
Cardiovascular Benefits from Gliflozins: Effects on Endothelial Function


 ,
, 
 , and
, and 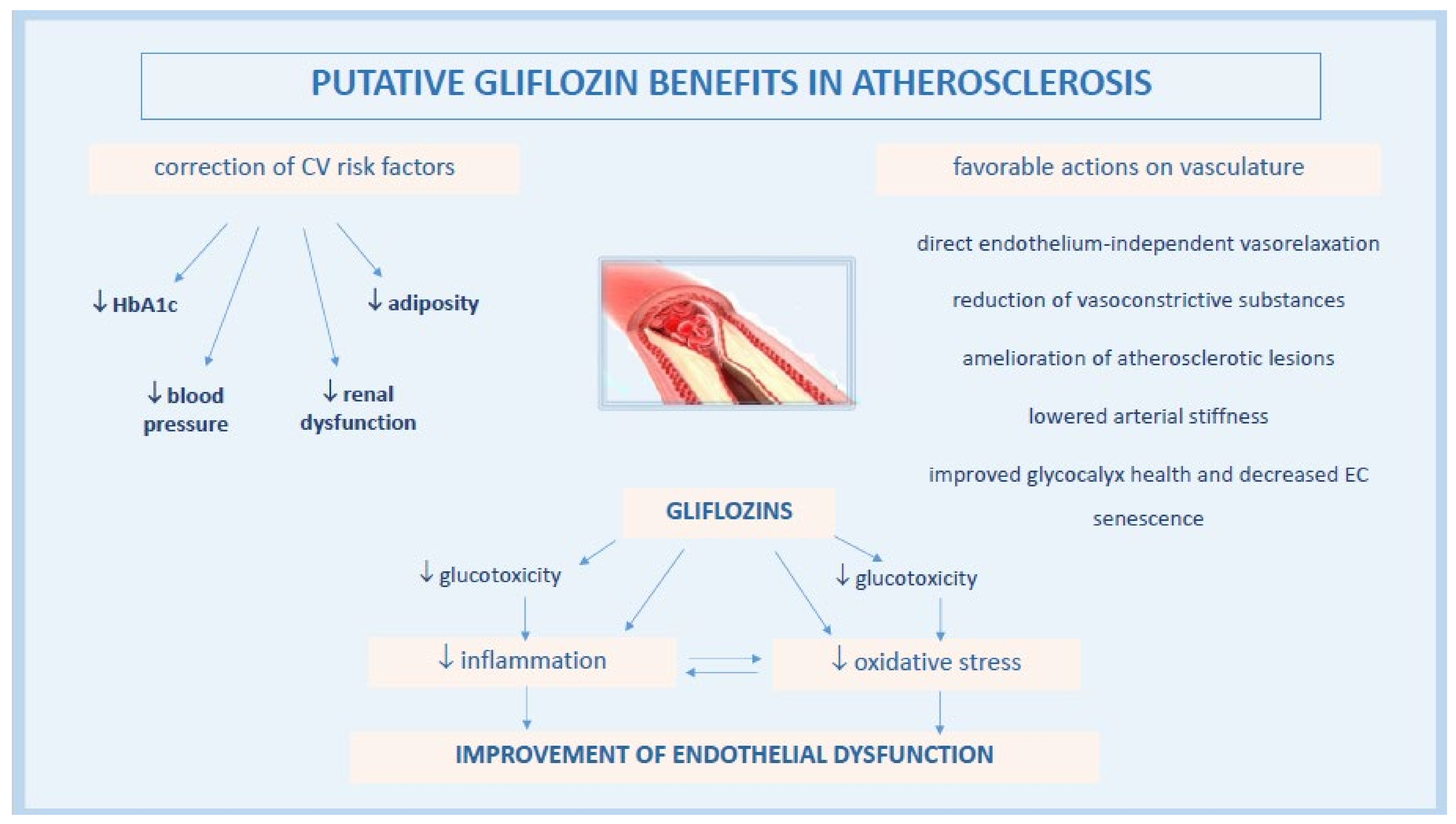{kind=link}
{kind=link}
Abstract
1. Introduction
2. Pathogenesis of Endothelial Dysfunction in Diabetes
3. Effects of Gliflozin on Vascular Endothelial Function
3.1. Gliflozins Modulate Endothelial Function by Attenuating Oxidative Stress and Inflammation
3.1.1. Correction of Glucotoxicity
3.1.2. Anti-Inflammatory Effects of Gliflozins
3.1.3. Reduction of ROS Levels by Gliflozins
3.1.4. Effects of Gliflozins on Glycocalyx Health and Endothelial Senescence
3.1.5. Gliflozin Effects on Angiotensin System
3.2. Results from Clinical Studies
4. Glifozins and Heart Failure
4.1. Gliflozins Correct Endothelial Dysfunction in Microvascular Coronary Bed
4.2. Gliflozin Benefit in HFpEF
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGE | advanced glycation end-product |
| Akt | protein kinase B |
| AMPK | AMP-protein kinase |
| Ang | angiotensin |
| BP | blood pressure |
| CHD | coronary heart disease |
| CVD | cardiovascular disease |
| CVOT | cardiovascular outcome trial |
| EC | endothelial cell |
| ED | endothelial dysfunction |
| EDHF | endothelium-derived hyperpolarizing factor |
| eNOS | endothelial nitric oxide synthase |
| FMD | flow mediated dilation |
| HAECs | human aortic endothelial cells |
| HbA1c | glycosylated hemoglobin |
| HCAECs | human coronary arterial ECs |
| HF | heart failure |
| HFpEF | HF with preserved ejection fraction |
| HFrEF | HF with reduced ejection fraction |
| HG | high glucose |
| HUVECs | human umbilical vein endothelial cells |
| ICAM-1 | intercellular adhesion molecule-1 |
| IL-1β | interleukin β |
| IL6 | interleukin 6 |
| I/R | ischemia/reperfusion |
| JNK | c-Jun N-terminal kinase |
| LV | left ventricle |
| MCP-1 | monocyte chemoattractant protein-1 |
| MPs | microparticles |
| NFkB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NHE1 | sodium-hydrogen exchanger 1 |
| NO | nitric oxide |
| NOX | NADPH oxidase |
| PI3K | phosphatidylinositol 3-kinase |
| PKC | protein kinase C |
| PKG | protein kinase G |
| PWV | pulse wave velocity |
| RAGE | receptor for AGE |
| ROS | reactive oxygen species |
| RH-PAT | reactive hyperemia peripheral arterial tonometry |
| SGLT2-I | sodium-glucose cotransporter-2 inhibitor |
| STZ | streptozotocin |
| T2DM | type 2 diabetes mellitus |
| TNFα | tumor necrosis factor α |
| VCAM-1 | vascular cell adhesion molecule-1 |
| ZDF rats | Zucker diabetic fatty rats |
References
- Sasso, F.C.; Pafundi, P.C.; Gelso, A.; Bono, V.; Costagliola, C.; Marfella, R.; Sardu, C.; Rinaldi, L.; Galiero, R.; Acierno, C.; et al. Applicability of telemedicine in the screening of diabetic retinopathy (DR): The first multicentre study in Italy. The No Blind Study. Diabetes/Metab. Res. Rev. 2018, 35, e3113. [Google Scholar] [CrossRef]
- Sasso, F.C.; Salvatore, T.; Tranchino, G.; Cozzolino, D.; Caruso, A.A.; Persico, M.; Gentile, S.; Torella, D.; Torella, R. Cochlear dysfunction in type 2 diabetes: A complication independent of neuropathy and acute hyperglycemia. Metabolism 1999, 48, 1346–1350. [Google Scholar] [CrossRef]
- Galiero, R.; Pafundi, P.C.; Nevola, R.; Rinaldi, L.; Acierno, C.; Caturano, A.; Salvatore, T.; Adinolfi, L.E.; Costagliola, C.; Sasso, F.C. The Importance of Telemedicine during COVID-19 Pandemic: A Focus on Diabetic Retinopathy. J. Diabetes Res. 2020, 2020, 9036847. [Google Scholar] [CrossRef] [PubMed]
- Masarone, M.; Rosato, V.; Aglitti, A.; Bucci, T.; Caruso, R.; Salvatore, T.; Sasso, F.C.; Tripodi, M.F.; Persico, M. Liver biopsy in type 2 diabetes mellitus: Steatohepatitis represents the sole feature of liver damage. PLoS ONE 2017, 12, e0178473. [Google Scholar] [CrossRef] [PubMed]
- Sasso, F.C.; De Nicola, L.; Carbonara, O.; Nasti, R.; Minutolo, R.; Salvatore, T.; Conte, G.; Torella, R.; for the NID-2 (Nephropathy in Diabetes-Type 2) Study Group. Cardiovascular Risk Factors and Disease Management in Type 2 Diabetic Patients with Diabetic Nephropathy. Diabetes Care 2006, 29, 498–503. [Google Scholar] [CrossRef]
- Torella, D.; Iaconetti, C.; Tarallo, R.; Marino, F.; Giurato, G.; Veneziano, C.; Aquila, I.; Scalise, M.; Mancuso, T.; Cianflone, E.; et al. miRNA Regulation of the Hyperproliferative Phenotype of Vascular Smooth Muscle Cells in Diabetes. Diabetes 2018, 67, 2554–2568. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovasc. Diabetol. 2018, 17, 1–19. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Albanese, G.; Di Martino, A.; Caturano, A.; Vetrano, E.; Rinaldi, L.; Sasso, F.C. The Diabetic Cardiomyopathy: The Contributing Pathophysiological Mechanisms. Front. Med. 2021, 8. [Google Scholar] [CrossRef]
- Kannel, W.B. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979, 241, 2035–2038. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K. Effect of Rosiglitazone on the Risk of Myocardial Infarction and Death from Cardiovascular Causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef]
- Guidance for Industry: Diabetes Mellitus—Evaluating Cardiovascular Risk in New Antidiabetic Therapies to Treat Type 2 Diabetes. Available online: https://www.federalregister.gov/documents/2008/12/19/E8-30086/guidance-for-industry-on-diabetes-mellitus-evaluating-cardiovascular-risk-in-new-antidiabetic (accessed on 10 August 2021).
- Frías, J.P.; Guja, C.; Hardy, E.; Ahmed, A.; Dong, F.; Öhman, P.; Jabbour, S.A. Exenatide once weekly plus dapagliflozin once daily versus exenatide or dapagliflozin alone in patients with type 2 diabetes inadequately controlled with metformin monotherapy (DURATION-8): A 28 week, multicentre, double-blind, phase 3, randomised controlled trial. Lancet Diabetes Endocrinol. 2016, 4, 1004–1016. [Google Scholar] [CrossRef]
- Fei, Y.; Tsoi, M.F.; Cheung, B.M.Y. Cardiovascular outcomes in trials of new antidiabetic drug classes: A network meta-analysis. Cardiovasc. Diabetol. 2019, 18, 1–13. [Google Scholar] [CrossRef]
- Wang, S.; Wu, T.; Zuo, Z.; Jin, P.; Luo, X.; Deng, M. Comparison of cardiovascular outcomes and cardiometabolic risk factors between patients with type 2 diabetes treated with sodium-glucose cotransporter-2 inhibitors and dipeptidyl peptidase-4 inhibitors: A meta-analysis. Eur. J. Prev. Cardiol. 2021. [Google Scholar] [CrossRef]
- Salvatore, T.; Carbonara, O.; Cozzolino, D.; Torella, R.; Nasti, R.; Lascar, N.; Sasso, F.C. Kidney in Diabetes: From Organ Damage Target to Therapeutic Target. Curr. Drug Metab. 2011, 12, 658–666. [Google Scholar] [CrossRef]
- Vrhovac, I.; Eror, D.B.; Klessen, D.; Burger, C.; Breljak, D.; Kraus, O.; Radović, N.; Jadrijević, S.; Aleksic, I.; Walles, T.; et al. Localizations of Na+-d-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflügers Arch.-Eur. J. Physiol. 2014, 467, 1881–1898. [Google Scholar] [CrossRef]
- List, J.F.; Woo, V.; Morales, E.; Tang, W.; Fiedorek, F.T. Sodium-Glucose Cotransport Inhibition With Dapagliflozin in Type 2 Diabetes. Diabetes Care 2009, 32, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Chao, E.C.; Henry, R.R. SGLT2 inhibition—A novel strategy for diabetes treatment. Nat. Rev. Drug Discov. 2010, 9, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Rieg, T.; Masuda, T.; Gerasimova, M.; Mayoux, E.; Platt, K.; Powell, D.R.; Thomson, S.C.; Koepsell, H.; Vallon, V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol. Physiol. 2014, 306, F188–F193. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, N. Place of sodium-glucose co-transporter type 2 inhibitors for treatment of type 2 diabetes. World J. Diabetes 2014, 5, 854–859. [Google Scholar] [CrossRef]
- Inzucchi, S.E.; Kosiborod, M.; Fitchett, D.; Wanner, C.; Hehnke, U.; Kaspers, S.; George, J.T.; Zinman, B. Improvement in Cardiovascular Outcomes With Empagliflozin Is Independent of Glycemic Control. Circulation 2018, 138, 1904–1907. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Trimarco, B.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Marfella, R.; Sasso, F.C.; Cacciapuoti, F.; Portoghese, M.; Rizzo, M.R.; Siniscalchi, M.; Carbonara, O.; Ferraraccio, F.; Torella, M.; Petrella, A.; et al. Tight Glycemic Control May Increase Regenerative Potential of Myocardium during Acute Infarction. J. Clin. Endocrinol. Metab. 2012, 97, 933–942. [Google Scholar] [CrossRef]
- Sasso, F.C.; Rinaldi, L.; Lascar, N.; Marrone, A.; Pafundi, P.C.; Adinolfi, L.E.; Marfella, R. Role of Tight Glycemic Control during Acute Coronary Syndrome on CV Outcome in Type 2 Diabetes. J. Diabetes Res. 2018, 2018, 3106056. [Google Scholar] [CrossRef]
- Staels, B. Cardiovascular Protection by Sodium Glucose Cotransporter 2 Inhibitors: Potential Mechanisms. Am. J. Med. 2017, 130, S30–S39. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Verma, S.; Connelly, K. Mechanistic insights regarding the role of SGLT2 inhibitors and GLP1 agonist drugs on cardiovascular disease in diabetes. Prog. Cardiovasc. Dis. 2019, 62, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Schork, A.; Saynisch, J.; Vosseler, A.; Jaghutriz, B.A.; Heyne, N.; Peter, A.; Häring, H.-U.; Stefan, N.; Fritsche, A.; Artunc, F. Effect of SGLT2 inhibitors on body composition, fluid status and renin–angiotensin–aldosterone system in type 2 diabetes: A prospective study using bioimpedance spectroscopy. Cardiovasc. Diabetol. 2019, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Uzu, K.; Hashimoto, N.; Onishi, T.; Takaya, T.; Shimane, A.; Taniguchi, Y.; Yasaka, Y.; Ohara, T.; Kawai, H. Empagliflozin’s Ameliorating Effect on Plasma Triglycerides: Association with Endothelial Function Recovery in Diabetic Patients with Coronary Artery Disease. J. Atheroscler. Thromb. 2020, 27, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Al-Sharea, A.; Murphy, A.; Huggins, L.; Hu, Y.; Goldberg, I.J.; Nagareddy, P.R. SGLT2 inhibition reduces atherosclerosis by enhancing lipoprotein clearance in Ldlr type 1 diabetic mice. Atherosclerosis 2018, 271, 166–176. [Google Scholar] [CrossRef]
- Ganbaatar, B.; Fukuda, D.; Shinohara, M.; Yagi, S.; Kusunose, K.; Yamada, H.; Soeki, T.; Hirata, K.-I.; Sata, M. Empagliflozin ameliorates endothelial dysfunction and suppresses atherogenesis in diabetic apolipoprotein E-deficient mice. Eur. J. Pharmacol. 2020, 875, 173040. [Google Scholar] [CrossRef]
- Ashry, N.A.; Abdelaziz, R.R.; Suddek, G.M.; Saleh, M.A. Canagliflozin ameliorates aortic and hepatic dysfunction in dietary-induced hypercholesterolemia in the rabbit. Life Sci. 2021, 280, 119731. [Google Scholar] [CrossRef]
- McMurray, J. EMPA-REG—The “diuretic hypothesis”. J. Diabetes Complicat. 2016, 30, 3–4. [Google Scholar] [CrossRef]
- Sasso, F.C.; Pafundi, P.C.; Simeon, V.; De Nicola, L.; Chiodini, P.; Galiero, R.; Rinaldi, L.; Nevola, R.; Salvatore, T.; Sardu, C.; et al. Efficacy and durability of multifactorial intervention on mortality and MACEs: A randomized clinical trial in type-2 diabetic kidney disease. Cardiovasc. Diabetol. 2021, 20, 145. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Khunti, K.; Fitchett, D.H.; Wanner, C.; Mattheus, M.; George, J.T.; Ofstad, A.P.; Zinman, B. Cardiovascular Benefit of Empagliflozin Across the Spectrum of Cardiovascular Risk Factor Control in the EMPA-REG OUTCOME Trial. J. Clin. Endocrinol. Metab. 2020, 105, 3025–3035. [Google Scholar] [CrossRef]
- Tziomalos, K.; Athyros, V.; Karagiannis, A.; Mikhailidis, D.P. Endothelial dysfunction in metabolic syndrome: Prevalence, pathogenesis and management. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.; Revin, V.; Sobenin, I.; Orekhov, A.; Bobryshev, Y. Vascular Endothelium: Functioning in Norm, Changes in Atherosclerosis and Current Dietary Approaches to Improve Endothelial Function. Mini-Rev. Med. Chem. 2015, 15, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Harrison, D.G.; Navas, J.P.; Fisher, A.A.; Dockery, S.P.; Uematsu, M.; Nerem, R.M.; Alexander, R.W.; Murphy, T.J. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J. Clin. Investig. 1992, 90, 2092–2096. [Google Scholar] [CrossRef] [PubMed]
- Tsihlis, N.D.; Oustwani, C.S.; Vavra, A.K.; Jiang, Q.; Keefer, L.K.; Kibbe, M.R. Nitric Oxide Inhibits Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia by Increasing the Ubiquitination and Degradation of UbcH10. Cell Biophys. 2011, 60, 89–97. [Google Scholar] [CrossRef]
- Wu, S.-J.; Soulez, M.; Yang, Y.-H.; Chu, C.-S.; Shih, S.-C.; Hébert, M.-J.; Kuo, M.-C.; Hsieh, Y.-J. Local Augmented Angiotensinogen Secreted from Apoptotic Vascular Endothelial Cells Is a Vital Mediator of Vascular Remodelling. PLoS ONE 2015, 10, e0132583. [Google Scholar] [CrossRef]
- Verma, S.; Buchanan, M.R.; Anderson, T.J. Endothelial Function Testing as a Biomarker of Vascular Disease. Circulation 2003, 108, 2054–2059. [Google Scholar] [CrossRef]
- Widlansky, M.E.; Gokce, N.; Keaney, J.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef]
- Suo, J.; Ferrara, D.E.; Sorescu, D.; Guldberg, R.E.; Taylor, W.R.; Giddens, D.P. Hemodynamic Shear Stresses in Mouse Aortas. Arter. Thromb. Vasc. Biol. 2007, 27, 346–351. [Google Scholar] [CrossRef]
- Tabit, C.E.; Chung, W.B.; Hamburg, N.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61–74. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Galiero, R.; Rinaldi, L.; Caturano, A.; Vetrano, E.; Aprea, C.; Albanese, G.; Di Martino, A.; Ricozzi, C.; et al. Can Metformin Exert as an Active Drug on Endothelial Dysfunction in Diabetic Subjects? Biomedicines 2020, 9, 3. [Google Scholar] [CrossRef]
- Pereira, C.A.; Carneiro, F.S.; Matsumoto, T.; Tostes, R.C. Bonus Effects of Antidiabetic Drugs: Possible Beneficial Effects on Endothelial Dysfunction, Vascular Inflammation and Atherosclerosis. Basic Clin. Pharmacol. Toxicol. 2018, 123, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.K.; Warnholtz, A.; et al. Mechanisms Underlying Endothelial Dysfunction in Diabetes Mellitus. Circ. Res. 2001, 88, E14–E22. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- James, A.M.; Murphy, M.P. How Mitochondrial Damage Affects Cell Function. J. Biomed. Sci. 2002, 9, 475–487. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, N.; Marchase, R.B.; Chatham, J.C. Role of protein O-linked N-acetyl-glucosamine in mediating cell function and survival in the cardiovascular system. Cardiovasc. Res. 2007, 73, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Pawate, S.; Bhat, N.R. C-Jun N-Terminal Kinase (JNK) Regulation of iNOS Expression in Glial Cells: Predominant Role of JNK1 Isoform. Antioxid. Redox Signal. 2006, 8, 903–909. [Google Scholar] [CrossRef]
- Jiang, B.-H.; Zheng, J.Z.; Aoki, M.; Vogt, P.K. Phosphatidylinositol 3-kinase signaling mediates angiogenesis and expression of vascular endothelial growth factor in endothelial cells. Proc. Natl. Acad. Sci. USA 2000, 97, 1749–1753. [Google Scholar] [CrossRef]
- Kuboki, K.; Jiang, Z.Y.; Takahara, N.; Ha, S.W.; Igarashi, M.; Yamauchi, T.; Feener, E.P.; Herbert, T.P.; Rhodes, C.J.; King, G.L. Regulation of Endothelial Constitutive Nitric Oxide Synthase Gene Expression in Endothelial Cells and In Vivo. Circulation 2000, 101, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.J.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1–associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, H.O.; Paradisi, G.; Hook, G.; Crowder, K.; Cronin, J.; Baron, A.D. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes 2000, 49, 1231–1238. [Google Scholar] [CrossRef]
- Matsumoto, T.; Kobayashi, T.; Kamata, K. Alterations in EDHF-type relaxation and phosphodiesterase activity in mesenteric arteries from diabetic rats. Am. J. Physiol. Circ. Physiol. 2003, 285, H283–H291. [Google Scholar] [CrossRef]
- Matsumoto, T.; Alves-Lopes, R.; Taguchi, K.; Kobayashi, T.; Tostes, R.C. Linking the beneficial effects of current therapeutic approaches in diabetes to the vascular endothelin system. Life Sci. 2014, 118, 129–135. [Google Scholar] [CrossRef]
- Matsumoto, T.; Noguchi, E.; Ishida, K.; Kobayashi, T.; Yamada, N.; Kamata, K. Metformin normalizes endothelial function by suppressing vasoconstrictor prostanoids in mesenteric arteries from OLETF rats, a model of type 2 diabetes. Am. J. Physiol. Circ. Physiol. 2008, 295, H1165–H1176. [Google Scholar] [CrossRef]
- Matsumoto, T.; Watanabe, S.; Kawamura, R.; Taguchi, K.; Kobayashi, T. Enhanced uridine adenosine tetraphosphate-induced contraction in renal artery from type 2 diabetic Goto-Kakizaki rats due to activated cyclooxygenase/thromboxane receptor axis. Pflügers Arch.-Eur. J. Physiol. 2014, 466, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Schächinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic Impact of Coronary Vasodilator Dysfunction on Adverse Long-Term Outcome of Coronary Heart Disease. Circulation 2000, 101, 1899–1906. [Google Scholar] [CrossRef] [PubMed]
- Vita, J.A.; Keaney, J. Endothelial Function. Circulation 2002, 106, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Krum, H.; Khan, T.; Knecht, M. Exercise-induced vasodilation in forearm circulation of normal subjects and patients with congestive heart failure: Role of endothelium-derived nitric oxide. J. Am. Coll. Cardiol. 1996, 28, 585–590. [Google Scholar] [CrossRef][Green Version]
- Borlaug, B.A.; Olson, T.P.; Lam, C.S.; Flood, K.S.; Lerman, A.; Johnson, B.D.; Redfield, M.M. Global Cardiovascular Reserve Dysfunction in Heart Failure With Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2010, 56, 845–854. [Google Scholar] [CrossRef]
- Batzias, K.; Antonopoulos, A.; Oikonomou, E.; Siasos, G.; Bletsa, E.; Stampouloglou, P.K.; Mistakidi, C.-V.; Noutsou, M.; Katsiki, N.; Karopoulos, P.; et al. Effects of Newer Antidiabetic Drugs on Endothelial Function and Arterial Stiffness: A Systematic Review and Meta-Analysis. J. Diabetes Res. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Alshnbari, A.S.; Millar, S.A.; O’Sullivan, S.E.; Idris, I. Effect of Sodium-Glucose Cotransporter-2 Inhibitors on Endothelial Function: A Systematic Review of Preclinical Studies. Diabetes Ther. 2020, 11, 1947–1963. [Google Scholar] [CrossRef]
- El-Daly, M.; Venu, V.K.P.; Saifeddine, M.; Mihara, K.; Kang, S.; Fedak, P.W.; Alston, L.A.; Hirota, S.A.; Ding, H.; Triggle, C.R.; et al. Hyperglycaemic impairment of PAR2-mediated vasodilation: Prevention by inhibition of aortic endothelial sodium-glucose-co-Transporter-2 and minimizing oxidative stress. Vasc. Pharmacol. 2018, 109, 56–71. [Google Scholar] [CrossRef]
- Park, S.-H.; Farooq, M.A.; Gaertner, S.; Bruckert, C.; Qureshi, A.W.; Lee, H.-H.; Benrahla, D.; Pollet, B.; Stephan, D.; Ohlmann, P.; et al. Empagliflozin improved systolic blood pressure, endothelial dysfunction and heart remodeling in the metabolic syndrome ZSF1 rat. Cardiovasc. Diabetol. 2020, 19, 1–14. [Google Scholar] [CrossRef]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; Preckel, B.; Koolwijk, P.; Van Hinsbergh, V.W.M.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor α-Stimulated Human Coronary Arterial Endothelial Cells. Cell Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Mancini, S.J.; Boyd, D.; Katwan, O.J.; Strembitska, A.; Almabrouk, T.A.; Kennedy, S.; Palmer, T.M.; Salt, I.P. Canagliflozin inhibits interleukin-1β-stimulated cytokine and chemokine secretion in vascular endothelial cells by AMP-activated protein kinase-dependent and -independent mechanisms. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, T.; Spizzo, I.; Liu, H.; Hu, Y.; Simpson, R.W.; Widdop, R.; Dear, A.E. Dapagliflozin attenuates human vascular endothelial cell activation and induces vasorelaxation: A potential mechanism for inhibition of atherogenesis. Diabetes Vasc. Dis. Res. 2017, 15, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shin, S.E.; Seo, M.S.; An, J.R.; Choi, I.-W.; Jung, W.-K.; Firth, A.L.; Lee, D.-S.; Yim, M.-J.; Choi, G.; et al. The anti-diabetic drug dapagliflozin induces vasodilation via activation of PKG and Kv channels. Life Sci. 2018, 197, 46–55. [Google Scholar] [CrossRef]
- Seo, M.S.; Jung, H.S.; An, J.R.; Kang, M.; Heo, R.; Li, H.; Han, E.-T.; Yang, S.-R.; Cho, E.-H.; Bae, Y.M.; et al. Empagliflozin dilates the rabbit aorta by activating PKG and voltage-dependent K+ channels. Toxicol. Appl. Pharmacol. 2020, 403, 115153. [Google Scholar] [CrossRef]
- Han, Y.; Cho, Y.-E.; Ayon, R.; Guo, R.; Youssef, K.D.; Pan, M.; Dai, A.; Yuan, J.X.-J.; Makino, A. SGLT inhibitors attenuate NO-dependent vascular relaxation in the pulmonary artery but not in the coronary artery. Am. J. Physiol. Cell. Mol. Physiol. 2015, 309, L1027–L1036. [Google Scholar] [CrossRef]
- Khat, D.Z.; Husain, M. Molecular Mechanisms Underlying the Cardiovascular Benefits of SGLT2i and GLP-1RA. Curr. Diabetes Rep. 2018, 18, 45. [Google Scholar] [CrossRef]
- Lastra, G.; Manrique, C. Perivascular adipose tissue, inflammation and insulin resistance: Link to vascular dysfunction and cardiovascular disease. Horm. Mol. Biol. Clin. Investig. 2015, 22, 19–26. [Google Scholar] [CrossRef]
- Oelze, M.; Kröller-Schön, S.; Welschof, P.; Jansen, T.; Hausding, M.; Mikhed, Y.; Stamm, P.; Mader, M.; Zinßius, E.; Agdauletova, S.; et al. The Sodium-Glucose Co-Transporter 2 Inhibitor Empagliflozin Improves Diabetes-Induced Vascular Dysfunction in the Streptozotocin Diabetes Rat Model by Interfering with Oxidative Stress and Glucotoxicity. PLoS ONE 2014, 9, e112394. [Google Scholar] [CrossRef]
- Juni, R.P.; Kuster, D.W.; Goebel, M.; Helmes, M.; Musters, R.J.; van der Velden, J.; Koolwijk, P.; Paulus, W.J.; van Hinsbergh, V.W. Cardiac Microvascular Endothelial Enhancement of Cardiomyocyte Function Is Impaired by Inflammation and Restored by Empagliflozin. JACC: Basic Transl. Sci. 2019, 4, 575–591. [Google Scholar] [CrossRef]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.M.; Battson, M.L.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Weir, T.L.; Gentile, C.L. SGLT2 inhibition via dapagliflozin improves generalized vascular dysfunction and alters the gut microbiota in type 2 diabetic mice. Cardiovasc. Diabetol. 2018, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, M.; Hiromura, M.; Mori, Y.; Kohashi, K.; Nagashima, M.; Kushima, H.; Watanabe, T.; Hirano, T. Amelioration of Hyperglycemia with a Sodium-Glucose Cotransporter 2 Inhibitor Prevents Macrophage-Driven Atherosclerosis through Macrophage Foam Cell Formation Suppression in Type 1 and Type 2 Diabetic Mice. PLoS ONE 2015, 10, e0143396. [Google Scholar] [CrossRef] [PubMed]
- Sayour, A.A.; Korkmaz-Icöz, S.; Loganathan, S.; Ruppert, M.; Sayour, V.N.; Oláh, A.; Benke, K.; Brune, M.; Benkő, R.; Horváth, E.M.; et al. Acute canagliflozin treatment protects against in vivo myocardial ischemia–reperfusion injury in non-diabetic male rats and enhances endothelium-dependent vasorelaxation. J. Transl. Med. 2019, 17, 1–14. [Google Scholar] [CrossRef]
- Han, J.H.; Oh, T.J.; Lee, G.; Maeng, H.J.; Lee, D.H.; Kim, K.M.; Choi, S.H.; Jang, H.C.; Lee, H.S.; Park, K.S.; et al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE −/− mice fed a western diet. Diabetologia 2017, 60, 364–376. [Google Scholar] [CrossRef]
- Leng, W.; Ouyang, X.; Lei, X.; Wu, M.; Chen, L.; Wu, Q.; Deng, W.; Liang, Z. The SGLT-2 Inhibitor Dapagliflozin Has a Therapeutic Effect on Atherosclerosis in Diabetic ApoE−/−Mice. Mediat. Inflamm. 2016, 2016, 1–13. [Google Scholar] [CrossRef]
- Nakatsu, Y.; Kokubo, H.; Bumdelger, B.; Yoshizumi, M.; Yamamotoya, T.; Matsunaga, Y.; Ueda, K.; Inoue, Y.; Inoue, M.-K.; Fujishiro, M.; et al. The SGLT2 Inhibitor Luseogliflozin Rapidly Normalizes Aortic mRNA Levels of Inflammation-Related but Not Lipid-Metabolism-Related Genes and Suppresses Atherosclerosis in Diabetic ApoE KO Mice. Int. J. Mol. Sci. 2017, 18, 1704. [Google Scholar] [CrossRef]
- Suzuki, J.-I.; Ogawa, M.; Watanabe, R.; Takayama, K.; Hirata, Y.; Nagai, R.; Isobe, M. Roles of Prostaglandin E2 in Cardiovascular Diseases. Int. Heart J. 2011, 52, 266–269. [Google Scholar] [CrossRef]
- Rajsheker, S.; Manka, D.; Blomkalns, A.L.; Chatterjee, T.K.; Stoll, L.L.; Weintraub, N. Crosstalk between perivascular adipose tissue and blood vessels. Curr. Opin. Pharmacol. 2010, 10, 191–196. [Google Scholar] [CrossRef]
- Li, T.; Liu, X.; Ni, L.; Wang, Z.; Wang, W.; Shi, T.; Liu, X.; Liu, C. Perivascular adipose tissue alleviates inflammatory factors and stenosis in diabetic blood vessels. Biochem. Biophys. Res. Commun. 2016, 480, 147–152. [Google Scholar] [CrossRef]
- Korkmaz-Icöz, S.; Kocer, C.; Sayour, A.; Kraft, P.; Benker, M.; Abulizi, S.; Georgevici, A.-I.; Brlecic, P.; Radovits, T.; Loganathan, S.; et al. The Sodium-Glucose Cotransporter-2 Inhibitor Canagliflozin Alleviates Endothelial Dysfunction Following In Vitro Vascular Ischemia/Reperfusion Injury in Rats. Int. J. Mol. Sci. 2021, 22, 7774. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ford, R.J.; Smith, B.K.; Gowans, G.J.; Mancini, S.J.; Pitt, R.D.; Day, E.A.; Salt, I.; Steinberg, G.R.; Hardie, D.G. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef]
- Aroor, A.R.; Das, N.A.; Carpenter, A.J.; Habibi, J.; Jia, G.; Ramirez-Perez, F.; Martinez-Lemus, L.; Manrique-Acevedo, C.M.; Hayden, M.R.; Duta, C.; et al. Glycemic control by the SGLT2 inhibitor empagliflozin decreases aortic stiffness, renal resistivity index and kidney injury. Cardiovasc. Diabetol. 2018, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Salim, H.M.; Fukuda, D.; Yagi, S.; Soeki, T.; Shimabukuro, M.; Sata, M. Glycemic Control with Ipragliflozin, a Novel Selective SGLT2 Inhibitor, Ameliorated Endothelial Dysfunction in Streptozotocin-Induced Diabetic Mouse. Front. Cardiovasc. Med. 2016, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Römer, G.; Kerindongo, R.; Hermanides, J.; Albrecht, M.; Hollmann, M.; Zuurbier, C.; Preckel, B.; Weber, N. Sodium Glucose Co-Transporter 2 Inhibitors Ameliorate Endothelium Barrier Dysfunction Induced by Cyclic Stretch through Inhibition of Reactive Oxygen Species. Int. J. Mol. Sci. 2021, 22, 6044. [Google Scholar] [CrossRef]
- Tarbell, J.M.; Cancel, L.M. The glycocalyx and its significance in human medicine. J. Intern. Med. 2016, 280, 97–113. [Google Scholar] [CrossRef]
- Florian, J.A.; Kosky, J.R.; Ainslie, K.; Pang, Z.; Dull, R.O.; Tarbell, J.M. Heparan Sulfate Proteoglycan Is a Mechanosensor on Endothelial Cells. Circ. Res. 2003, 93, e136–e142. [Google Scholar] [CrossRef]
- McDonald, K.K.; Cooper, S.; Danielzak, L.; Leask, R.L. Glycocalyx Degradation Induces a Proinflammatory Phenotype and Increased Leukocyte Adhesion in Cultured Endothelial Cells under Flow. PLoS ONE 2016, 11, e0167576. [Google Scholar] [CrossRef]
- Nieuwdorp, M.; Van Haeften, T.W.; Gouverneur, M.C.L.G.; Mooij, H.L.; Van Lieshout, M.H.P.; Levi, M.; Meijers, J.C.M.; Holleman, F.; Hoekstra, J.B.L.; Vink, H.; et al. Loss of Endothelial Glycocalyx During Acute Hyperglycemia Coincides With Endothelial Dysfunction and Coagulation Activation In Vivo. Diabetes 2006, 55, 480–486. [Google Scholar] [CrossRef]
- Rao, G.; Ding, H.G.; Huang, W.; Le, D.; Maxhimer, J.B.; Oosterhof, A.; Van Kuppevelt, T.; Lum, H.; Lewis, E.J.; Reddy, V.; et al. Reactive oxygen species mediate high glucose-induced heparanase-1 production and heparan sulphate proteoglycan degradation in human and rat endothelial cells: A potential role in the pathogenesis of atherosclerosis. Diabetologia 2011, 54, 1527–1538. [Google Scholar] [CrossRef]
- Cooper, S.; Teoh, H.; Campeau, M.A.; Verma, S.; Leask, R.L. Empagliflozin restores the integrity of the endothelial glycocalyx in vitro. Mol. Cell. Biochem. 2019, 459, 121–130. [Google Scholar] [CrossRef]
- Ikonomidis, I.; Pavlidis, G.; Thymis, J.; Birba, D.; Kalogeris, A.; Kousathana, F.; Kountouri, A.; Balampanis, K.; Parissis, J.; Andreadou, I.; et al. Effects of Glucagon-Like Peptide-1 Receptor Agonists, Sodium-Glucose Cotransporter-2 Inhibitors, and Their Combination on Endothelial Glycocalyx, Arterial Function, and Myocardial Work Index in Patients with Type 2 Diabetes Mellitus After 12-Month Treatment. J. Am. Heart Assoc. 2020, 9, e015716. [Google Scholar] [CrossRef]
- Chen, J.; Brodsky, S.V.; Goligorsky, D.M.; Hampel, D.J.; Li, H.; Gross, S.S.; Goligorsky, M.S. Glycated Collagen I Induces Premature Senescence-Like Phenotypic Changes in Endothelial Cells. Circ. Res. 2002, 90, 1290–1298. [Google Scholar] [CrossRef]
- Hayashi, T.; Matsui-Hirai, H.; Miyazaki-Akita, A.; Fukatsu, A.; Funami, J.; Ding, Q.-F.; Kamalanathan, S.; Hattori, Y.; Ignarro, L.J.; Iguchi, A. Endothelial cellular senescence is inhibited by nitric oxide: Implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. USA 2006, 103, 17018–17023. [Google Scholar] [CrossRef]
- Kumar, A.; Kim, C.-S.; Hoffman, T.A.; Naqvi, A.; DeRicco, J.; Jung, S.-B.; Lin, Z.; Jain, M.K.; Irani, K. p53 Impairs Endothelial Function by Transcriptionally Repressing Kruppel-Like Factor 2. Arter. Thromb. Vasc. Biol. 2011, 31, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Khemais-Benkhiat, S.; Belcastro, E.; Idris-Khodja, N.; Park, S.-H.; Amoura, L.; Abbas, M.; Auger, C.; Kessler, L.; Mayoux, E.; Toti, F.; et al. Angiotensin II-induced redox-sensitive SGLT1 and 2 expression promotes high glucose-induced endothelial cell senescence. J. Cell. Mol. Med. 2019, 24, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Chobanian, A.V.; Alexander, R.W. Exacerbation of atherosclerosis by hypertension. Potential mechanisms and clinical implications. Arch. Intern. Med. 1996, 156, 8823148. [Google Scholar] [CrossRef]
- Khodja, N.I.; Chataigneau, T.; Auger, C.; Schini-Kerth, V.B. Grape-Derived Polyphenols Improve Aging-Related Endothelial Dysfunction in Rat Mesenteric Artery: Role of Oxidative Stress and the Angiotensin System. PLoS ONE 2012, 7, e32039. [Google Scholar] [CrossRef]
- Harrison, D.G.; Cai, H.; Landmesser, U.; Griendling, K.K. The Pickering Lecture British Hypertension Society, 10th September 2002. J. Renin-Angiotensin-Aldosterone Syst. 2003, 4, 51–61. [Google Scholar] [CrossRef]
- Imanishi, T.; Hano, T.; Nishio, I. Angiotensin II accelerates endothelial progenitor cell senescence through induction of oxidative stress. J. Hypertens. 2005, 23, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.; Jesel, L.; Auger, C.; Amoura, L.K.; Messas, N.; Manin, G.; Rumig, C.; León-González, A.J.; Ribeiro, T.P.; Silva, G.C.; et al. Endothelial Microparticles From Acute Coronary Syndrome Patients Induce Premature Coronary Artery Endothelial Cell Aging and Thrombogenicity: Role of the ang II/AT1 receptor/NADPH oxidase-mediated activation of MAPKs and PI3-Kinase pathways. Circulation 2017, 135, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; He, K.; Sun, H.; Zhou, X.; Chang, L.; Li, X.; Chu, W.; Qiao, G.; Lu, Y. Hydrogen peroxide induces overexpression of angiotensin-converting enzyme in human umbilical vein endothelial cells. Free Radic. Res. 2012, 47, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Belcastro, E.; Hasan, H.; Matsushita, K.; Marchandot, B.; Abbas, M.; Toti, F.; Auger, C.; Jesel, L.; Ohlmann, P.; et al. Angiotensin II-induced upregulation of SGLT1 and 2 contributes to human microparticle-stimulated endothelial senescence and dysfunction: Protective effect of gliflozins. Cardiovasc. Diabetol. 2021, 20, 1–17. [Google Scholar] [CrossRef]
- Majowicz, M.; Bosc, L.G.; Borghese, M.A.; Delgado, M.; Ortiz, M.; Speziale, N.S.; Vidal, N. Atrial natriuretic peptide and endothelin-3 target renal sodium-glucose cotransporter. Peptides 2003, 24, 1971–1976. [Google Scholar] [CrossRef]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Lüscher, T.F.; Shechter, M.; Taddei, S.; et al. The Assessment of Endothelial Function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef]
- Ferrannini, E.; Ramos, S.J.; Salsali, A.; Tang, W.; List, J.F. Dapagliflozin Monotherapy in Type 2 Diabetic Patients With Inadequate Glycemic Control by Diet and Exercise: A randomized, double-blind, placebo-controlled, phase 3 trial. Diabetes Care 2010, 33, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Giannini, L.; Seghieri, M.; Vitolo, E.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. Dapagliflozin acutely improves endothelial dysfunction, reduces aortic stiffness and renal resistive index in type 2 diabetic patients: A pilot study. Cardiovasc. Diabetol. 2017, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Seghieri, M.; Giannini, L.; Biancalana, E.; Parolini, F.; Rossi, C.; Dardano, A.; Taddei, S.; Ghiadoni, L.; Bruno, R.M. The Effects of Dapagliflozin on Systemic and Renal Vascular Function Display an Epigenetic Signature. J. Clin. Endocrinol. Metab. 2019, 104, 4253–4263. [Google Scholar] [CrossRef] [PubMed]
- Shigiyama, F.; Kumashiro, N.; Miyagi, M.; Ikehara, K.; Kanda, E.; Uchino, H.; Hirose, T. Effectiveness of dapagliflozin on vascular endothelial function and glycemic control in patients with early-stage type 2 diabetes mellitus: DEFENCE study. Cardiovasc. Diabetol. 2017, 16, 1–12. [Google Scholar] [CrossRef]
- Sugiyama, S.; Jinnouchi, H.; Kurinami, N.; Hieshima, K.; Yoshida, A.; Jinnouchi, K.; Nishimura, H.; Suzuki, T.; Miyamoto, F.; Kajiwara, K.; et al. The SGLT2 Inhibitor Dapagliflozin Significantly Improves the Peripheral Microvascular Endothelial Function in Patients with Uncontrolled Type 2 Diabetes Mellitus. Intern. Med. 2018, 57, 2147–2156. [Google Scholar] [CrossRef]
- Zainordin, N.A.; Hatta, S.F.W.M.; Shah, F.Z.M.; Rahman, T.A.; Ismail, N.; Ismail, Z.; Ghani, R.A. Effects of Dapagliflozin on Endothelial Dysfunction in Type 2 Diabetes With Established Ischemic Heart Disease (EDIFIED). J. Endocr. Soc. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.-Y.; Park, K.-Y.; Kim, J.-D.; Hwang, W.-M.; Lim, D.-M. Effects of 6 Months of Dapagliflozin Treatment on Metabolic Profile and Endothelial Cell Dysfunction for Obese Type 2 Diabetes Mellitus Patients without Atherosclerotic Cardiovascular Disease. J. Obes. Metab. Syndr. 2020, 29, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.K. Letter: Effects of 6 Months of Dapagliflozin Treatment on Metabolic Profile and Endothelial Cell Dysfunction for Obese Type 2 Diabetes Mellitus Patients without Atherosclerotic Cardiovascular Disease (J Obes Metab Syndr 2020;29:215-21). J. Obes. Metab. Syndr. 2021, 30, 72–73. [Google Scholar] [CrossRef] [PubMed]
- Sposito, A.C.; Breder, I.; Soares, A.A.S.; Kimura-Medorima, S.T.; Munhoz, D.B.; Cintra, R.M.R.; Bonilha, I.; Oliveira, D.C.; Breder, J.C.; Cavalcante, P.; et al. Dapagliflozin effect on endothelial dysfunction in diabetic patients with atherosclerotic disease: A randomized active-controlled trial. Cardiovasc. Diabetol. 2021, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Har, R.; Fagan, N.; Johansen, O.E.; Woerle, H.-J.; Von Eynatten, M.; Broedl, U.C. The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc. Diabetol. 2014, 13, 28. [Google Scholar] [CrossRef]
- Lunder, M.; Janić, M.; Japelj, M.; Juretič, A.; Janež, A.; Šabovič, M. Empagliflozin on top of metformin treatment improves arterial function in patients with type 1 diabetes mellitus. Cardiovasc. Diabetol. 2018, 17, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Fasano, M.; Della Corte, C.M.; Sasso, F.C.; Papaccio, F.; Viscardi, G.; Esposito, G.; DI Liello, R.; Normanno, N.; Capuano, A.; et al. Results of the safety run-in part of the METAL (METformin in Advanced Lung cancer) study: A multicentre, open-label phase I–II study of metformin with erlotinib in second-line therapy of patients with stage IV non-small-cell lung cancer. ESMO Open 2017, 2, e000132. [Google Scholar] [CrossRef]
- Salvatore, T.; Pafundi, P.C.; Morgillo, F.; Di Liello, R.; Galiero, R.; Nevola, R.; Marfella, R.; Monaco, L.; Rinaldi, L.; Adinolfi, L.E.; et al. Metformin: An old drug against old age and associated morbidities. Diabetes Res. Clin. Pr. 2020, 160, 108025. [Google Scholar] [CrossRef]
- Irace, C.; Cutruzzolà, A.; Parise, M.; Fiorentino, R.; Frazzetto, M.; Gnasso, C.; Casciaro, F.; Gnasso, A. Effect of empagliflozin on brachial artery shear stress and endothelial function in subjects with type 2 diabetes: Results from an exploratory study. Diabetes Vasc. Dis. Res. 2019, 17. [Google Scholar] [CrossRef]
- Tanaka, A.; Shimabukuro, M.; Machii, N.; Teragawa, H.; Okada, Y.; Shima, K.R.; Takamura, T.; Taguchi, I.; Hisauchi, I.; Toyoda, S.; et al. Effect of Empagliflozin on Endothelial Function in Patients With Type 2 Diabetes and Cardiovascular Disease: Results from the Multicenter, Randomized, Placebo-Controlled, Double-Blind EMBLEM Trial. Diabetes Care 2019, 42, e159–e161. [Google Scholar] [CrossRef]
- Tanaka, A.; Shimabukuro, M.; Machii, N.; Teragawa, H.; Okada, Y.; Shima, K.R.; Takamura, T.; Taguchi, I.; Hisauchi, I.; Toyoda, S.; et al. Secondary analyses to assess the profound effects of empagliflozin on endothelial function in patients with type 2 diabetes and established cardiovascular diseases: The placebo-controlled double-blind randomized effect of empagliflozin on endothelial function in cardiovascular high risk diabetes mellitus: Multi-center placebo-controlled double-blind randomized trial. J. Diabetes Investig. 2020, 11, 1551–1563. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Shimabukuro, M.; Okada, Y.; Sugimoto, K.; Kurozumi, A.; Torimoto, K.; Hirai, H.; Node, K.; the PROCEED trial investigators. Rationale and design of an investigator-initiated, multicenter, prospective open-label, randomized trial to evaluate the effect of ipragliflozin on endothelial dysfunction in type 2 diabetes and chronic kidney disease: The PROCEED trial. Cardiovasc. Diabetol. 2020, 19, 1–11. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Steg, P.G.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Voors, A.A.; Metra, M.; et al. Sotagliflozin in Patients with Diabetes and Recent Worsening Heart Failure. N. Engl. J. Med. 2021, 384, 117–128. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Vargas-Delgado, A.P.; Requena-Ibanez, J.A.; Garcia-Ropero, A.; Mancini, D.; Pinney, S.; Macaluso, F.; Sartori, S.; Roque, M.; Sabatel-Perez, F.; et al. Randomized Trial of Empagliflozin in Nondiabetic Patients With Heart Failure and Reduced Ejection Fraction. J. Am. Coll. Cardiol. 2021, 77, 243–255. [Google Scholar] [CrossRef]
- Kosiborod, M.; Cavender, M.A.; Fu, A.Z.; Wilding, J.; Khunti, K.; Holl, R.W.; Norhammar, A.; Birkeland, K.I.; Jørgensen, M.E.; Thuresson, M.; et al. Lower Risk of Heart Failure and Death in Patients Initiated on Sodium-Glucose Cotransporter-2 Inhibitors Versus Other Glucose-Lowering Drugs. Circulation 2017, 136, 249–259. [Google Scholar] [CrossRef]
- Kosiborod, M.; Lam, C.S.; Kohsaka, S.; Kim, D.J.; Karasik, A.; Shaw, J.; Tangri, N.; Goh, S.-Y.; Thuresson, M.; Chen, H.; et al. Cardiovascular Events Associated With SGLT-2 Inhibitors Versus Other Glucose-Lowering Drugs. J. Am. Coll. Cardiol. 2018, 71, 2628–2639. [Google Scholar] [CrossRef] [PubMed]
- Rosengren, A.; Edqvist, J.; Rawshani, A.; Sattar, N.; Franzén, S.; Adiels, M.; Svensson, A.-M.; Lind, M.; Gudbjörnsdottir, S. Excess risk of hospitalisation for heart failure among people with type 2 diabetes. Diabetologia 2018, 61, 2300–2309. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; McMurray, J.J.V.; Cherney, D.Z.I. The Metabolodiuretic Promise of Sodium-Dependent Glucose Cotransporter 2 Inhibition. JAMA Cardiol. 2017, 2, 939–940. [Google Scholar] [CrossRef] [PubMed]
- Hallow, K.M.; Helmlinger, G.; Greasley, P.J.; McMurray, J.J.V.; Boulton, D.W. Why do SGLT2 inhibitors reduce heart failure hospitalization? A differential volume regulation hypothesis. Diabetes Obes. Metab. 2017, 20, 479–487. [Google Scholar] [CrossRef]
- Chilton, R.; Tikkanen, I.; Cannon, C.P.; Crowe, S.; Woerle, H.J.; Broedl, U.C.; Johansen, O.E. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 1180–1193. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z.I. Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus. Circulation 2016, 134, 752–772. [Google Scholar] [CrossRef]
- Bautista, R.; Manning, R.; Martínez, F.; Avila-Casado, M.D.C.; Soto, V.; Medina, A.; Escalante, B. Angiotensin II-dependent increased expression of Na+-glucose cotransporter in hypertension. Am. J. Physiol. Physiol. 2004, 286, F127–F133. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Zinman, B.; Fitchett, D.; Wanner, C.; Ferrannini, E.; Schumacher, M.; Schmoor, C.; Ohneberg, K.; Johansen, O.E.; George, J.T.; et al. How Does Empagliflozin Reduce Cardiovascular Mortality? Insights from a Mediation Analysis of the EMPA-REG OUTCOME Trial. Diabetes Care 2018, 41, 356–363. [Google Scholar] [CrossRef]
- Von Lewinski, D.; Gasser, R.; Rainer, P.P.; Huber, M.-S.; Wilhelm, B.; Roessl, U.; Haas, T.; Wasler, A.; Grimm, M.; Bisping, E.; et al. Functional effects of glucose transporters in human ventricular myocardium. Eur. J. Heart Fail. 2010, 12, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Van Steenbergen, A.; Balteau, M.; Ginion, A.; Ferté, L.; Battault, S.; Ravenstein, C.D.M.D.; Balligand, J.-L.; Daskalopoulos, E.-P.; Gilon, P.; Despa, F.; et al. Sodium-myoinositol cotransporter-1, SMIT1, mediates the production of reactive oxygen species induced by hyperglycemia in the heart. Sci. Rep. 2017, 7, srep41166. [Google Scholar] [CrossRef]
- Berezin, A.E.; Kremzer, A.A.; Cammarota, G.; Zulli, A.; Petrovič, D.; Martell-Claros, N.; Sabo, J.; Kruzliak, P. Circulating endothelial-derived apoptotic microparticles and insulin resistance in non-diabetic patients with chronic heart failure. Clin. Chem. Lab. Med. 2015, 54, 1259–1267. [Google Scholar] [CrossRef]
- Kishimoto, S.; Kajikawa, M.; Maruhashi, T.; Iwamoto, Y.; Matsumoto, T.; Iwamoto, A.; Oda, N.; Matsui, S.; Hidaka, T.; Kihara, Y.; et al. Endothelial dysfunction and abnormal vascular structure are simultaneously present in patients with heart failure with preserved ejection fraction. Int. J. Cardiol. 2017, 231, 181–187. [Google Scholar] [CrossRef]
- Uthman, L.; Baartscheer, A.; Schumacher, C.A.; Fiolet, J.W.T.; Kuschma, M.C.; Hollmann, M.W.; Coronel, R.; Weber, N.C.; Zuurbier, C.J. Direct Cardiac Actions of Sodium Glucose Cotransporter 2 Inhibitors Target Pathogenic Mechanisms Underlying Heart Failure in Diabetic Patients. Front. Physiol. 2018, 9, 1575. [Google Scholar] [CrossRef] [PubMed]
- Baartscheer, A.; Schumacher, C.A.; Wust, R.C.; Fiolet, J.W.T.; Stienen, G.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017, 60, 568–573. [Google Scholar] [CrossRef]
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na+/H+ exchanger, lowering of cytosolic Na+ and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, S.; Zhu, P.; Hu, S.; Chen, Y.; Ren, J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018, 15, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Adingupu, D.D.; Göpel, S.O.; Grönros, J.; Behrendt, M.; Sotak, M.; Miliotis, T.; Dahlqvist, U.; Gan, L.-M.; Jönsson-Rylander, A.-C. SGLT2 inhibition with empagliflozin improves coronary microvascular function and cardiac contractility in prediabetic ob/ob−/− mice. Cardiovasc. Diabetol. 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Juni, R.P.; Al-Shama, R.; Kuster, D.W.; van der Velden, J.; Hamer, H.M.; Vervloet, M.G.; Eringa, E.C.; Koolwijk, P.; van Hinsbergh, V.W. Empagliflozin restores chronic kidney disease–induced impairment of endothelial regulation of cardiomyocyte relaxation and contraction. Kidney Int. 2021, 99, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschöpe, C. A Novel Paradigm for Heart Failure With Preserved Ejection Fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Lam, C.S.P.; Svedlund, S.; Saraste, A.; Hage, C.; Tan, R.S.; Beussink-Nelson, M.L.; Faxén, U.L.; Fermer, M.L.; Broberg, M.A.; et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur. Heart J. 2018, 39, 3439–3450. [Google Scholar] [CrossRef] [PubMed]
- Taqueti, V.R.; Solomon, S.D.; Shah, A.M.; Desai, A.S.; Groarke, J.D.; Osborne, M.; Hainer, J.; Bibbo, C.F.; Dorbala, S.; Blankstein, R.; et al. Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur. Heart J. 2017, 39, 840–849. [Google Scholar] [CrossRef]
- Sakai, T.; Miura, S. Effects of Sodium-Glucose Cotransporter 2 Inhibitor on Vascular Endothelial and Diastolic Function in Heart Failure With Preserved Ejection Fraction—Novel Prospective Cohort Study. Circ. Rep. 2019, 1, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nat. Cell Biol. 2019, 568, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Cappetta, D.; De Angelis, A.; Ciuffreda, L.P.; Coppini, R.; Cozzolino, A.; Miccichè, A.; Dell’Aversana, C.; D’Amario, D.; Cianflone, E.; Scavone, C.; et al. Amelioration of diastolic dysfunction by dapagliflozin in a non-diabetic model involves coronary endothelium. Pharmacol. Res. 2020, 157, 104781. [Google Scholar] [CrossRef] [PubMed]
- Tochiya, M.; Makino, H.; Tamanaha, T.; Matsuo, M.; Hishida, A.; Koezuka, R.; Ohata, Y.; Tomita, T.; Son, C.; Miyamoto, Y.; et al. Effect of tofogliflozin on cardiac and vascular endothelial function in patients with type 2 diabetes and heart diseases: A pilot study. J. Diabetes Investig. 2019, 11, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, M.; Franchi, C.; Nobili, A.; Mannucci, P.M.; Ardoino, I.; REPOSI Investigators. Defining Aging Phenotypes and Related Outcomes: Clues to Recognize Frailty in Hospitalized Older Patients. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2017, 72, 395–402. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvatore, T.; Caturano, A.; Galiero, R.; Di Martino, A.; Albanese, G.; Vetrano, E.; Sardu, C.; Marfella, R.; Rinaldi, L.; Sasso, F.C. Cardiovascular Benefits from Gliflozins: Effects on Endothelial Function. Biomedicines 2021, 9, 1356. https://doi.org/10.3390/biomedicines9101356
Salvatore T, Caturano A, Galiero R, Di Martino A, Albanese G, Vetrano E, Sardu C, Marfella R, Rinaldi L, Sasso FC. Cardiovascular Benefits from Gliflozins: Effects on Endothelial Function. Biomedicines. 2021; 9(10):1356. https://doi.org/10.3390/biomedicines9101356
Chicago/Turabian StyleSalvatore, Teresa, Alfredo Caturano, Raffaele Galiero, Anna Di Martino, Gaetana Albanese, Erica Vetrano, Celestino Sardu, Raffaele Marfella, Luca Rinaldi, and Ferdinando Carlo Sasso. 2021. "Cardiovascular Benefits from Gliflozins: Effects on Endothelial Function" Biomedicines 9, no. 10: 1356. https://doi.org/10.3390/biomedicines9101356
APA StyleSalvatore, T., Caturano, A., Galiero, R., Di Martino, A., Albanese, G., Vetrano, E., Sardu, C., Marfella, R., Rinaldi, L., & Sasso, F. C. (2021). Cardiovascular Benefits from Gliflozins: Effects on Endothelial Function. Biomedicines, 9(10), 1356. https://doi.org/10.3390/biomedicines9101356