
IL-20 Cytokines Are Involved in Epithelial Lesions Associated with Virus-Induced COPD Exacerbation in Mice

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Viral Challenge
2.3. Sample Collection and Processing
2.4. Flow Cytometry
2.5. Lung Histology
2.6. Cytokine Quantification by ELISA
2.7. mRNA Expression Quantification by Reverse Transcription-Polymerase Chain Reaction (RT-qPCR)
2.8. Statistical Analysis
3. Results
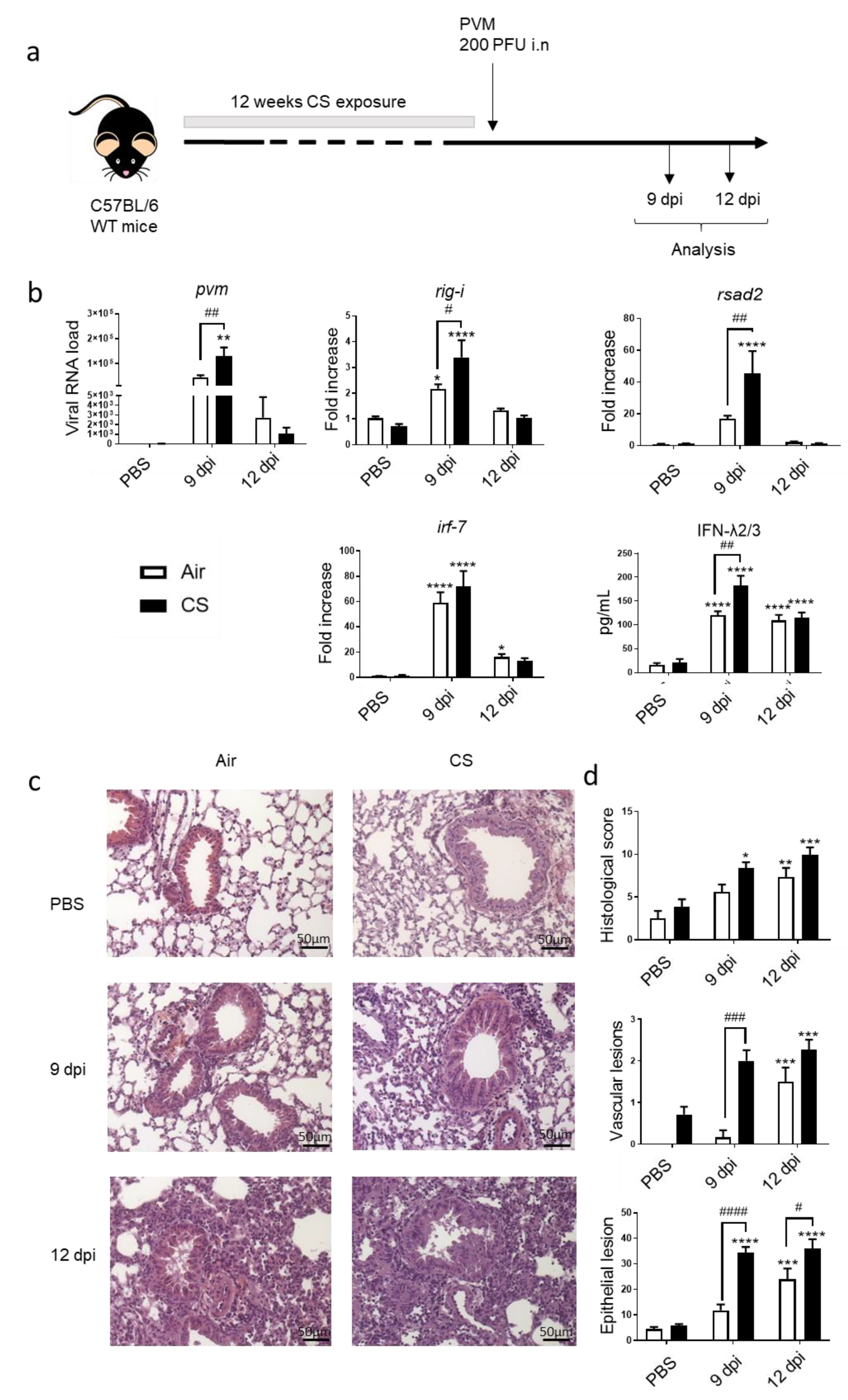
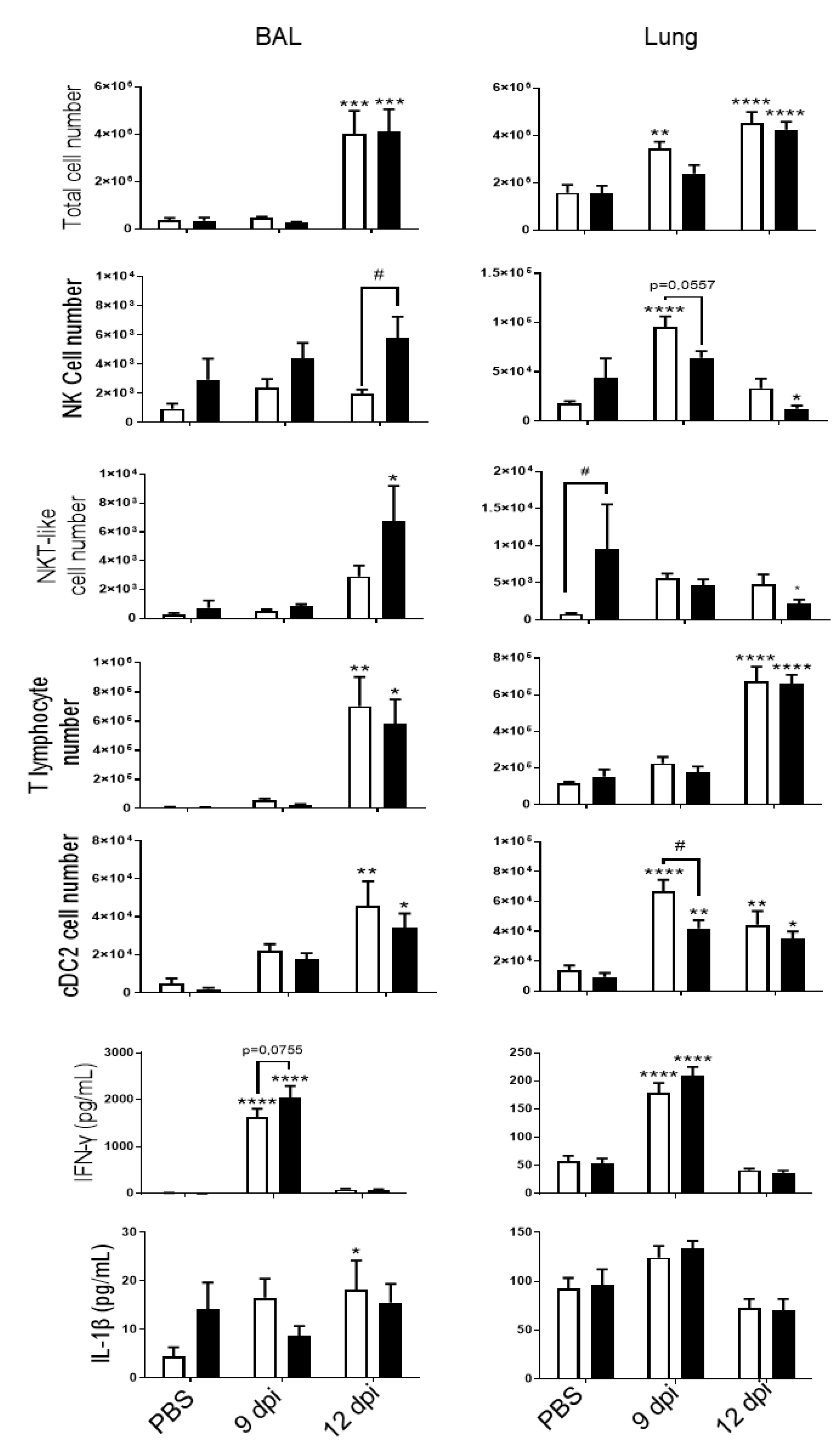
3.1. PVM Infection Exacerbates CS-Induced Inflammation and Antiviral Response
3.2. PVM Infection Modulates IL-20 Pathway
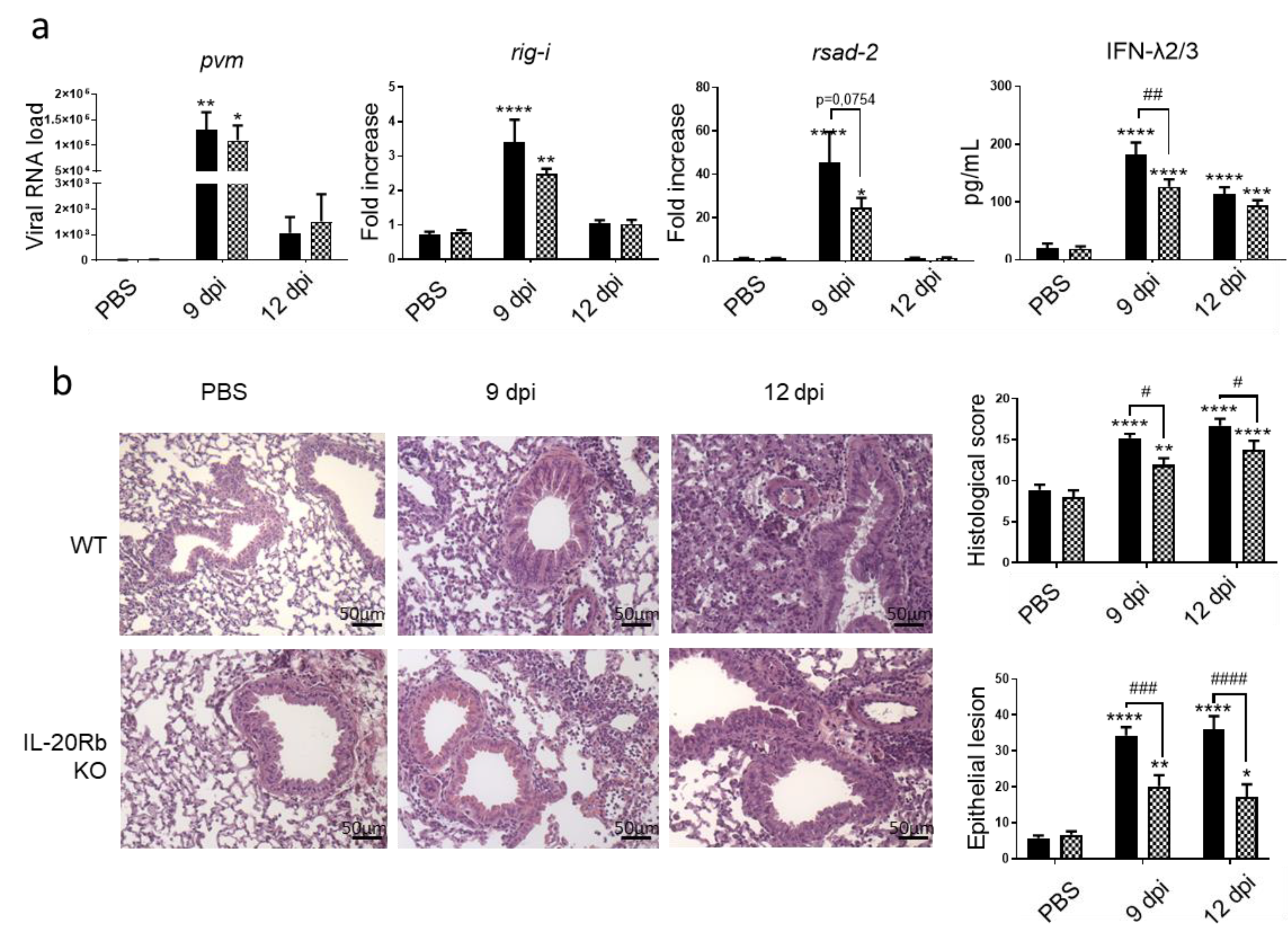
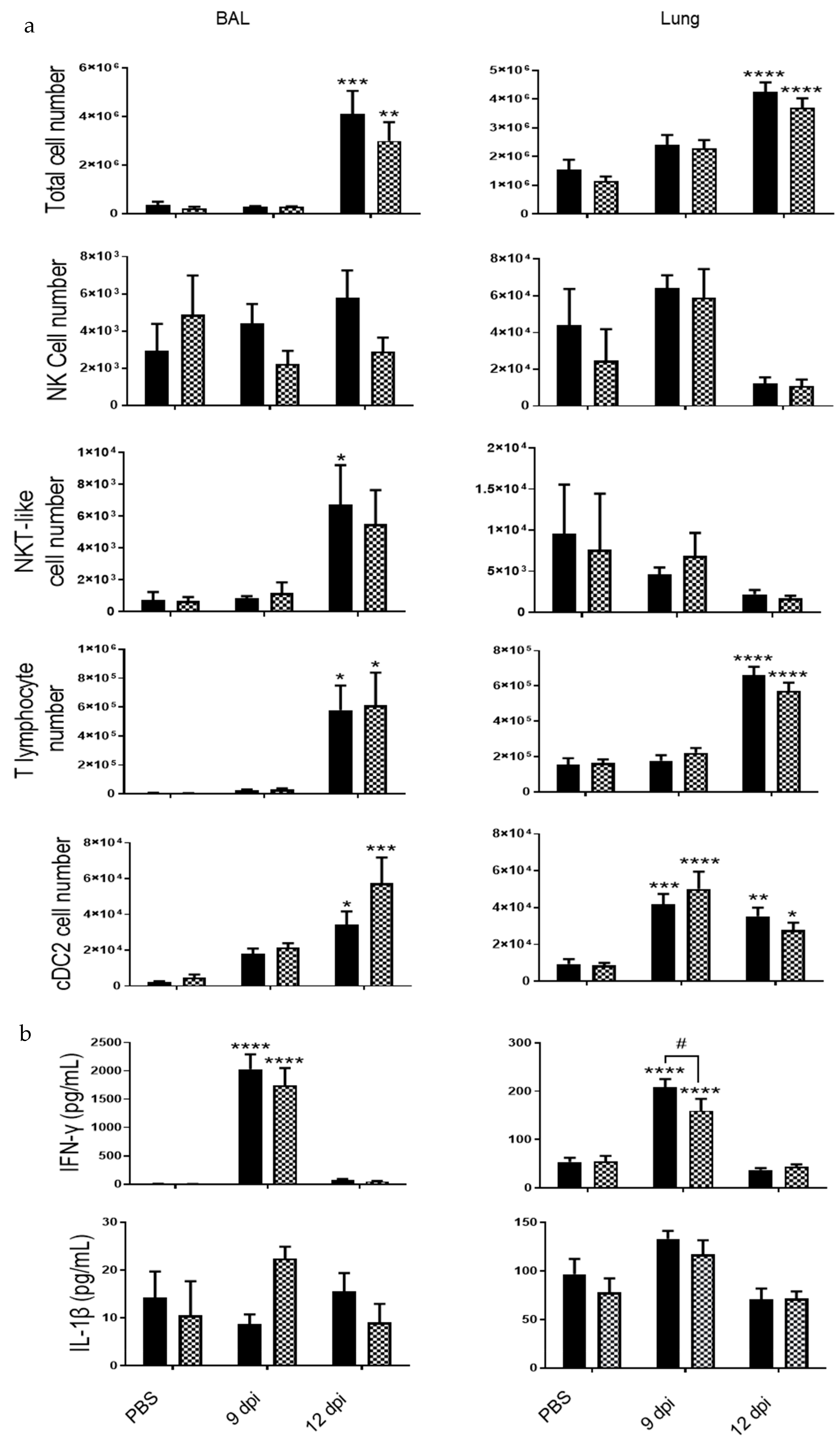
3.3. IL-20 Cytokines Have a Deletorious Effect during PVM Infection in CS-Exposed Mice
3.4. Implication of IL-20 Cytokines during PVM Infection in Control Mice
3.5. IL-20 Cytokines Play a Role in Lung Permeability during PVM Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo-Parke, H.; Linden, D.; Weldon, S.; Kidney, J.C.; Taggart, C.C. Mechanisms of Virus-Induced Airway Immunity Dysfunction in the Pathogenesis of COPD Disease, Progression, and Exacerbation. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Soler-Cataluña, J.J.; Martínez-García, M.Á.; Sánchez, P.R.; Salcedo, E.; Navarro, M.; Ochando, R. Yy. Thorax 2005, 60, 925–931. [Google Scholar] [CrossRef] [Green Version]
- Majzoub, K.; Wrensch, F.; Baumert, T.F. The Innate Antiviral Response in Animals: An Evolutionary Perspective from Flagellates to Humans. Viruses 2019, 11, 758. [Google Scholar] [CrossRef] [Green Version]
- Egli, A.; Santer, D.M.; O’Shea, D.; Tyrrell, D.L.; Houghton, M. The Impact of the Interferon-Lambda Family on the Innate and Adaptive Immune Response to Viral Infections. Emerg. Microbes Infect. 2014, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Okabayashi, T.; Kojima, T.; Masaki, T.; Yokota, S.; Imaizumi, T.; Tsutsumi, H.; Himi, T.; Fujii, N.; Sawada, N. Type-III Interferon, Not Type-I, Is the Predominant Interferon Induced by Respiratory Viruses in Nasal Epithelial Cells. Virus Res. 2011, 160, 360–366. [Google Scholar] [CrossRef]
- Kumar, P.; Thakar, M.S.; Ouyang, W.; Malarkannan, S. IL-22 from Conventional NK Cells Is Epithelial Regenerative and Inflammation Protective during Influenza Infection. Mucosal Immunol. 2013, 6, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Barthelemy, A.; Sencio, V.; Soulard, D.; Deruyter, L.; Faveeuw, C.; Le Goffic, R.; Trottein, F. Interleukin-22 Immunotherapy during Severe Influenza Enhances Lung Tissue Integrity and Reduces Secondary Bacterial Systemic Invasion. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Koné, B.; Pérez-Cruz, M.; Porte, R.; Hennegrave, F.; Carnoy, C.; Gosset, P.; Trottein, F.; Sirard, J.-C.; Pichavant, M.; Gosset, P. Boosting the IL-22 Response Using Flagellin Prevents Bacterial Infection in Cigarette Smoke-Exposed Mice. Clin. Exp. Immunol. 2020, 201, 171–186. [Google Scholar] [CrossRef]
- Rutz, S.; Wang, X.; Ouyang, W. The IL-20 Subfamily of Cytokines--from Host Defence to Tissue Homeostasis. Nat. Rev. Immunol. 2014, 14, 783–795. [Google Scholar] [CrossRef]
- Madouri, F.; Barada, O.; Kervoaze, G.; Trottein, F.; Pichavant, M.; Gosset, P. Production of Interleukin-20 Cytokines Limits Bacterial Clearance and Lung Inflammation during Infection by Streptococcus Pneumoniae. EBioMedicine 2018, 37, 417–427. [Google Scholar] [CrossRef]
- Myles, I.A.; Fontecilla, N.M.; Valdez, P.A.; Vithayathil, P.J.; Naik, S.; Belkaid, Y.; Ouyang, W.; Datta, S.K. Signaling via the IL-20 Receptor Inhibits Cutaneous Production of IL-1β and IL-17A to Promote Infection with Methicillin-Resistant Staphylococcus Aureus. Nat. Immunol. 2013, 14, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Rong, B.; Liu, Y.; Li, M.; Fu, T.; Gao, W.; Liu, H. Correlation of Serum Levels of HIF-1α and IL-19 with the Disease Progression of COPD: A Retrospective Study. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 3791–3803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermers, M.J.J.; Janssen, R.; Onland-Moret, N.C.; Hodemaekers, H.M.; Rovers, M.M.; Houben, M.L.; Kimpen, J.L.L.; Bont, L.J. IL10 Family Member Genes IL19 and IL20 Are Associated with Recurrent Wheeze after Respiratory Syncytial Virus Bronchiolitis. Pediatr. Res. 2011, 70, 518–523. [Google Scholar] [CrossRef] [Green Version]
- Truelove, A.L.; Oleksyk, T.K.; Shrestha, S.; Thio, C.L.; Goedert, J.J.; Donfield, S.M.; Kirk, G.D.; Thomas, D.L.; O’Brien, S.J.; Smith, M.W. Evaluation of IL10, IL19 and IL20 Gene Polymorphisms and Chronic Hepatitis B Infection Outcome. Int. J. Immunogenet. 2008, 35, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, H.F.; Domachowske, J.B. Pneumonia Virus of Mice: Severe Respiratory Infection in a Natural Host. Immunol. Lett. 2008, 118, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, H.F.; Bonville, C.A.; Easton, A.J.; Domachowske, J.B. The Pneumonia Virus of Mice Infection Model for Severe Respiratory Syncytial Virus Infection: Identifying Novel Targets for Therapeutic Intervention. Pharmacol. Ther. 2005, 105, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wahl, C.; Müller, W.; Leithäuser, F.; Adler, G.; Oswald, F.; Reimann, J.; Schirmbeck, R.; Seier, A.; Weiss, J.M.; Prochnow, B.; et al. IL-20 Receptor 2 Signaling down-Regulates Antigen-Specific T Cell Responses. J. Immunol. Baltim. Md 1950 2009, 182, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Pichavant, M.; Rémy, G.; Bekaert, S.; Le Rouzic, O.; Kervoaze, G.; Vilain, E.; Just, N.; Tillie-Leblond, I.; Trottein, F.; Cataldo, D.; et al. Oxidative Stress-Mediated INKT-Cell Activation Is Involved in COPD Pathogenesis. Mucosal Immunol. 2014, 7, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Sharan, R.; Perez-Cruz, M.; Kervoaze, G.; Gosset, P.; Weynants, V.; Godfroid, F.; Hermand, P.; Trottein, F.; Pichavant, M.; Gosset, P. Interleukin-22 Protects against Non-Typeable Haemophilus Influenzae Infection: Alteration during Chronic Obstructive Pulmonary Disease. Mucosal Immunol. 2017, 10, 139–149. [Google Scholar] [CrossRef]
- Bosteels, C.; Neyt, K.; Vanheerswynghels, M.; van Helden, M.J.; Sichien, D.; Debeuf, N.; De Prijck, S.; Bosteels, V.; Vandamme, N.; Martens, L.; et al. Inflammatory Type 2 CDCs Acquire Features of CDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 2020, 52, 1039–1056.e9. [Google Scholar] [CrossRef]
- Hogea, S.-P.; Tudorache, E.; Fildan, A.P.; Fira-Mladinescu, O.; Marc, M.; Oancea, C. Risk Factors of Chronic Obstructive Pulmonary Disease Exacerbations. Clin. Respir. J. 2020, 14, 183–197. [Google Scholar] [CrossRef] [PubMed]
- MOHAN, A.; CHANDRA, S.; AGARWAL, D.; GULERIA, R.; BROOR, S.; GAUR, B.; PANDEY, R.M. Prevalence of Viral Infection Detected by PCR and RT-PCR in Patients with Acute Exacerbation of COPD: A Systematic Review. Respirol. Carlton Vic 2010, 15, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Rohde, G.; Wiethege, A.; Borg, I.; Kauth, M.; Bauer, T.T.; Gillissen, A.; Bufe, A.; Schultze-Werninghaus, G. Respiratory Viruses in Exacerbations of Chronic Obstructive Pulmonary Disease Requiring Hospitalisation: A Case-Control Study. Thorax 2003, 58, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, T.M.A.; Donaldson, G.C.; Johnston, S.L.; Openshaw, P.J.M.; Wedzicha, J.A. Respiratory Syncytial Virus, Airway Inflammation, and FEV1 Decline in Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2006, 173, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and Viruses: An Interplay between Induction, Signalling, Antiviral Responses and Virus Countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef]
- Singanayagam, A.; Loo, S.-L.; Calderazzo, M.; Finney, L.J.; Trujillo Torralbo, M.-B.; Bakhsoliani, E.; Girkin, J.; Veerati, P.; Pathinayake, P.S.; Nichol, K.S.; et al. Antiviral Immunity Is Impaired in COPD Patients with Frequent Exacerbations. Am. J. Physiol. - Lung Cell. Mol. Physiol. 2019, 317, L893–L903. [Google Scholar] [CrossRef]
- García-Valero, J.; Olloquequi, J.; Montes, J.F.; Rodríguez, E.; Martín-Satué, M.; Texidó, L.; Ferrer Sancho, J. Deficient Pulmonary IFN-β Expression in COPD Patients. PLoS ONE 2019, 14, e0217803. [Google Scholar] [CrossRef]
- Hsu, A.C.-Y.; Dua, K.; Starkey, M.R.; Haw, T.-J.; Nair, P.M.; Nichol, K.; Zammit, N.; Grey, S.T.; Baines, K.J.; Foster, P.S.; et al. MicroRNA-125a and -b Inhibit A20 and MAVS to Promote Inflammation and Impair Antiviral Response in COPD. JCI Insight 2017, 2, e90443. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Patel, K.B.; Booth, J.L.; Zhang, W.; Metcalf, J.P. Cigarette Smoke Extract Suppresses the RIG-I-Initiated Innate Immune Response to Influenza Virus in the Human Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L821–L830. [Google Scholar] [CrossRef] [Green Version]
- Hilzendeger, C.; da Silva, J.; Henket, M.; Schleich, F.; Corhay, J.L.; Kebadze, T.; Edwards, M.R.; Mallia, P.; Johnston, S.L.; Louis, R. Reduced Sputum Expression of Interferon-Stimulated Genes in Severe COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 2016, 11, 1485–1494. [Google Scholar] [CrossRef] [Green Version]
- Baines, K.J.; Hsu, A.C.-Y.; Tooze, M.; Gunawardhana, L.P.; Gibson, P.G.; Wark, P.A. Novel Immune Genes Associated with Excessive Inflammatory and Antiviral Responses to Rhinovirus in COPD. Respir. Res. 2013, 14, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collinson, N.; Snape, N.; Beagley, K.; Fantino, E.; Spann, K. COPD Is Associated with Elevated IFN-β Production by Bronchial Epithelial Cells Infected with RSV or HMPV. Viruses 2021, 13, 911. [Google Scholar] [CrossRef] [PubMed]
- Foronjy, R.F.; Dabo, A.J.; Taggart, C.C.; Weldon, S.; Geraghty, P. Respiratory Syncytial Virus Infections Enhance Cigarette Smoke Induced COPD in Mice. PLoS ONE 2014, 9, e90567. [Google Scholar] [CrossRef] [Green Version]
- Watkiss, E.R.T.; Shrivastava, P.; Arsic, N.; Gomis, S.; Van Drunen Littel-van den Hurk, S. Innate and Adaptive Immune Response to Pneumonia Virus of Mice in a Resistant and a Susceptible Mouse Strain. Viruses 2013, 5, 295–320. [Google Scholar] [CrossRef]
- Van Helden, M.J.G.; van Kooten, P.J.S.; Bekker, C.P.J.; Gröne, A.; Topham, D.J.; Easton, A.J.; Boog, C.J.P.; Busch, D.H.; Zaiss, D.M.W.; Sijts, A.J.A.M. Pre-Existing Virus-Specific CD8+ T-Cells Provide Protection against Pneumovirus-Induced Disease in Mice. Vaccine 2012, 30, 6382–6388. [Google Scholar] [CrossRef] [Green Version]
- Van Leuven, J.T.; Gonzalez, A.J.; Ijezie, E.C.; Wixom, A.Q.; Clary, J.L.; Naranjo, M.N.; Ridenhour, B.J.; Miller, C.R.; Miura, T.A. Rhinovirus Reduces the Severity of Subsequent Respiratory Viral Infections by Interferon-Dependent and -Independent Mechanisms. mSphere 2021, 6, e0047921. [Google Scholar] [CrossRef]
- Seong, R.-K.; Choi, Y.-K.; Shin, O.S. MDA7/IL-24 Is an Anti-Viral Factor That Inhibits Influenza Virus Replication. J. Microbiol. Seoul Korea 2016, 54, 695–700. [Google Scholar] [CrossRef]
- Strumillo, S.T.; Curcio, M.F.; de Carvalho, F.F., Jr.; Sucupira, M.A.; Diaz, R.S.; Monteiro, H.P.; Janini, L.M.R. HIV-1 Infection Modulates IL-24 Expression Which Contributes to Cell Apoptosis in Vitro. Cell Biol. Int. 2019, 43, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-H.; Wu, C.-Y.; Hsing, C.-H.; Lai, W.-T.; Wu, L.-W.; Chang, M.-S. Anti-IL-20 Monoclonal Antibody Suppresses Prostate Cancer Growth and Bone Osteolysis in Murine Models. PLoS ONE 2015, 10, e0139871. [Google Scholar] [CrossRef] [Green Version]
- Nakada, T.-A.; Wacharasint, P.; Russell, J.A.; Boyd, J.H.; Nakada, E.; Thair, S.A.; Shimada, T.; Walley, K.R. The IL20 Genetic Polymorphism Is Associated with Altered Clinical Outcome in Septic Shock. J. Innate Immun. 2018, 10, 181–188. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequences |
|---|---|
| hprt1 | Sens: 5′ TCC TCC TCA GAC CGC TTT T 3′ Antisens: 5′ CCT GGT TCA TCA TCG CTA ATC 3′ |
| il-19 | Sens: 5′ TGT GTG CTG CAT GAC CAA CAA 3′ Antisens: 5′ GGC AAT GCT GCT GAT TCT CCT 3′ |
| il-20 | Sens: 5′ TCT TGC CTT TGG ACT GTT CTC 3′ Antisens: 5′ GTT TGC AGT AAT CAC ACA GCT TC 3′ |
| il-24 | Sens: 5′ AGC ACT GGC CCT TTC TTC AA 3′ Antisens: 5′ TGG CAA GAC CCA AAT CGG AA 3′ |
| rig-i | Sens: 5′ TGC GGA AAT ACA ACG ATG CA 3′ Antisens: 5′ GCT CGG TCT CAT CGA ATG CTG 3′ |
| rsad-2 | Sens: 5′ TGC TGG CTG AGA ATA GCA TTA GG 3′ Antisens: 5′ GCT GAG TGC TGT TCC CAT CT 3′ |
| pvm | Sens: 5′ GCC GTC ATC AAC ACA GTG TGT 3′ Antisens: 5′ GCC TGA TGT GGC AGT GCT T 3′ Probe: 5′[FAM] C GCT GAT AAT GGC CTG AG CA [TAM] 3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le Roux, M.; Ollivier, A.; Kervoaze, G.; Beke, T.; Gillet, L.; Pichavant, M.; Gosset, P. IL-20 Cytokines Are Involved in Epithelial Lesions Associated with Virus-Induced COPD Exacerbation in Mice. Biomedicines 2021, 9, 1838. https://doi.org/10.3390/biomedicines9121838
Le Roux M, Ollivier A, Kervoaze G, Beke T, Gillet L, Pichavant M, Gosset P. IL-20 Cytokines Are Involved in Epithelial Lesions Associated with Virus-Induced COPD Exacerbation in Mice. Biomedicines. 2021; 9(12):1838. https://doi.org/10.3390/biomedicines9121838
Chicago/Turabian StyleLe Roux, Mélina, Anaïs Ollivier, Gwenola Kervoaze, Timothé Beke, Laurent Gillet, Muriel Pichavant, and Philippe Gosset. 2021. "IL-20 Cytokines Are Involved in Epithelial Lesions Associated with Virus-Induced COPD Exacerbation in Mice" Biomedicines 9, no. 12: 1838. https://doi.org/10.3390/biomedicines9121838
APA StyleLe Roux, M., Ollivier, A., Kervoaze, G., Beke, T., Gillet, L., Pichavant, M., & Gosset, P. (2021). IL-20 Cytokines Are Involved in Epithelial Lesions Associated with Virus-Induced COPD Exacerbation in Mice. Biomedicines, 9(12), 1838. https://doi.org/10.3390/biomedicines9121838