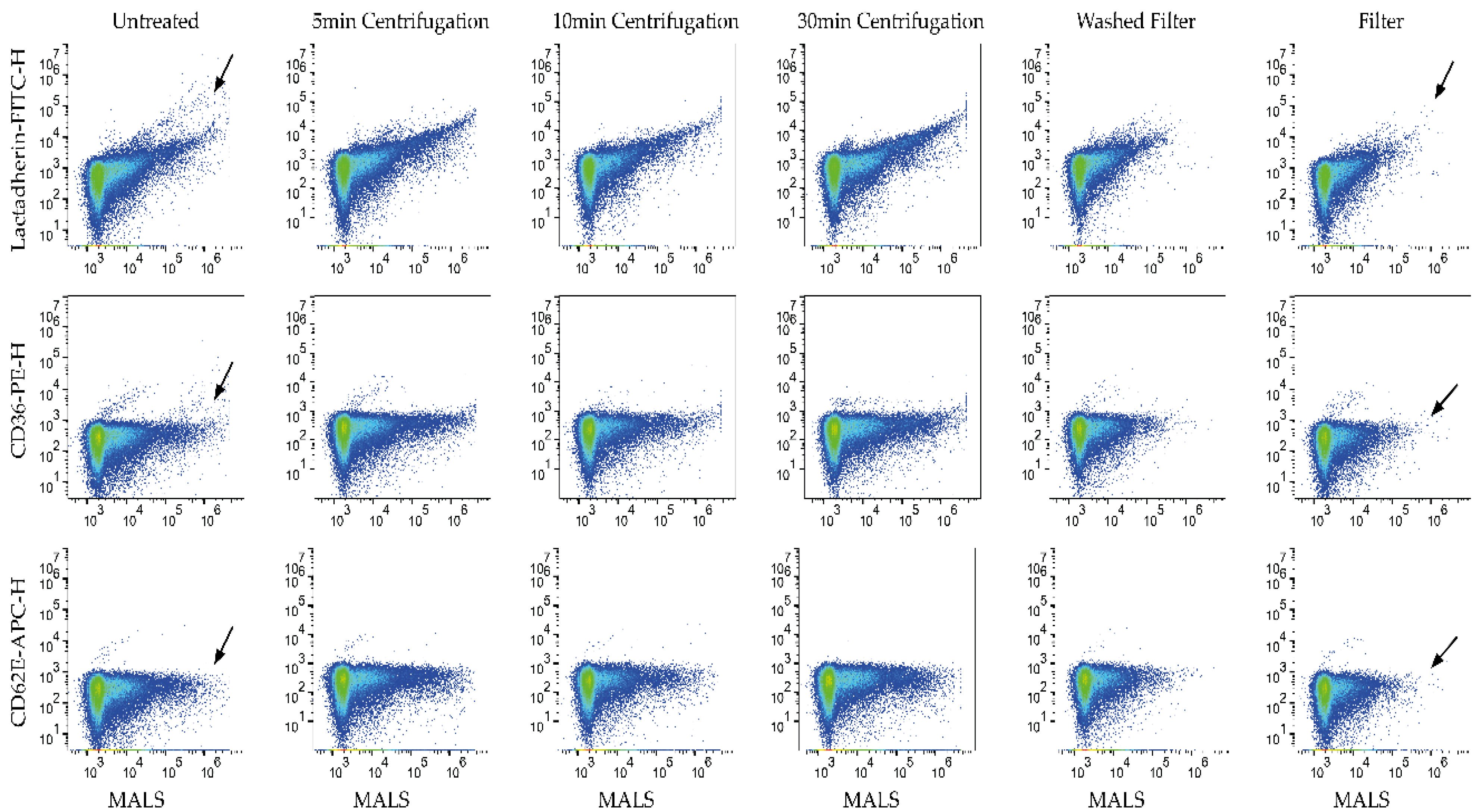
3.1. Aggregates Were Present in Untreated Labels
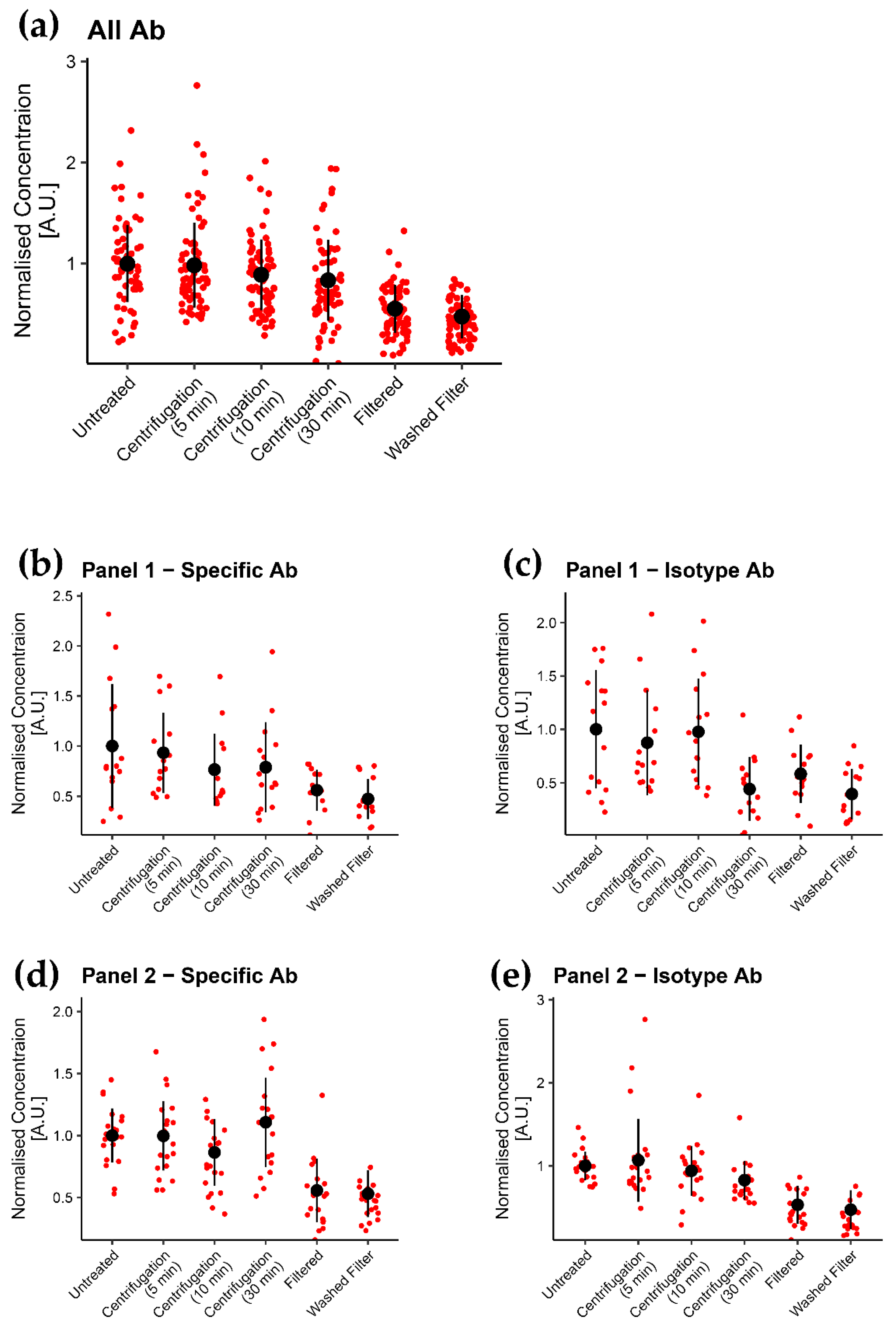
3.2. Filtration of Labels Is the Most Efficient of the Tested Methods in Removing Label Aggregates
3.3. Aggregates Were Considerably Reduced by Using Filter
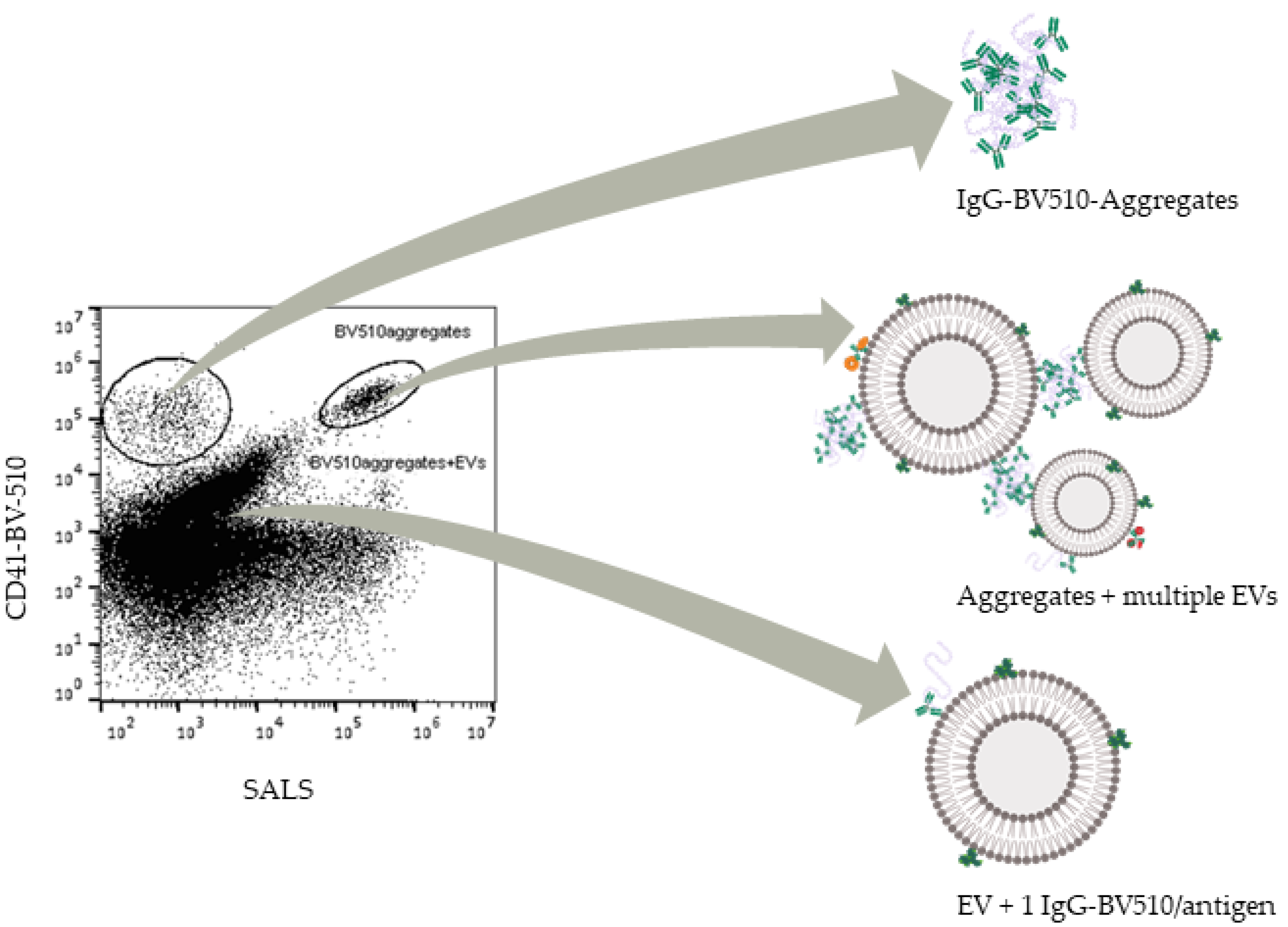
3.4. Filtrated Labels Are Functional; However, Some Aggregates Pass through the Filter
4. Discussion
4.1. Aggregates in the Size Range of EVs Are Present in Labelled PBS
4.2. Efficacy of Treatments
4.3. Reasons for Aggregation and Implications in EV Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.; Vizio, D.D.; Sahoo, S.; Théry, C.; Witwer, K.W.; Wauben, M.; Hill, A.F. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. J. Extracell. Vesicles 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Coumans, F.A.W.; Grootemaat, A.E.; Gardiner, C.; Sargent, I.L.; Harrison, P.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J. Thromb. Haemost. 2014, 12, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- György, B.; Módos, K.; Pállinger, É.; Pálóczi, K.; Pásztói, M.; Misják, P.; Deli, M.A.; Sipos, Á.; Szalai, A.; Voszka, I.; et al. Detection and isolation of cell-derived microparticles are compromised by protein complexes resulting from shared biophysical parameters. Blood 2011, 117, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Prabakaran, P.; Chen, W.; Zhu, Z.; Feng, Y.; Dimitrov, D. Antibody Aggregation: Insights from Sequence and Structure. Antibodies 2016, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nema, S.; Teagarden, D. Protein aggregation—Pathways and influencing factors. Int. J. Pharm. 2010, 390, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Aass, H.C.D.; Øvstebø, R.; Trøseid, A.M.S.; Kierulf, P.; Berg, J.P.; Henriksson, C.E. Fluorescent particles in the antibody solution result in false TF- and CD14-positive microparticles in flow cytometric analysis. Cytom. Part A 2011, 79, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Inglis, H.; Norris, P.; Danesh, A. Techniques for the analysis of extracellular vesicles using flow cytometry. J. Vis. Exp. 2015, 2015, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Erdbrügger, U.; Lannigan, J. Analytical challenges of extracellular vesicle detection: A comparison of different techniques. Cytom. Part A 2016, 89, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Görgens, A.; Bremer, M.; Ferrer-Tur, R.; Murke, F.; Tertel, T.; Horn, P.A.; Thalmann, S.; Welsh, J.A.; Probst, C.; Guerin, C.; et al. Optimisation of imaging flow cytometry for the analysis of single extracellular vesicles by using fluorescence-tagged vesicles as biological reference material. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Erdbrügger, U.; Rudy, C.K.; Etter, M.E.; Dryden, K.A.; Yeager, M.; Klibanov, A.L.; Lannigan, J. Imaging flow cytometry elucidates limitations of microparticle analysis by conventional flow cytometry. Cytom. Part A 2014, 85, 756–770. [Google Scholar] [CrossRef]
- Lacroix, R.; Robert, S.; Poncelet, P.; Dignat-George, F. Overcoming limitations of microparticle measurement by flow cytometry. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2010. [Google Scholar] [CrossRef]
- Ayers, L.; Kohler, M.; Harrison, P.; Sargent, I.; Dragovic, R.; Schaap, M.; Nieuwland, R.; Brooks, S.A.; Ferry, B. Measurement of circulating cell-derived microparticles by flow cytometry: Sources of variability within the assay. Thromb. Res. 2011, 127, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Van der Vlist, E.J.; Nolte-’t Hoen, E.N.M.; Stoorvogel, W.; Arkesteijn, G.J.A.; Wauben, M.H.M. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat. Protoc. 2012, 7, 1311–1326. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, R.; Judicone, C.; Poncelet, P.; Robert, S.; Arnaud, L.; Sampol, J.; Dignat-George, F. Impact of pre-analytical parameters on the measurement of circulating microparticles: Towards standardization of protocol. J. Thromb. Haemost. 2012, 10, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Dragulescu, A.A. xlsx: Read, Write, Format Excel 2007 and Excel 97/2000/XP/2003 Files. 2014. Available online: https://github.com/colearendt/xlsx (accessed on 8 February 2021).
- Wickham, H. Reshaping Data with the reshape Package. J. Stat. Softw. 2007, 21. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Murell, P. R Graphics; Lafferty, J., Madigan, D., Murtagh, F., Smyth, P., Eds.; Chapman & Hall/CRC: London, UK, 2006; ISBN 978-1439831762. [Google Scholar]
- Inglis, H.C.; Danesh, A.; Shah, A.; Lacroix, J.; Spinella, P.C.; Norris, P.J. Techniques to improve detection and analysis of extracellular vesicles using flow cytometry. Cytom. Part A 2015, 87, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Arraud, N.; Gounou, C.; Linares, R.; Brisson, A.R. A simple flow cytometry method improves th17e detection of phosphatidylserine-exposing extracellular vesicles. J. Thromb. Haemost. 2015, 13, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Botha, J.; Nielsen, M.H.; Christensen, M.H.; Vestergaard, H.; Handberg, A. Bariatric surgery reduces CD36-bearing microvesicles of endothelial and monocyte origin 11 Medical and Health Sciences 1103 Clinical Sciences. Nutr. Metab. 2018, 15, 1–9. [Google Scholar] [CrossRef]
- Berg, J.; Tymoczko, J.; Stryer, L. Biochemistry; Freeman and Company: New York, NY, USA, 2002; ISBN 0-7167-3051-0. [Google Scholar]
- Van Der Pol, E.; Van Gemert, M.J.C.; Sturk, A.; Nieuwland, R.; Van Leeuwen, T.G. Single vs. swarm detection of microparticles and exosomes by flow cytometry. J. Thromb. Haemost. 2012, 10, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Xu R, Simpson RJ, Greening DW. A protocol for isolation and proteomic characterization of distinct extracellular vesicle subtypes by sequential centrifugal ultrafiltration. Methods Mol. Biol. 2017, 1545, 91–116. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Panel | Specific Label | Isotype Control Label |
|---|---|---|
| 1 | 5 µL bovine lactadherin-FITC, Cat. #BLAC-FITC, lot #GG0420 | 5 µL bovine lactadherin-FITC, Cat. #BLAC-FITC, lot #GG0420 |
| 2.5 µL mouse monoclonal anti-humanCD36-PE, Cat. #336206, lot #B219025 | 2.5 µL mouse monoclonal IgG2a, κ-PE, Cat. #400214, lot #B213581 | |
| 2.5 µL mouse monoclonal anti-human CD62E-APC, Cat. #336012, lot #B194637 | 2.5 µL mouse monoclonal IgG1, κ-APC, Cat. #400122, lot #B210432 1 | |
| 2 | 5 µL bovine lactadherin-FITC, Cat. #BLAC-FITC, lot #HH0216 | 5 µL bovine lactadherin-FITC, Cat. #BLAC-FITC, lot #HH0216 |
| 2.5 µL mouse monoclonal anti-human CD36-PE, Cat. #336206, lot #B219025 | 2.5 µL mouse monoclonal IgG2a, κ-PE, Cat. #400214, lot #B213581 | |
| 2.5 µL mouse monoclonal anti-human CD14-APC, Cat. #367118, lot #B230117 | 2.5 µL mouse monoclonal IgG1, κ-APC, Cat. #400122, lot #B216781 | |
| 2.5 µL mouse monoclonal anti-human Leukotrine B4 R1-AF700, Cat. #NB100-64831AF700, lot #0709-052517 2 | 2.5 µL mouse monoclonal IgG2a-AF700, Cat. #IC003N, lot #ACIT025061 | |
| 3 | 5 µL bovine lactadherin-FITC, Cat. #BLAC-FITC, lot #HH0216 | 5 µL bovine lactadherin-FITC, Cat. #BLAC-FITC, lot #HH0216 |
| 2.5 µL mouse monoclonal anti-human CD41a-BV510, Cat. #563250, lot #7164967 | 2.5 µL mouse monoclonal IgG1, κ-BV510, Cat. #562946, lot #7282966 3 | |
| 5 µL mouse monoclonal anti-human CD9-PE, Cat. #312106, lot #B251912 | 5 µL mouse monoclonal IgG1, κ-PE, Cat. #400112, lot #B245983 4 |
| Laser | Collected Signal | PMT 1 | LP/BP 6 Filter |
|---|---|---|---|
| 405 nm (190 mW) | SALS 2 | 545 | LP415 |
| - | MALS 3 | 405 | - |
| - | LALS 4 | 410 | - |
| - | BV510 5 | 500 | BP 525/40 |
| 488 nm (100 mW) | FITC 5 | 480 | BP 530/40 |
| - | PE 5 | 407 | BP 575/30 |
| 638 nm (100 mW) | APC 5 | 450 | BP 680/35 |
| - | AF700 5 | 520 | LP 740 |
| Panel | Label | Fluorophore | Mean 3 Conc. ± SD (Events/µL) | FC ± SD 4 | p-Value |
|---|---|---|---|---|---|
| PBS | Unlabelled | FITC (n = 13) | 3798 ± 2360 | - | - |
| - | - | PE (n = 13) | 92 ± 103 | - | - |
| - | - | APC (n = 13) | 35 ± 64 | - | - |
| - | - | AF700 (n = 13) | 6642 ± 1898 | - | - |
| P1(U) 1 | Specific | FITC (n = 5) | 66,234 ± 52,782 | 17.4 ± 13.9 | 0.0021 |
| - | - | PE (n = 5) | 10,141 ± 3558 | 110.1 ± 38.6 | 0.0018 |
| - | - | APC (n = 5) | 1844 ± 1401 | 52.0 ± 39.5 | 0.26 |
| P1(U) 1 | Isotype | FITC (n = 5) | 69,816 ± 44,351 | 18.4 ± 11.7 | 0.0021 |
| - | - | PE (n = 5) | 10,069 ± 5897 | 109.3 ± 64 | 0.0087 |
| - | - | APC (n = 5) | 1736 ± 990 | 48.9 ± 27.9 | 0.31 |
| P2(U) 2 | Specific | FITC (n = 5) | 38,179 ± 5098 | 10.1 ± 1.3 | 0.0021 |
| - | - | PE (n = 5) | 37,458 ± 10,780 | 406.6 ± 117.0 | 0.0005 |
| - | - | APC (n = 5) | 24,647 ± 9529 | 694.9 ± 268.7 | 0.0022 |
| - | - | AF700 (n = 5) | 25,931 ± 3521 | 3.9 ± 0.5 | 0.0018 |
| P2(U) 2 | Isotype | FITC (n = 5) | 106,293 ± 21,650 | 28.0 ± 5.7 | 0.0021 |
| - | - | PE (n = 5) | 67,109 ± 9426 | 728.4 ± 102.3 | 0.0005 |
| - | - | APC (n = 5) | 63,642 ± 18,649 | 1794.3 ± 525.8 | 0.0022 |
| - | - | AF700 (n = 5) | 48907 ± 6233 | 7.4 ± 0.9 | 0.0018 |
| Mean U total | N.A. | FITC (n = 20) | 70,130 ± 41,492 | 18.4 ± 10.9 | <0.001 |
| - | - | PE (n = 20) | 31,194 ± 25,239 | 338.6 ± 273.9 | <0.001 |
| - | - | APC (n = 10) | 22,967 ± 27,660 | 648 ± 780 | 0.0021 |
| - | - | AF700 (n = 10) | 37,419 ± 13,016 | 5.6 ± 2.0 | <0.001 |
| Treatment | Fluorophore (All Panels) | Mean Conc. ± SD (Events/µL) | Reduction % (Compared to U) ± SD | CV % (Reduction) |
|---|---|---|---|---|
| C5 | FITC (n = 20) | 98,485 ± 72,816 | −31 ± 61 | - |
| - | PE (n = 20) | 29,665 ± 24,401 | 8 ± 27 | - |
| - | APC (n = 20) | 19,178 ± 22,428 | 25 ± 30 | - |
| - | AF700 (n = 10) | 34,983 ± 12,557 | 6 ± 16 | - |
| C5 total | All (n = 70) | 47,091 ± 53,701 | 2 ± 45 | 2477 |
| C10 | FITC (n = 20) | 78,410 ± 46,615 | −9 ± 47 | - |
| - | PE (n = 20) | 26,480 ± 25,301 | 16 ± 31 | - |
| - | APC (n = 20) | 18,287 ± 23,235 | 33 ± 25 | - |
| - | AF700 (n = 10) | 32,627 ± 12,169 | 12 ± 18 | - |
| C10 total | All (n = 70) | 39,845 ± 39,594 | 13 ± 37 | 281 |
| C30 | FITC (n = 20) | 65,824 ± 37,851 | 5 ± 42 | - |
| - | PE (n = 20) | 24,564 ± 21,156 | 30 ± 30 | - |
| - | APC (n = 20) | 19,953 ± 21,083 | 29 ± 53 | - |
| - | AF700 (n = 10) | 33,404 ± 9282 | 6 ± 25 | - |
| C30 total | All (n = 70) | 36,298 ± 31,979 | 19 ± 42 | 216 |
| F | FITC (n = 20) | 25,340 ± 16,192 | 63 ± 22 | - |
| - | PE (n = 20) | 15,281 ± 12,283 | 49 ± 16 | - |
| - | APC (n = 20) | 13,875 ± 16,425 | 33 ± 26 | - |
| - | AF700 (n = 10) | 22,630 ± 10,008 | 40 ± 16 | - |
| F total | All (n = 70) | 18,803 ± 15,041 | 47 ± 24 | 51 |
| WF | FITC (n = 20) | 26,350 ± 15,525 | 62 ± 20 | - |
| - | PE (n = 20) | 12,586 ± 11,454 | 59 ± 18 | - |
| - | APC (n = 20) | 8470 ± 9490 | 57 ± 17 | - |
| - | AF700 (n = 10) | 20,911 ± 7871 | 43 ± 13 | - |
| WF total | All (n = 70) | 16,532 ± 13,764 | 57 ± 18 | 32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasmussen, R.W.; Botha, J.; Prip, F.; Sanden, M.; Nielsen, M.H.; Handberg, A. Zoom in on Antibody Aggregates: A Potential Pitfall in the Search of Rare EV Populations. Biomedicines 2021, 9, 206. https://doi.org/10.3390/biomedicines9020206
Rasmussen RW, Botha J, Prip F, Sanden M, Nielsen MH, Handberg A. Zoom in on Antibody Aggregates: A Potential Pitfall in the Search of Rare EV Populations. Biomedicines. 2021; 9(2):206. https://doi.org/10.3390/biomedicines9020206
Chicago/Turabian StyleRasmussen, Rikke W., Jaco Botha, Frederik Prip, Mathilde Sanden, Morten H. Nielsen, and Aase Handberg. 2021. "Zoom in on Antibody Aggregates: A Potential Pitfall in the Search of Rare EV Populations" Biomedicines 9, no. 2: 206. https://doi.org/10.3390/biomedicines9020206
APA StyleRasmussen, R. W., Botha, J., Prip, F., Sanden, M., Nielsen, M. H., & Handberg, A. (2021). Zoom in on Antibody Aggregates: A Potential Pitfall in the Search of Rare EV Populations. Biomedicines, 9(2), 206. https://doi.org/10.3390/biomedicines9020206