
The Morphological Spectrum of Papillary Renal Cell Carcinoma and Prevalence of Provisional/Emerging Renal Tumor Entities with Papillary Growth

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
4.1. Classic Papillary RCC
4.2. FH Deficient RCC and Tubulocystic RCC
4.3. Collecting Duct Carcinoma and SMARCB1 Deficient Medullary RCC
4.4. Clear Cell Papillary RCC and Acquired Cystic Disease-Associated RCC
4.5. Mixed Epithelial and Stromal Tumors
4.6. Provisional/Emerging Renal Tumor Entities with Papillary Growth
4.7. Molecularly Defined RCC with Papillary Growth
4.8. Solid Renal Tumors Showing Areas with Papillary Differentiation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Glossary
| 2SC | S-(2succino)cysteine |
| ACD-associated RCC | Acquired cystic disease-associated renal cell carcinoma |
| BHP RCC | Biphasic hyalinizing psammomatous renal cell carcinoma |
| BSA RCC | Biphasic squamoid/alveolar renal cell carcinoma |
| ccpRCC | Clear cell papillary RCC |
| ccRCC | Clear cell renal cell carcinoma |
| chRCC | Chromophobe renal cell carcinoma |
| ESC RCC | Eosinophilic solid and cystic renal cell carcinoma |
| EVT | Eosinophilic vacuolated tumor |
| FH | Fumarate hydratase |
| FISH | Fluorescence in situ hybridization |
| HLRCC | Hereditary leiomyomatosis and renal cell carcinoma |
| HOCT | Hybrid oncocytic-chromophobe tumor |
| ISUP | International Society of Urological Pathology |
| MEST | Mixed epithelial and stromal tumor |
| MTSCC | Mucinous tubular and spindle carcinoma |
| NET | Neuroendocrine tumor |
| pRCC | Papillary renal cell carcinoma |
| PRNRP | Papillary renal neoplasm with reversed polarity |
| RCC | Renal cell carcinoma |
| RCC FMS | Renal cell carcinoma with fibromyomatous stroma |
| SDH | Succinate dehydrogenase |
| TLF RCC | Thyroid-like follicular renal cell carcinoma |
| WHO | World Health Organization |
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F.; International Agency for Research on Cancer (IARC). Global Cancer Observatory: Cancer Today. Available online: https://gco.iarc.fr/today (accessed on 15 January 2021).
- Capitanio, U.; Bensalah, K.; Bex, A.; Boorjian, S.A.; Bray, F.; Coleman, J.; Gore, J.L.; Sun, M.; Wood, C.; Russo, P. Epidemiology of Renal Cell Carcinoma. Eur. Urol. 2019, 75, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Beksac, A.T.; Paulucci, D.J.; Blum, K.A.; Yadav, S.S.; Sfakianos, J.P.; Badani, K.K. Heterogeneity in renal cell carcinoma. Urol. Oncol. 2017, 35, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Vera-Badillo, F.E.; Templeton, A.J.; Duran, I.; Ocana, A.; de Gouveia, P.; Aneja, P.; Knox, J.J.; Tannock, I.F.; Escudier, B.; Amir, E. Systemic therapy for non-clear cell renal cell carcinomas: A systematic review and meta-analysis. Eur. Urol. 2015, 67, 740–749. [Google Scholar] [CrossRef]
- Ciccarese, C.; Iacovelli, R.; Brunelli, M.; Massari, F.; Bimbatti, D.; Fantinel, E.; De Marco, V.; Porcaro, A.B.; Martignoni, G.; Artibani, W.; et al. Addressing the best treatment for non-clear cell renal cell carcinoma: A meta-analysis of randomised clinical trials comparing VEGFR-TKis versus mTORi-targeted therapies. Eur. J. Cancer 2017, 83, 237–246. [Google Scholar] [CrossRef]
- Nahouraii, L.M.; Allen, J.L.; Merrill, S.B.; Lehman, E.; Kaag, M.G.; Raman, J.D. Histologic Heterogeneity of Extirpated Renal Cell Carcinoma Specimens: Implications for Renal Mass Biopsy. J. Kidney Cancer VHL 2020, 7, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Gao, R.; Wu, H.; Wang, Z.; Han, G. Single-cell analysis reveals metastatic cell heterogeneity in clear cell renal cell carcinoma. J. Cell Mol. Med. 2021, 25, 4260–4274. [Google Scholar] [CrossRef] [PubMed]
- Raspollini, M.R.; Montagnani, I.; Montironi, R.; Castiglione, F.; Martignoni, G.; Cheng, L.; Lopez-Beltran, A. Intratumoural heterogeneity may hinder precision medicine strategies in patients with clear cell renal cell carcinoma. J. Clin. Pathol. 2018, 71, 467–471. [Google Scholar] [CrossRef]
- Trpkov, K.; Williamson, S.R.; Gill, A.J.; Adeniran, A.J.; Agaimy, A.; Alaghehbandan, R.; Amin, M.B.; Argani, P.; Chen, Y.B.; Cheng, L.; et al. Novel, emerging and provisional renal entities: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod. Pathol. 2021, 34, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Cimadamore, A.; Cheng, L.; Scarpelli, M.; Massari, F.; Mollica, V.; Santoni, M.; Lopez-Beltran, A.; Montironi, R.; Moch, H. Towards a new WHO classification of renal cell tumor: What the clinician needs to know-a narrative review. Transl. Androl. Urol. 2021, 10, 1506–1520. [Google Scholar] [CrossRef]
- Trpkov, K.; Hes, O.; Williamson, S.R.; Adeniran, A.J.; Agaimy, A.; Alaghehbandan, R.; Amin, M.B.; Argani, P.; Chen, Y.B.; Cheng, L.; et al. New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod. Pathol. 2021, 34, 1392–1424. [Google Scholar] [CrossRef]
- Trpkov, K.; Hes, O. New and emerging renal entities: A perspective post-WHO 2016 classification. Histopathology 2019, 74, 31–59. [Google Scholar] [CrossRef] [Green Version]
- Moch, H.; Humphrey, P.; Ulbright, T.; Reuter, V. WHO Classification of Tumours of the Urinary System and Male Genital Organs, 4th ed.; International Agency for Research on Cancer (IARC): Lyon, France, 2016. [Google Scholar]
- Sirohi, D.; Smith, S.C.; Agarwal, N.; Maughan, B.L. Unclassified renal cell carcinoma: Diagnostic difficulties and treatment modalities. Res. Rep. Urol. 2018, 10, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Perrino, C.M.; Grignon, D.J.; Williamson, S.R.; Idrees, M.T.; Eble, J.N.; Cheng, L. Morphological spectrum of renal cell carcinoma, unclassified: An analysis of 136 cases. Histopathology 2018, 72, 305–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delahunt, B.; Eble, J.N. Papillary renal cell carcinoma: A clinicopathologic and immunohistochemical study of 105 tumors. Mod. Pathol. 1997, 10, 537–544. [Google Scholar] [PubMed]
- Jiang, F.; Richter, J.; Schraml, P.; Bubendorf, L.; Gasser, T.; Sauter, G.; Mihatsch, M.J.; Moch, H. Chromosomal imbalances in papillary renal cell carcinoma: Genetic differences between histological subtypes. Am. J. Pathol. 1998, 153, 1467–1473. [Google Scholar] [CrossRef]
- Saleeb, R.M.; Brimo, F.; Farag, M.; Rompre-Brodeur, A.; Rotondo, F.; Beharry, V.; Wala, S.; Plant, P.; Downes, M.R.; Pace, K.; et al. Toward Biological Subtyping of Papillary Renal Cell Carcinoma With Clinical Implications Through Histologic, Immunohistochemical, and Molecular Analysis. Am. J. Surg. Pathol. 2017, 41, 1618–1629. [Google Scholar] [CrossRef]
- Ross, H.; Martignoni, G.; Argani, P. Renal cell carcinoma with clear cell and papillary features. Arch. Pathol. Lab. Med. 2012, 136, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delahunt, B.; Eble, J.N.; Egevad, L.; Yaxley, J.; Thunders, M.; Samaratunga, H. Emerging entities of renal cell neoplasia. Surg. Exp. Pathol. 2019, 2, 1–7. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Linehan, W.M.; Spellman, P.T.; Ricketts, C.J.; Creighton, C.J.; Fei, S.S.; Davis, C.; Wheeler, D.A.; Murray, B.A.; Schmidt, L.; et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Marsaud, A.; Dadone, B.; Ambrosetti, D.; Baudoin, C.; Chamorey, E.; Rouleau, E.; Lefol, C.; Roussel, J.F.; Fabas, T.; Cristofari, G.; et al. Dismantling papillary renal cell carcinoma classification: The heterogeneity of genetic profiles suggests several independent diseases. Genes Chromosomes Cancer 2015, 54, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; Diorio, G.; Brito, J.; Zargar, K.; Pareek, G.; Renzulli, J.F.; Dickinson, S.I.; Amin, A.; Sexton, W.J.; Spiess, P.E.; et al. Presence and percentage of type 2 papillary RCC in mixed (type 1 and type 2) papillary renal cell carcinoma does not portend worse prognosis in patients treated by partial/radical nephrectomy in non-metastatic disease. J. Clin. Oncol. 2016, 34, e16126. [Google Scholar] [CrossRef]
- Stempel, K.; Bencherki, A.; Tedehammar, N.; Sagen, N.E.; Elzanaty, S. True collision renal tumour of oncocytoma and papillary Renal cell carcinoma: Case Report and Review of the Literature. Arch. Urol. Res. 2020, 4, 080–084. [Google Scholar]
- Kinney, S.N.; Eble, J.N.; Hes, O.; Williamson, S.R.; Grignon, D.J.; Wang, M.; Zhang, S.; Baldrige, L.A.; Martignoni, G.; Brunelli, M.; et al. Metanephric adenoma: The utility of immunohistochemical and cytogenetic analyses in differential diagnosis, including solid variant papillary renal cell carcinoma and epithelial-predominant nephroblastoma. Mod. Pathol. 2015, 28, 1236–1248. [Google Scholar] [CrossRef] [PubMed]
- Moch, H. Neuroendocrine tumors of the kidneys. Pathologe 2015, 36, 278–282. [Google Scholar] [CrossRef]
- Mantoan Padilha, M.; Billis, A.; Allende, D.; Zhou, M.; Magi-Galluzzi, C. Metanephric adenoma and solid variant of papillary renal cell carcinoma: Common and distinctive features. Histopathology 2013, 62, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Pivovarcikova, K.; Peckova, K.; Martinek, P.; Montiel, D.P.; Kalusova, K.; Pitra, T.; Hora, M.; Skenderi, F.; Ulamec, M.; Daum, O.; et al. “Mucin”-secreting papillary renal cell carcinoma: Clinicopathological, immunohistochemical, and molecular genetic analysis of seven cases. Virchows Arch. 2016, 469, 71–80. [Google Scholar] [CrossRef]
- Ren, Q.; Wang, L.; Al-Ahmadie, H.A.; Fine, S.W.; Gopalan, A.; Sirintrapun, S.J.; Tickoo, S.K.; Reuter, V.E.; Chen, Y.B. Distinct Genomic Copy Number Alterations Distinguish Mucinous Tubular and Spindle Cell Carcinoma of the Kidney From Papillary Renal Cell Carcinoma With Overlapping Histologic Features. Am. J. Surg. Pathol. 2018, 42, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Peckova, K.; Martinek, P.; Sperga, M.; Montiel, D.P.; Daum, O.; Rotterova, P.; Kalusova, K.; Hora, M.; Pivovarcikova, K.; Rychly, B.; et al. Mucinous spindle and tubular renal cell carcinoma: Analysis of chromosomal aberration pattern of low-grade, high-grade, and overlapping morphologic variant with papillary renal cell carcinoma. Ann. Diagn. Pathol. 2015, 19, 226–231. [Google Scholar] [CrossRef]
- Delahunt, B.; Cheville, J.C.; Martignoni, G.; Humphrey, P.A.; Magi-Galluzzi, C.; McKenney, J.; Egevad, L.; Algaba, F.; Moch, H.; Grignon, D.J.; et al. The International Society of Urological Pathology (ISUP) grading system for renal cell carcinoma and other prognostic parameters. Am. J. Surg. Pathol. 2013, 37, 1490–1504. [Google Scholar] [CrossRef] [Green Version]
- Delahunt, B.; Eble, J.N.; McCredie, M.R.; Bethwaite, P.B.; Stewart, J.H.; Bilous, A.M. Morphologic typing of papillary renal cell carcinoma: Comparison of growth kinetics and patient survival in 66 cases. Hum. Pathol. 2001, 32, 590–595. [Google Scholar] [CrossRef]
- Le, X.; Wang, X.B.; Zhao, H.; Chen, R.F.; Ge, P. Comparison of clinicopathologic parameters and oncologic outcomes between type 1 and type 2 papillary renal cell carcinoma. BMC Urol. 2020, 20, 148. [Google Scholar] [CrossRef]
- Yang, C.; Shuch, B.; Kluger, H.; Humphrey, P.A.; Adeniran, A.J. High WHO/ISUP Grade and Unfavorable Architecture, Rather Than Typing of Papillary Renal Cell Carcinoma, May Be Associated With Worse Prognosis. Am. J. Surg. Pathol. 2020, 44, 582–593. [Google Scholar] [CrossRef]
- Iczkowski, K.A.; Czaja, R.C. Eosinophilic Kidney Tumors: Old and New. Arch. Pathol. Lab. Med. 2019, 143, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Wyvekens, N.; Valtcheva, N.; Mischo, A.; Helmchen, B.; Hermanns, T.; Choschzick, M.; Hotker, A.M.; Rauch, A.; Muhleisen, B.; Akhoundova, D.; et al. Novel morphological and genetic features of fumarate hydratase deficient renal cell carcinoma in HLRCC syndrome patients with a tailored therapeutic approach. Genes Chromosomes Cancer 2020, 59, 611–619. [Google Scholar] [CrossRef]
- Trpkov, K.; Hes, O.; Agaimy, A.; Bonert, M.; Martinek, P.; Magi-Galluzzi, C.; Kristiansen, G.; Luders, C.; Nesi, G.; Comperat, E.; et al. Fumarate Hydratase-deficient Renal Cell Carcinoma Is Strongly Correlated With Fumarate Hydratase Mutation and Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome. Am. J. Surg. Pathol. 2016, 40, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Zhang, X.; Liang, J.; Pan, X.; Zhu, S.; Liu, Z.; Armstrong, C.M.; Chen, J.; Lin, W.; Liao, B.; et al. Integrated Molecular Characterization of Fumarate Hydratase-deficient Renal Cell Carcinoma. Clin. Cancer Res. 2021, 27, 1734–1743. [Google Scholar] [CrossRef]
- Sarungbam, J.; Mehra, R.; Tomlins, S.A.; Smith, S.C.; Jayakumaran, G.; Al-Ahmadie, H.; Gopalan, A.; Sirintrapun, S.J.; Fine, S.W.; Zhang, Y.; et al. Tubulocystic renal cell carcinoma: A distinct clinicopathologic entity with a characteristic genomic profile. Mod. Pathol. 2019, 32, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Ohe, C.; Smith, S.C.; Sirohi, D.; Divatia, M.; de Peralta-Venturina, M.; Paner, G.P.; Agaimy, A.; Amin, M.B.; Argani, P.; Chen, Y.-B. Reappraisal of morphological differences between renal medullary carcinoma, collecting duct carcinoma, and fumarate hydratase-deficient renal cell carcinoma. Am. J. Surg. Pathol. 2018, 42, 279. [Google Scholar] [CrossRef]
- Orsola, A.; Trias, I.; Raventos, C.X.; Espanol, I.; Cecchini, L.; Orsola, I. Renal collecting (Bellini) duct carcinoma displays similar characteristics to upper tract urothelial cell carcinoma. Urology 2005, 65, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Karth, J.; Schneider, D.T.; Rodriguez, R.; Beckwith, J.B.; Perlman, E.J. Renal medullary carcinoma: Clinical, pathologic, immunohistochemical, and genetic analysis with pathogenetic implications. Urology 2002, 60, 1083–1089. [Google Scholar] [CrossRef]
- Calderaro, J.; Masliah-Planchon, J.; Richer, W.; Maillot, L.; Maille, P.; Mansuy, L.; Bastien, C.; de la Taille, A.; Boussion, H.; Charpy, C. Balanced translocations disrupting SMARCB1 are hallmark recurrent genetic alterations in renal medullary carcinomas. Eur. Urol. 2016, 69, 1055–1061. [Google Scholar] [CrossRef]
- Tickoo, S.K.; de Peralta-Venturina, M.N.; Harik, L.R.; Worcester, H.D.; Salama, M.E.; Young, A.N.; Moch, H.; Amin, M.B. Spectrum of epithelial neoplasms in end-stage renal disease: An experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am. J. Surg. Pathol. 2006, 30, 141–153. [Google Scholar] [CrossRef]
- Aydin, H.; Chen, L.; Cheng, L.; Vaziri, S.; He, H.; Ganapathi, R.; Delahunt, B.; Magi-Galluzzi, C.; Zhou, M. Clear cell tubulopapillary renal cell carcinoma: A study of 36 distinctive low-grade epithelial tumors of the kidney. Am. J. Surg. Pathol. 2010, 34, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zheng, S.; Truong, L.D.; Ro, J.Y.; Ayala, A.G.; Shen, S.S. Clear cell papillary renal cell carcinoma is the fourth most common histologic type of renal cell carcinoma in 290 consecutive nephrectomies for renal cell carcinoma. Hum. Pathol. 2014, 45, 59–64. [Google Scholar] [CrossRef]
- Weng, S.; DiNatale, R.G.; Silagy, A.; Mano, R.; Attalla, K.; Kashani, M.; Weiss, K.; Benfante, N.E.; Winer, A.G.; Coleman, J.A.; et al. The Clinicopathologic and Molecular Landscape of Clear Cell Papillary Renal Cell Carcinoma: Implications in Diagnosis and Management. Eur. Urol. 2021, 79, 468–477. [Google Scholar] [CrossRef]
- Dhakal, H.P.; McKenney, J.K.; Khor, L.Y.; Reynolds, J.P.; Magi-Galluzzi, C.; Przybycin, C.G. Renal Neoplasms With Overlapping Features of Clear Cell Renal Cell Carcinoma and Clear Cell Papillary Renal Cell Carcinoma: A Clinicopathologic Study of 37 Cases From a Single Institution. Am. J. Surg. Pathol. 2016, 40, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Goyal, R.; Yang, X.J. Pathologic characterization of renal epithelial neoplasms arising in nonfunctioning kidneys. Hum. Pathol. 2020, 97, 1–7. [Google Scholar] [CrossRef]
- Przybycin, C.G.; Harper, H.L.; Reynolds, J.P.; Magi-Galluzzi, C.; Nguyen, J.K.; Wu, A.; Sangoi, A.R.; Liu, P.S.; Umar, S.; Mehra, R.; et al. Acquired Cystic Disease-associated Renal Cell Carcinoma (ACD-RCC): A Multiinstitutional Study of 40 Cases With Clinical Follow-up. Am. J. Surg. Pathol. 2018, 42, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.C.; Hruban, R.H.; Horton, K.M.; Fishman, E.K. Mixed epithelial and stromal tumor of the kidney: Radiologic-pathologic correlation. Radiographics 2010, 30, 1541–1551. [Google Scholar] [CrossRef]
- Mehra, R.; Vats, P.; Cao, X.; Su, F.; Lee, N.D.; Lonigro, R.; Premkumar, K.; Trpkov, K.; McKenney, J.K.; Dhanasekaran, S.M.; et al. Somatic Bi-allelic Loss of TSC Genes in Eosinophilic Solid and Cystic Renal Cell Carcinoma. Eur. Urol. 2018, 74, 483–486. [Google Scholar] [CrossRef]
- Palsgrove, D.N.; Li, Y.; Pratilas, C.A.; Lin, M.T.; Pallavajjalla, A.; Gocke, C.; De Marzo, A.M.; Matoso, A.; Netto, G.J.; Epstein, J.I.; et al. Eosinophilic Solid and Cystic (ESC) Renal Cell Carcinomas Harbor TSC Mutations: Molecular Analysis Supports an Expanding Clinicopathologic Spectrum. Am. J. Surg. Pathol. 2018, 42, 1166–1181. [Google Scholar] [CrossRef]
- Tretiakova, M.S. Eosinophilic solid and cystic renal cell carcinoma mimicking epithelioid angiomyolipoma: Series of 4 primary tumors and 2 metastases. Hum. Pathol. 2018, 80, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Kiyozawa, D.; Kohashi, K.; Takamatsu, D.; Yamamoto, T.; Eto, M.; Iwasaki, T.; Motoshita, J.; Shimokama, T.; Kinjo, M.; Oshiro, Y.; et al. Morphological, immunohistochemical, and genomic analyses of papillary renal neoplasm with reverse polarity. Hum. Pathol. 2021, 112, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Hang, J.F.; Wu, C.Y.; Lin, T.P.; Chung, H.J.; Chang, Y.H.; Pan, C.C. Clinicopathological and molecular characterisation of papillary renal neoplasm with reverse polarity and its renal papillary adenoma analogue. Histopathology 2021, 78, 1019–1031. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, J.; Wang, S.; Yang, X.; Li, C.; Zhou, J.; Zhang, P.; Xu, H.; Wang, C. Papillary Renal Neoplasm With Reverse Polarity: A Clinicopathologic Study of 7 Cases. Int. J. Surg. Pathol. 2020, 28, 728–734. [Google Scholar] [CrossRef]
- Al-Obaidy, K.I.; Eble, J.N.; Cheng, L.; Williamson, S.R.; Sakr, W.A.; Gupta, N.; Idrees, M.T.; Grignon, D.J. Papillary Renal Neoplasm With Reverse Polarity: A Morphologic, Immunohistochemical, and Molecular Study. Am. J. Surg. Pathol. 2019, 43, 1099–1111. [Google Scholar] [CrossRef]
- Kim, S.S.; Cho, Y.M.; Kim, G.H.; Kee, K.H.; Kim, H.S.; Kim, K.M.; Kim, J.H.; Choi, C. Recurrent KRAS mutations identified in papillary renal neoplasm with reverse polarity-a comparative study with papillary renal cell carcinoma. Mod. Pathol. 2020, 33, 690–699. [Google Scholar] [CrossRef]
- Hes, O.; Condom Mundo, E.; Peckova, K.; Lopez, J.I.; Martinek, P.; Vanecek, T.; Falconieri, G.; Agaimy, A.; Davidson, W.; Petersson, F.; et al. Biphasic Squamoid Alveolar Renal Cell Carcinoma: A Distinctive Subtype of Papillary Renal Cell Carcinoma? Am. J. Surg. Pathol. 2016, 40, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Kum, J.B.; Goheen, M.P.; Cheng, L.; Grignon, D.J.; Idrees, M.T. Clear cell renal cell carcinoma with a syncytial-type multinucleated giant tumor cell component: Implications for differential diagnosis. Hum. Pathol. 2014, 45, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Vilela, D.; Izquierdo, F.M. CD57 in biphasic squamoid alveolar renal cell carcinoma. Pathol. Int. 2020, 70, 56–57. [Google Scholar] [CrossRef]
- Chartier, S.; Mejean, A.; Richard, S.; Thiounn, N.; Vasiliu, V.; Verkarre, V. Biphasic Squamoid Alveolar Renal Cell Carcinoma: 2 Cases in a Family Supporting a Continuous Spectrum With Papillary Type I Renal Cell Carcinoma. Am. J. Surg. Pathol. 2017, 41, 1011–1012. [Google Scholar] [CrossRef]
- Denize, T.; Just, P.A.; Sibony, M.; Blons, H.; Timsit, M.O.; Drossart, T.; Jakubowicz, D.; Broudin, C.; Morini, A.; Molina, T.; et al. MET alterations in biphasic squamoid alveolar papillary renal cell carcinomas and clinicopathological features. Mod. Pathol. 2021, 34, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Reuter, V.E.; Eble, J.N.; Vlatkovic, L.; Yaskiv, O.; Swanson, D.; Dickson, B.C.; Antonescu, C.R.; Matoso, A.; Gagan, J.; et al. Biphasic Hyalinizing Psammomatous Renal Cell Carcinoma (BHP RCC): A Distinctive Neoplasm Associated With Somatic NF2 Mutations. Am. J. Surg. Pathol. 2020, 44, 901–916. [Google Scholar] [CrossRef] [PubMed]
- Skenderi, F.; Ulamec, M.; Vanecek, T.; Martinek, P.; Alaghehbandan, R.; Foix, M.P.; Babankova, I.; Montiel, D.P.; Alvarado-Cabrero, I.; Svajdler, M.; et al. Warthin-like papillary renal cell carcinoma: Clinicopathologic, morphologic, immunohistochemical and molecular genetic analysis of 11 cases. Ann. Diagn. Pathol. 2017, 27, 48–56. [Google Scholar] [CrossRef]
- Srigley, J.R.; Delahunt, B.; Eble, J.N.; Egevad, L.; Epstein, J.I.; Grignon, D.; Hes, O.; Moch, H.; Montironi, R.; Tickoo, S.K.; et al. The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am. J. Surg. Pathol. 2013, 37, 1469–1489. [Google Scholar] [CrossRef]
- Michalova, K.; Steiner, P.; Alaghehbandan, R.; Trpkov, K.; Martinek, P.; Grossmann, P.; Montiel, D.P.; Sperga, M.; Straka, L.; Prochazkova, K.; et al. Papillary renal cell carcinoma with cytologic and molecular genetic features overlapping with renal oncocytoma: Analysis of 10 cases. Ann. Diagn. Pathol. 2018, 35, 1–6. [Google Scholar] [CrossRef]
- Argani, P. MiT family translocation renal cell carcinoma. Semin. Diagn. Pathol. 2015, 32, 103–113. [Google Scholar] [CrossRef]
- Green, W.M.; Yonescu, R.; Morsberger, L.; Morris, K.; Netto, G.J.; Epstein, J.I.; Illei, P.B.; Allaf, M.; Ladanyi, M.; Griffin, C.A.; et al. Utilization of a TFE3 break-apart FISH assay in a renal tumor consultation service. Am. J. Surg. Pathol. 2013, 37, 1150–1163. [Google Scholar] [CrossRef]
- Argani, P.; Ladanyi, M. The evolving story of renal translocation carcinomas. Am. J. Clin. Pathol. 2006, 126, 332–334. [Google Scholar] [CrossRef]
- Craig, K.M.; Poppas, D.P.; Akhavan, A. Pediatric renal cell carcinoma. Curr. Opin. Urol. 2019, 29, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.T.; Xia, Q.Y.; Ye, S.B.; Wang, X.; Li, R.; Fang, R.; Shi, S.S.; Zhang, R.S.; Tan, X.; Chen, J.Y.; et al. RNA sequencing of Xp11 translocation-associated cancers reveals novel gene fusions and distinctive clinicopathologic correlations. Mod. Pathol. 2018, 31, 1346–1360. [Google Scholar] [CrossRef]
- Ellis, C.L.; Eble, J.N.; Subhawong, A.P.; Martignoni, G.; Zhong, M.; Ladanyi, M.; Epstein, J.I.; Netto, G.J.; Argani, P. Clinical heterogeneity of Xp11 translocation renal cell carcinoma: Impact of fusion subtype, age, and stage. Mod. Pathol. 2014, 27, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Argani, P.; Zhang, L.; Reuter, V.E.; Tickoo, S.K.; Antonescu, C.R. RBM10-TFE3 Renal Cell Carcinoma: A Potential Diagnostic Pitfall Due to Cryptic Intrachromosomal Xp11.2 Inversion Resulting in False-negative TFE3 FISH. Am. J. Surg. Pathol. 2017, 41, 655–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersson, F.; Vanecek, T.; Michal, M.; Martignoni, G.; Brunelli, M.; Halbhuber, Z.; Spagnolo, D.; Kuroda, N.; Yang, X.; Cabrero, I.A.; et al. A distinctive translocation carcinoma of the kidney; ‘rosette forming,’ t(6;11), HMB45-positive renal tumor: A histomorphologic, immunohistochemical, ultrastructural, and molecular genetic study of 4 cases. Hum. Pathol. 2012, 43, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, G.; Pea, M.; Gobbo, S.; Brunelli, M.; Bonetti, F.; Segala, D.; Pan, C.C.; Netto, G.; Doglioni, C.; Hes, O.; et al. Cathepsin-K immunoreactivity distinguishes MiTF/TFE family renal translocation carcinomas from other renal carcinomas. Mod. Pathol. 2009, 22, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Calio, A.; Brunelli, M.; Segala, D.; Pedron, S.; Tardanico, R.; Remo, A.; Gobbo, S.; Meneghelli, E.; Doglioni, C.; Hes, O.; et al. t(6;11) renal cell carcinoma: A study of seven cases including two with aggressive behavior, and utility of CD68 (PG-M1) in the differential diagnosis with pure epithelioid PEComa/epithelioid angiomyolipoma. Mod. Pathol. 2018, 31, 474–487. [Google Scholar] [CrossRef]
- Calio, A.; Segala, D.; Munari, E.; Brunelli, M.; Martignoni, G. MiT Family Translocation Renal Cell Carcinoma: From the Early Descriptions to the Current Knowledge. Cancers 2019, 11, 1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.E.; Parilla, M.; Wanjari, P.; Segal, J.P.; Antic, T. A Tale of 2 Morphologies: Diagnostic Pitfalls in TFEB-Associated Renal Cell Carcinomas, Including a Novel NEAT1-TFEB Fusion. Int. J. Surg. Pathol. 2021, 29, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Reuter, V.E.; Zhang, L.; Sung, Y.S.; Ning, Y.; Epstein, J.I.; Netto, G.J.; Antonescu, C.R. TFEB-amplified Renal Cell Carcinomas: An Aggressive Molecular Subset Demonstrating Variable Melanocytic Marker Expression and Morphologic Heterogeneity. Am. J. Surg. Pathol. 2016, 40, 1484–1495. [Google Scholar] [CrossRef]
- Martignoni, G.; Gobbo, S.; Camparo, P.; Brunelli, M.; Munari, E.; Segala, D.; Pea, M.; Bonetti, F.; Illei, P.B.; Netto, G.J.; et al. Differential expression of cathepsin K in neoplasms harboring TFE3 gene fusions. Mod. Pathol. 2011, 24, 1313–1319. [Google Scholar] [CrossRef] [Green Version]
- Williamson, S.R.; Cheng, L.; Eble, J.N.; True, L.D.; Gupta, N.S.; Wang, M.; Zhang, S.; Grignon, D.J. Renal cell carcinoma with angioleiomyoma-like stroma: Clinicopathological, immunohistochemical, and molecular features supporting classification as a distinct entity. Mod. Pathol. 2015, 28, 279–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.B.; Stohr, B.A.; Tu, Z.J.; Gao, Y.; Przybycin, C.G.; Nguyen, J.; Cox, R.M.; Rashid-Kolvear, F.; Weindel, M.D.; Farkas, D.H.; et al. “Renal Cell Carcinoma With Leiomyomatous Stroma” Harbor Somatic Mutations of TSC1, TSC2, MTOR, and/or ELOC (TCEB1): Clinicopathologic and Molecular Characterization of 18 Sporadic Tumors Supports a Distinct Entity. Am. J. Surg. Pathol. 2020, 44, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Deml, K.F.; Schildhaus, H.U.; Comperat, E.; von Teichman, A.; Storz, M.; Schraml, P.; Bonventre, J.V.; Fend, F.; Fleige, B.; Nerlich, A.; et al. Clear cell papillary renal cell carcinoma and renal angiomyoadenomatous tumor: Two variants of a morphologic, immunohistochemical, and genetic distinct entity of renal cell carcinoma. Am. J. Surg. Pathol. 2015, 39, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakimi, A.A.; Tickoo, S.K.; Jacobsen, A.; Sarungbam, J.; Sfakianos, J.P.; Sato, Y.; Morikawa, T.; Kume, H.; Fukayama, M.; Homma, Y.; et al. TCEB1-mutated renal cell carcinoma: A distinct genomic and morphological subtype. Mod. Pathol. 2015, 28, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Al-Obaidy, K.I.; Bridge, J.A.; Cheng, L.; Sumegi, J.; Reuter, V.E.; Benayed, R.; Hameed, M.; Williamson, S.R.; Hes, O.; Alruwaii, F.I.; et al. EWSR1-PATZ1 fusion renal cell carcinoma: A recurrent gene fusion characterizing thyroid-like follicular renal cell carcinoma. Mod. Pathol. 2021, 34, 1921–1934. [Google Scholar] [CrossRef]
- Ohe, C.; Kuroda, N.; Pan, C.C.; Yang, X.J.; Hes, O.; Michal, M.; Uehara, H.; Hamada, S.; Kirime, S.; Senzaki, H. A unique renal cell carcinoma with features of papillary renal cell carcinoma and thyroid-like carcinoma: A morphological, immunohistochemical and genetic study. Histopathology 2010, 57, 494–497. [Google Scholar] [CrossRef]
- Hang, J.F.; Chung, H.J.; Pan, C.C. ALK-rearranged renal cell carcinoma with a novel PLEKHA7-ALK translocation and metanephric adenoma-like morphology. Virchows Arch. 2020, 476, 921–929. [Google Scholar] [CrossRef]
- Rupp, N.J.; Moch, H. ALK-rearranged renal cell carcinoma-different morphological faces of a rare tumor: Editorial to Hang et al.: ALK-rearranged renal cell carcinoma with a novel PLEKHA7-ALK translocation and metanephric adenoma-like morphology. Virchows Arch. 2020, 477, 759–760. [Google Scholar] [CrossRef]
- Tao, J.J.; Wei, G.; Patel, R. ALK fusions in renal cell carcinoma: Response to entrectinib. JCO Precis. Oncol. 2018, 2, 1–8. [Google Scholar] [CrossRef]
- Al-Delfi, F.; Karaman, U.; Chen, S.; Abdulsattar, J.; Isac, W.; Elmajian, D.; Gomelsky, A.; Herrera, G. Oncocytoma, Tubulocystic Pattern with Papillary Features: A Diagnostic Challenge. Am. J. Clin. Pathol. 2016, 146, 282. [Google Scholar] [CrossRef] [Green Version]
- Michalova, K.; Tretiakova, M.; Pivovarcikova, K.; Alaghehbandan, R.; Perez Montiel, D.; Ulamec, M.; Osunkoya, A.; Trpkov, K.; Yuan, G.; Grossmann, P.; et al. Expanding the morphologic spectrum of chromophobe renal cell carcinoma: A study of 8 cases with papillary architecture. Ann. Diagn. Pathol. 2020, 44, 151448. [Google Scholar] [CrossRef]
- Chevarie-Davis, M.; Riazalhosseini, Y.; Arseneault, M.; Aprikian, A.; Kassouf, W.; Tanguay, S.; Latour, M.; Brimo, F. The morphologic and immunohistochemical spectrum of papillary renal cell carcinoma: Study including 132 cases with pure type 1 and type 2 morphology as well as tumors with overlapping features. Am. J. Surg. Pathol. 2014, 38, 887–894. [Google Scholar] [CrossRef]
- Williamson, S.R.; Eble, J.N.; Amin, M.B.; Gupta, N.S.; Smith, S.C.; Sholl, L.M.; Montironi, R.; Hirsch, M.S.; Hornick, J.L. Succinate dehydrogenase-deficient renal cell carcinoma: Detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod. Pathol. 2015, 28, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Gill, A.J.; Hes, O.; Papathomas, T.; Sedivcova, M.; Tan, P.H.; Agaimy, A.; Andresen, P.A.; Kedziora, A.; Clarkson, A.; Toon, C.W.; et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: A morphologically distinct entity: A clinicopathologic series of 36 tumors from 27 patients. Am. J. Surg. Pathol. 2014, 38, 1588–1602. [Google Scholar] [CrossRef] [Green Version]
- Kapur, P.; Gao, M.; Zhong, H.; Rakheja, D.; Cai, Q.; Pedrosa, I.; Margulis, V.; Xu, L.; Kinch, L.; Brugarolas, J. Eosinophilic Vacuolated Tumor of the Kidney: A Review of Evolving Concepts in This Novel Subtype With Additional Insights From a Case With MTOR Mutation and Concomitant Chromosome 1 Loss. Adv. Anat. Pathol. 2021, 28, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Siadat, F.; Trpkov, K. ESC, ALK, HOT and LOT: Three Letter Acronyms of Emerging Renal Entities Knocking on the Door of the WHO Classification. Cancers 2020, 12, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Gruenvald, V.; Horwich, A.; Committee, E.G. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v58–v68. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B. Combination Therapy as First-Line Treatment in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1176–1178. [Google Scholar] [CrossRef] [PubMed]












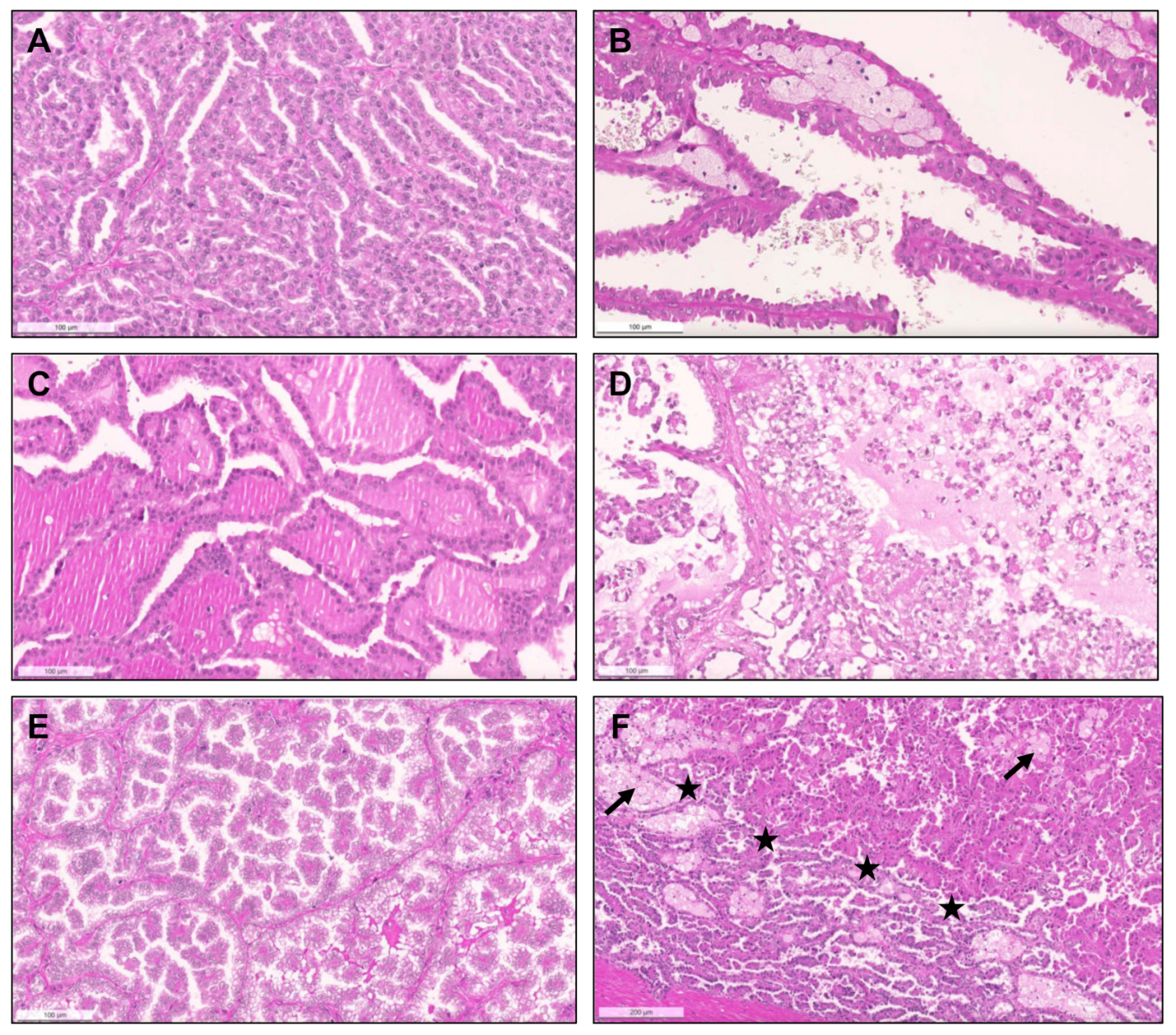{kind=link}
{kind=link}
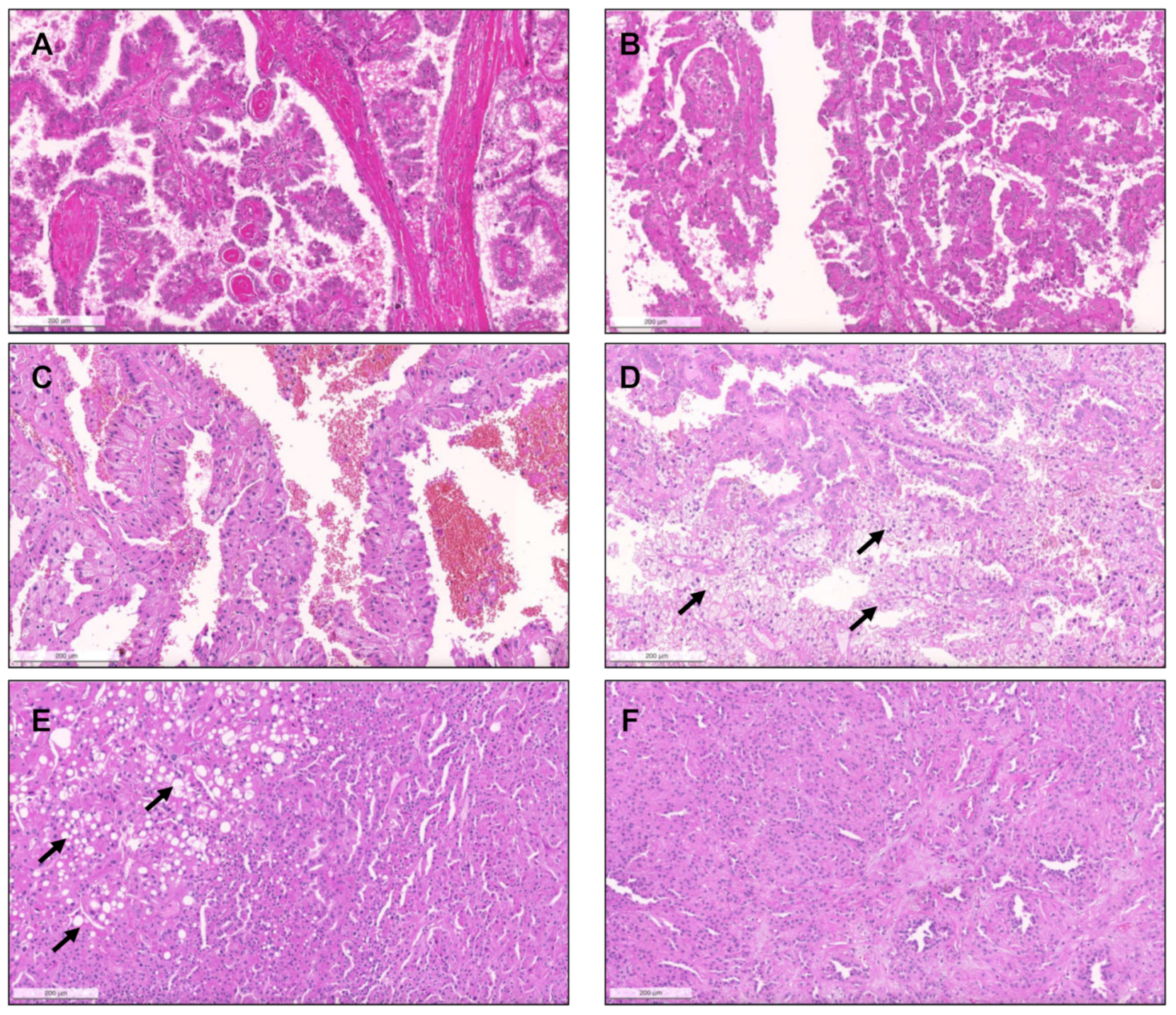{kind=link}
{kind=link}
{kind=link}
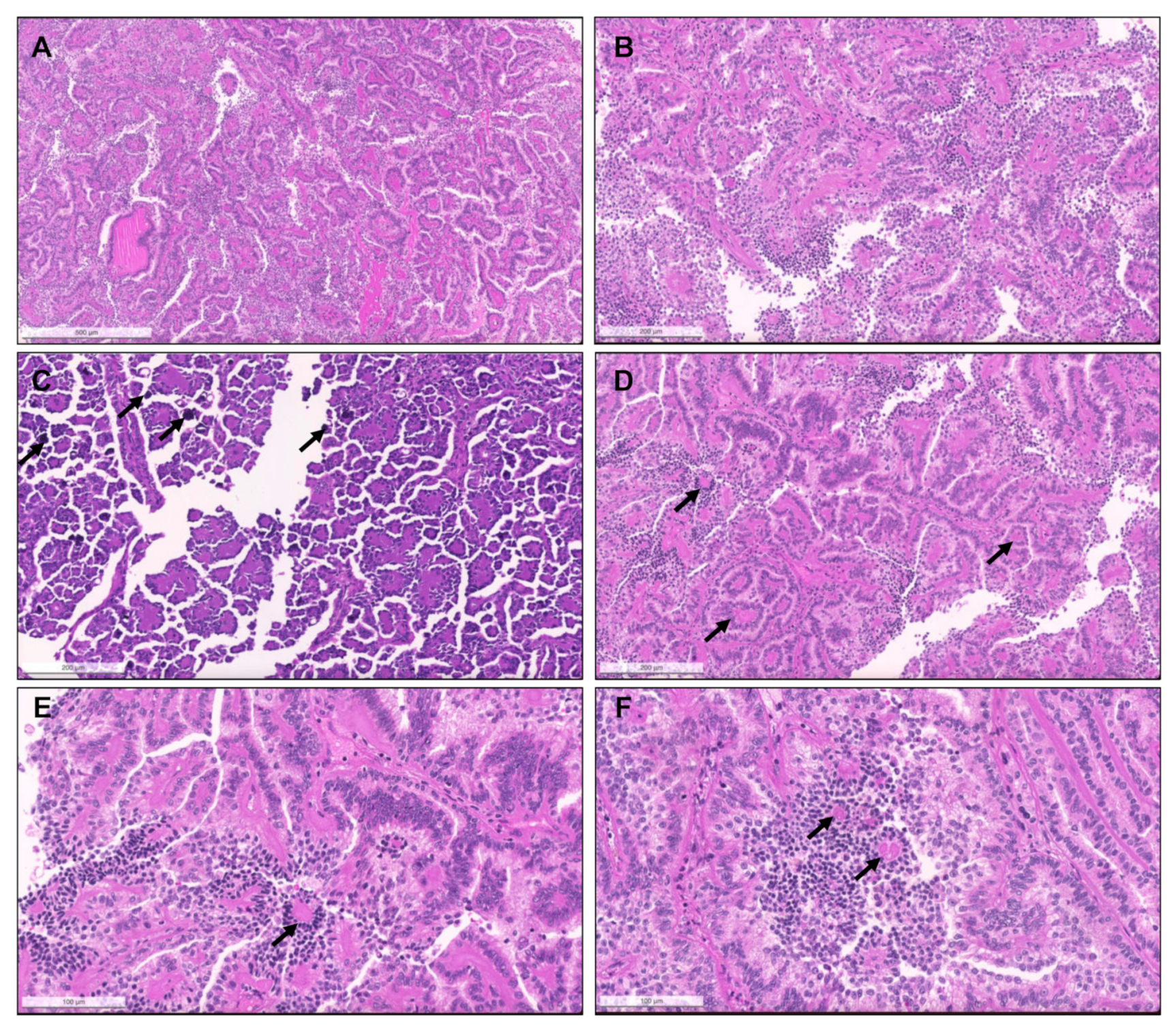{kind=link}
{kind=link}
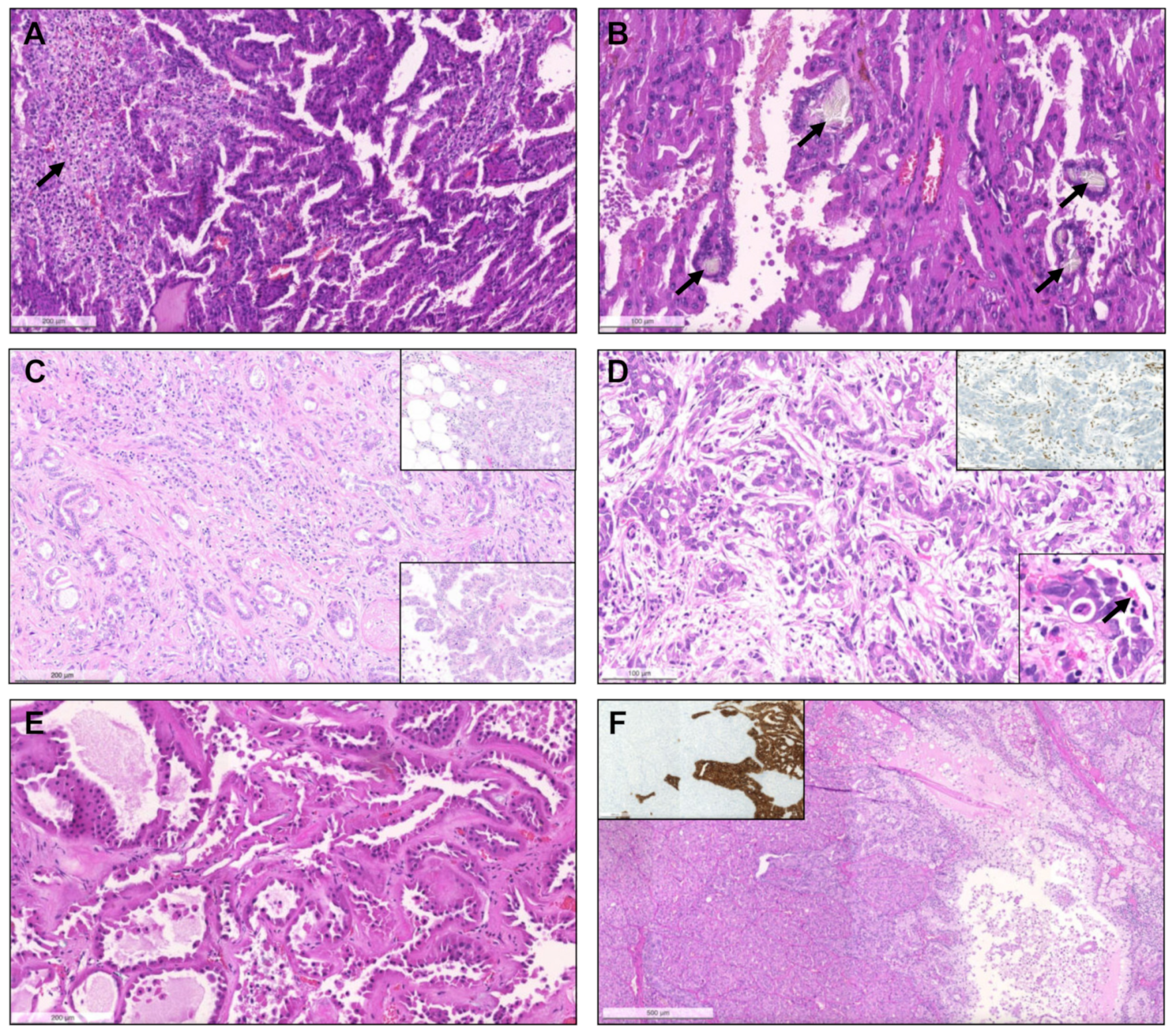{kind=link}
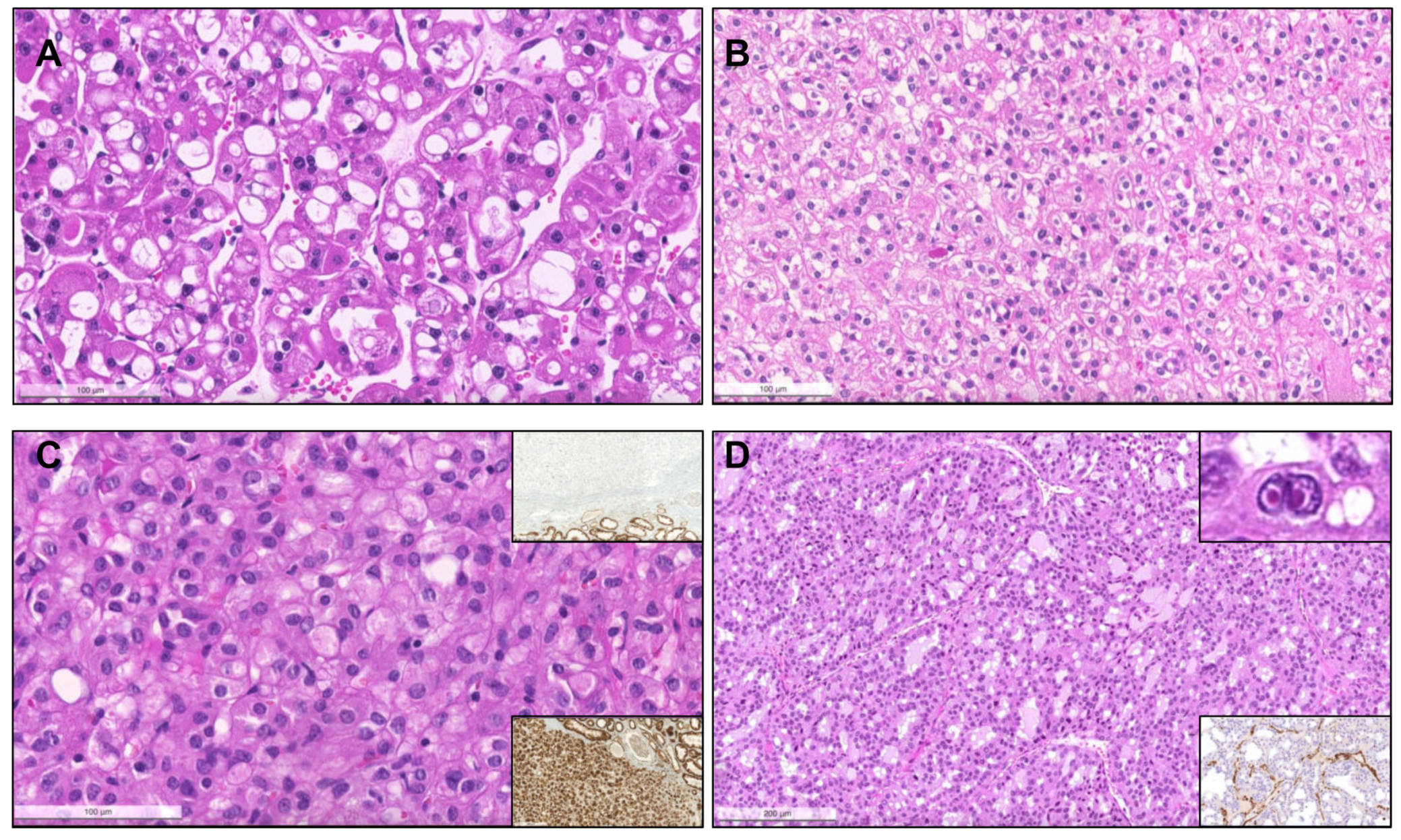{kind=link}
{kind=link}
{kind=link}
| Antibody | Device | Dilution/Clone | Producer | Detection |
|---|---|---|---|---|
| AMACR | Ventana | 1:30 [monoclonal] 13H4 | Biologo | OptiView DAB |
| CAIX | BOND | 1:3000 [polyclonal] | Abcam Limited | IHC Refine-30 min |
| CD10 | Ventana | 1:25 [monoclonal] 56C6 | Novocastra Laboratories Ltd. | OptiView DAB |
| CK20 | Ventana | Prediluted [monoclonal] SP33 | Ventana-Roche | OptiView DAB |
| CK7 | Ventana | 1:100 [monoclonal] SP52 | Abcam Limited | OptiView DAB |
| CyclinD1 | BOND | 1:40 [monoclonal] SP4 | Labvision | IHC Refine-30 min |
| FH | BOND | 1:1000 [monoclonal] J-13 | Santa Cruz Biotechnology, Inc. | IHC Refine-30 min |
| GATA3 | Ventana | 1:200 [monoclonal] L50-823 | Cell Marque Lifescreen Ltd. | OptiView DAB |
| Melan-A | Ventana | 1:30 [monoclonal] A103 | DAKO A/S | UView mono AP |
| PAX8 | Ventana | 1:100 [monoclonal] SP348 | Abcam Limited | OptiView DAB |
| SDHB | BOND | 1:200 [monoclonal] 21A11AE7 | Abcam Limited | IHC Refine-30 min |
| TFE3 | Ventana | Prediluted [monoclonal] MRQ-37 | Cell Marque Lifescreen Ltd. | OptiView DAB |
| Renal Tumor Subtype | n (%) |
|---|---|
| pRCC | |
| type 1 (classic) | 89 (57.8) |
| type 2 | 53 (34.4) |
| papillary renal neoplasm with reversed polarity | 2 (1.3) |
| biphasic squamoid/alveolar | 7 (4.5) |
| biphasic hyalinizing psammomatous | 2 (1.3) |
| thyroid-like follicular | 1 (0.7) |
| Warthin-like | 0 |
| TOTAL | 154 (100) |
| Diagnosis | N |
|---|---|
| ccRCC | 58 |
| chRCC | 48 |
| of which, eosinophilic variant | 23 |
| Oncocytoma | 9 |
| HOCT | 2 |
| EVT | 1 |
| SDH-deficient RCC | 4 |
| pRCC | 56 |
| type 1 (classic) | 12 |
| type 2 | 23 |
| mixed type 1/2 | 17 |
| biphasic squamoid/alveolar | 2 |
| papillary renal neoplasm with reversed polarity | 2 |
| ccpRCC | 9 |
| Acquired cystic disease-associated RCC | 1 |
| MTSCC | 13 |
| Multilocular cystic renal neoplasm of low malignant potential | 2 |
| Collecting duct carcinoma | 5 |
| SMARCB1 deficient medullary RCC | 1 |
| Tubulocystic RCC | 1 |
| FH-deficient RCC | 2 |
| ESC-RCC | 3 |
| MiT family translocation RCC | 18 |
| of which, TFE3-translocated | 11 |
| of which, TFEB-translocated | 6 |
| of which, TFEB-amplified | 1 |
| RCC with fibromyomatous stroma | 2 |
| MEST/cystic nephroma | 6 |
| Metanephric adenoma | 1 |
| Wilms’ tumor of the adult | 1 |
| Primary kidney NET, well differentiated | 1 |
| Collision tumor * | 5 |
| Angiomyolipoma | 5 |
| Angiosarcoma | 1 |
| Capillary hemangioma | 1 |
| Juxtaglomerular tumor | 2 |
| Liposarcoma | 1 |
| Synovial sarcoma | 1 |
| Epithelioid sarcoma | 1 |
| Myofibroblastic inflammatory tumor | 1 |
| Solitary fibrous tumor | 1 |
| Xanthogranulomatous pyelonephritis | 1 |
| IgG4 kidney disease | 1 |
| RCC, unclassified | 16 |
| TOTAL | 281 |
| Architecturally/Cytologically Defined | Molecularly Defined | Anatomically Defined | With Associated Diseases |
|---|---|---|---|
| ccRCC | TFE3-translocated RCC | Collecting duct carcinoma | Acquired cystic disease-associated RCC |
| ccpRCC | TFEB-translocated RCC | ||
| TFEB-amplified RCC | |||
| pRCC: | ALK rearrangement-associated RCC | ||
| Classic (type 1) | SMARCB1-deficient medullary RCC | ||
| Solid | TCEB1-mutated RCC | ||
| Warthin-like | |||
| BSA RCC | |||
| BPH RCC * | |||
| PRNRP * | |||
| MTSCC | |||
| ESC RCC | |||
| Tubulocystic RCC | |||
| TLF RCC * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lobo, J.; Ohashi, R.; Helmchen, B.M.; Rupp, N.J.; Rüschoff, J.H.; Moch, H. The Morphological Spectrum of Papillary Renal Cell Carcinoma and Prevalence of Provisional/Emerging Renal Tumor Entities with Papillary Growth. Biomedicines 2021, 9, 1418. https://doi.org/10.3390/biomedicines9101418
Lobo J, Ohashi R, Helmchen BM, Rupp NJ, Rüschoff JH, Moch H. The Morphological Spectrum of Papillary Renal Cell Carcinoma and Prevalence of Provisional/Emerging Renal Tumor Entities with Papillary Growth. Biomedicines. 2021; 9(10):1418. https://doi.org/10.3390/biomedicines9101418
Chicago/Turabian StyleLobo, João, Riuko Ohashi, Birgit M. Helmchen, Niels J. Rupp, Jan H. Rüschoff, and Holger Moch. 2021. "The Morphological Spectrum of Papillary Renal Cell Carcinoma and Prevalence of Provisional/Emerging Renal Tumor Entities with Papillary Growth" Biomedicines 9, no. 10: 1418. https://doi.org/10.3390/biomedicines9101418
APA StyleLobo, J., Ohashi, R., Helmchen, B. M., Rupp, N. J., Rüschoff, J. H., & Moch, H. (2021). The Morphological Spectrum of Papillary Renal Cell Carcinoma and Prevalence of Provisional/Emerging Renal Tumor Entities with Papillary Growth. Biomedicines, 9(10), 1418. https://doi.org/10.3390/biomedicines9101418