Considering the Experimental Use of Temozolomide in Glioblastoma Research

 ,
, 
 and
and
Abstract
1. Introduction
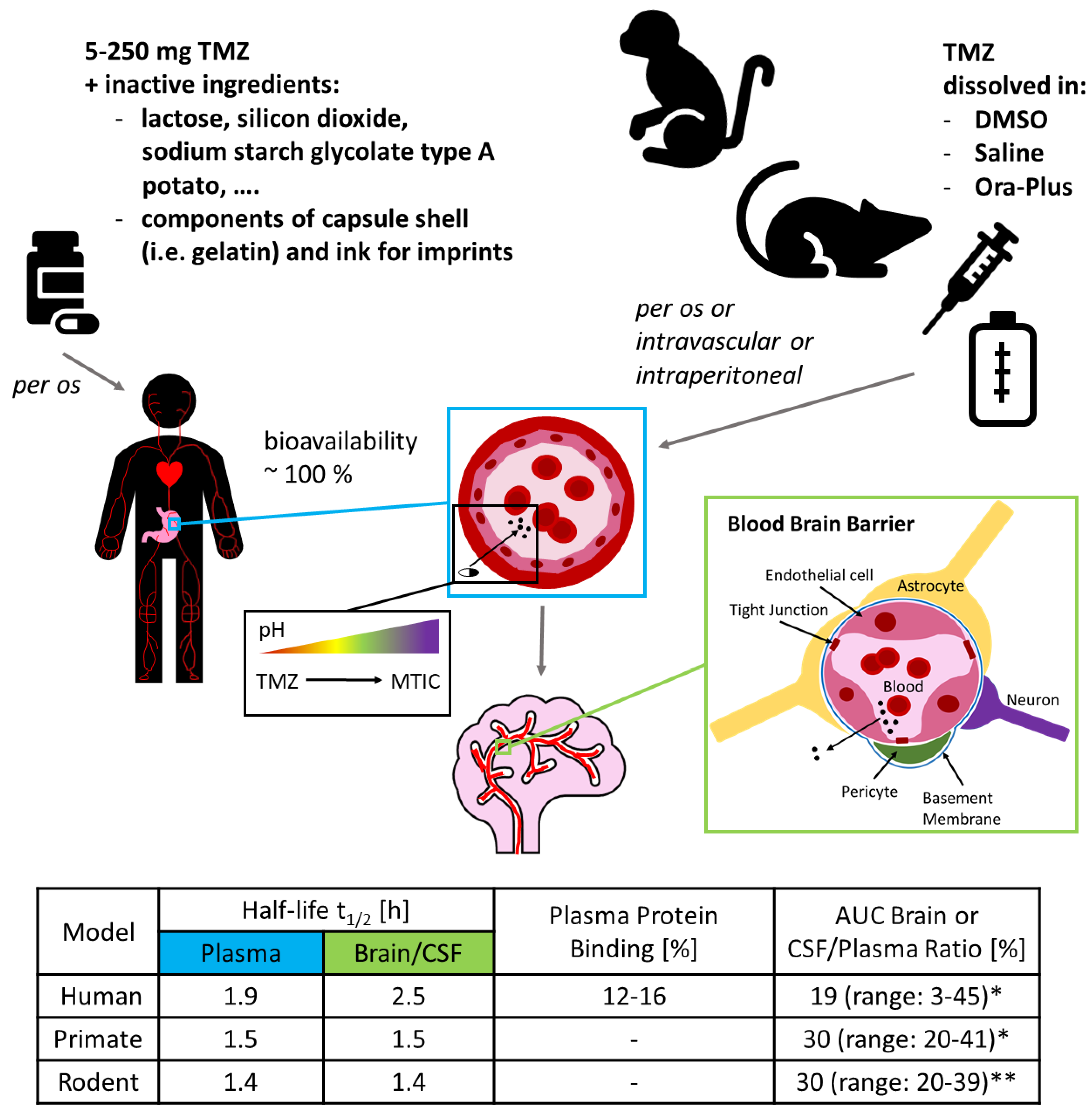
2. Experimental Limitations 1: Finding Your Rhythm
“Alle Dinge sind Gift, und nichts ist ohne Gift; allein die Dosis machts, daß ein Ding kein Gift sei.”Theophrastus Bombast von Hohenheim
3. Experimental Limitations 2: There is no Alkahest
“There is not more neutrality in the world. You either have to be part of the solution or you’re going to be part of the problem.”Eldridge Cleaver
4. Experimental Limitations 3: GB’s Next Top Model
“… der denkende treue Beobachter lernt immer mehr seine Beschränkung kennen, er sieht: je weiter sich das Wissen ausbreitet, desto mehr Probleme kommen zum Vorschein.”Johann Wolfgang von Goethe
4.1. ‘Classic’ GB Cell Lines
4.2. Primary Cultured, Patient-Derived GB Stem Cell-Like Cells and Their Differentiated Progeny
4.3. Genetically Identical Cells Responding Differently to TMZ
4.4. Co-Culture Systems Mimicking the TME
5. Experimental Limitations 4: Of Mice and Men
“The best-laid schemes o’ mice an’ men // Gang aft agley”Robert Burns
6. Experimental Limitations in the Context of TMZ’s Effect on the Immune System
7. Conclusions and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Robertson, F.L.; Marqués-Torrejón, M.-A.; Morrison, G.M.; Pollard, S.M. Experimental models and tools to tackle glioblastoma. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncology 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Dolecek, T.A.; Propp, J.M.; Stroup, N.E.; Kruchko, C. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2005–2009. Neuro-Oncology 2012, 14 (Suppl. 5), v1–v49. [Google Scholar] [CrossRef] [PubMed]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. 2014, 9, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Bjerkvig, R.; Berens, M.E.; Westphal, M. Cost of migration: Invasion of malignant gliomas and implications for treatment. J. Clin. Oncol. 2003, 21, 1624–1636. [Google Scholar] [CrossRef]
- Louis, D.N. Molecular pathology of malignant gliomas. Annu. Rev. Pathol. 2006, 1, 97–117. [Google Scholar] [CrossRef]
- Rong, Y.; Durden, D.L.; van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’ necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef]
- Persano, L.; Rampazzo, E.; Della Puppa, A.; Pistollato, F.; Basso, G. The three-layer concentric model of glioblastoma: Cancer stem cells, microenvironmental regulation, and therapeutic implications. Science 2011, 11, 1829–1841. [Google Scholar] [CrossRef] [PubMed]
- Beauchesne, P. Letter to the editor: The natural history of extra-cranial metastasis from glioblastoma multiform. J. Neuro. Oncol. 2012, 109, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Da Ros, M.; de Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; de Martino, M.; Genitori, L.; Sardi, I. Glioblastoma Chemoresistance: The Double Play by Microenvironment and Blood-Brain Barrier. Int. J. Mol. Sci. 2018, 19, 2879. [Google Scholar] [CrossRef]
- Strobel, H.; Baisch, T.; Fitzel, R.; Schilberg, K.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.-M.; Westhoff, M.-A. Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 69. [Google Scholar] [CrossRef]
- Kumari, S.; Ahsan, S.M.; Kumar, J.M.; Kondapi, A.K.; Rao, N.M. Overcoming blood brain barrier with a dual purpose Temozolomide loaded Lactoferrin nanoparticles for combating glioma (SERP-17-12433). Sci. Rep. 2017, 7, 6602. [Google Scholar] [CrossRef]
- Fang, C.; Wang, K.; Stephen, Z.R.; Mu, Q.; Kievit, F.M.; Chiu, D.T.; Press, O.W.; Zhang, M. Temozolomide nanoparticles for targeted glioblastoma therapy. ACS Appl. Mater. Interfaces 2015, 7, 6674–6682. [Google Scholar] [CrossRef]
- Annovazzi, L.; Mellai, M.; Schiffer, D. Chemotherapeutic Drugs: DNA Damage and Repair in Glioblastoma. Cancers (Basel) 2017, 9, 57. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Chekhonin, V.P. On the Critical Issues in Temozolomide Research in Glioblastoma: Clinically Relevant Concentrations and MGMT-independent Resistance. Biomedicines 2019, 7, 92. [Google Scholar] [CrossRef]
- Nonnenmacher, L.; Westhoff, M.-A.; Fulda, S.; Karpel-Massler, G.; Halatsch, M.-E.; Engelke, J.; Simmet, T.; Corbacioglu, S.; Debatin, K.-M. RIST: A potent new combination therapy for glioblastoma. Int. J. Cancer 2015, 136, E173–E187. [Google Scholar] [CrossRef]
- Kast, R.E.; Boockvar, J.A.; Brüning, A.; Cappello, F.; Chang, W.-W.; Cvek, B.; Dou, Q.P.; Duenas-Gonzalez, A.; Efferth, T.; Focosi, D.; et al. A conceptually new treatment approach for relapsed glioblastoma: Coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget 2013, 4, 502–530. [Google Scholar] [CrossRef] [PubMed]
- Langhans, J.; Schneele, L.; Trenkler, N.; von Bandemer, H.; Nonnenmacher, L.; Karpel-Massler, G.; Siegelin, M.D.; Zhou, S.; Halatsch, M.-E.; Debatin, K.-M.; et al. The effects of PI3K-mediated signalling on glioblastoma cell behaviour. Oncogenesis 2017, 6, 398. [Google Scholar] [CrossRef] [PubMed]
- Liston, D.R.; Davis, M. Clinically Relevant Concentrations of Anticancer Drugs: A Guide for Nonclinical Studies. Clin. Cancer Res. 2017, 23, 3489–3498. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, M.-A.; Baisch, T.; Herbener, V.J.; Karpel-Massler, G.; Debatin, K.-M.; Strobel, H. Comment in Response to “Temozolomide in Glioblastoma Therapy: Role of Apoptosis, Senescence and Autophagy etc. by B. Kaina”. Biomedicines 2020, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Houghton, P. A proposal regarding reporting of in vitro testing results. Clin. Cancer Res. 2013, 19, 2828–2833. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Temodal: Temozolomide. EPAR summary for the public. Available online: https://www.ema.europa.eu/en/documents/overview/temodal-epar-summary-public_en.pdf (accessed on 2 April 2020).
- Stevens, M.F.G.; Newlands, E.S. From triazines and triazenes to temozolomide. Eur. J. Cancer 1993, 29, 1045–1047. [Google Scholar] [CrossRef]
- Tsang, L.L.H.; Farmer, P.B.; Gescher, A.; Slack, J.A. Characterisation of urinary metabolites of temozolomide in humans and mice and evaluation of their cytotoxicity. Cancer Chemother. Pharm. 1990, 26, 429–436. [Google Scholar] [CrossRef]
- Newlands, E.S.; Blackledge, G.R.; Slack, J.A.; Rustin, G.J.; Smith, D.B.; Stuart, N.S.; Quarterman, C.P.; Hoffman, R.; Stevens, M.F.; Brampton, M.H. Phase I trial of temozolomide (CCRG 81045: M&B 39831: NSC 362856). Br. J. Cancer 1992, 65, 287–291. [Google Scholar] [CrossRef]
- Brock, C.S.; Matthews, J.C.; Brown, G.; Newlands, E.S. Molecular pathology of cancer: In vivo demonstration of 11C-Temozolomide uptake by human recurrent high grade astrocytomas. Br. J. Cancer 1997, 75, 1226–1245. [Google Scholar]
- Baker, S.D.; Wirth, M.; Statkevich, P.; Reidenberg, P.; Alton, K.; Sartorius, S.E.; Dugan, M.; Cutler, D.; Batra, V.; Grochow, L.B.; et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin. Cancer Res. 1999, 5, 309–317. [Google Scholar]
- Panetta, J.C.; Kirstein, M.N.; Gajjar, A.; Nair, G.; Fouladi, M.; Heideman, R.L.; Wilkinson, M.; Stewart, C.F. Population pharmacokinetics of temozolomide and metabolites in infants and children with primary central nervous system tumors. Cancer Chemother. Pharm. 2003, 52, 435–441. [Google Scholar] [CrossRef]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef]
- Jackson, S.; Weingart, J.; Nduom, E.K.; Harfi, T.T.; George, R.T.; McAreavey, D.; Ye, X.; Anders, N.M.; Peer, C.; Figg, W.D.; et al. The effect of an adenosine A2A agonist on intra-tumoral concentrations of temozolomide in patients with recurrent glioblastoma. Fluids Barriers CNS 2018, 15, 2. [Google Scholar] [CrossRef]
- Urso, R.; Blardi, P.; Giorgi, G. A short introduction to pharmacokinetics. Eur. Rev. Med. Pharm. Sci. 2002, 6, 33–44. [Google Scholar]
- Spilker, M.E.; Chen, X.; Visswanathan, R.; Vage, C.; Yamazaki, S.; Li, G.; Lucas, J.; Bradshaw-Pierce, E.L.; Vicini, P. Found in Translation: Maximizing the Clinical Relevance of Nonclinical Oncology Studies. Clin. Cancer Res. 2017, 23, 1080–1090. [Google Scholar] [CrossRef]
- Beier, D.; Röhrl, S.; Pillai, D.R.; Schwarz, S.; Kunz-Schughart, L.A.; Leukel, P.; Proescholdt, M.; Brawanski, A.; Bogdahn, U.; Trampe-Kieslich, A.; et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008, 68, 5706–5715. [Google Scholar] [CrossRef]
- Beier, D.; Schriefer, B.; Brawanski, K.; Hau, P.; Weis, J.; Schulz, J.B.; Beier, C.P. Efficacy of clinically relevant temozolomide dosing schemes in glioblastoma cancer stem cell lines. J. Neurooncol. 2012, 109, 45–52. [Google Scholar] [CrossRef]
- Patel, M.; McCully, C.; Godwin, K.; Balis, F.M. Plasma and cerebrospinal fluid pharmacokinetics of intravenous temozolomide in non-human primates. J. Neurooncol. 2003, 61, 203–207. [Google Scholar] [CrossRef]
- Baisiwala, S.; Auffinger, B.; Caragher, S.P.; Shireman, J.M.; Ahsan, R.; Lee, G.; Hasan, T.; Park, C.; Saathoff, M.R.; Christensen, A.C.; et al. Chemotherapeutic Stress Induces Transdifferentiation of Glioblastoma Cells to Endothelial Cells and Promotes Vascular Mimicry. Stem Cells Int. 2019, 2019, 6107456. [Google Scholar] [CrossRef]
- Towner, R.A.; Smith, N.; Saunders, D.; Brown, C.A.; Cai, X.; Ziegler, J.; Mallory, S.; Dozmorov, M.G.; Coutinho De Souza, P.; Wiley, G.; et al. OKN-007 Increases temozolomide (TMZ) Sensitivity and Suppresses TMZ-Resistant Glioblastoma (GBM) Tumor Growth. Transl. Oncol. 2019, 12, 320–335. [Google Scholar] [CrossRef]
- Da Ros, M.; Iorio, A.L.; de Gregorio, V.; Fantappiè, O.; Laffi, G.; de Martino, M.; Pisano, C.; Genitori, L.; Sardi, I. Aldoxorubicin and Temozolomide combination in a xenograft mice model of human glioblastoma. Oncotarget 2018, 9, 34935–34944. [Google Scholar] [CrossRef]
- Carlson, B.L.; Grogan, P.T.; Mladek, A.C.; Schroeder, M.A.; Kitange, G.J.; Decker, P.A.; Giannini, C.; Wu, W.; Ballman, K.A.; James, C.D.; et al. Radiosensitizing effects of temozolomide observed in vivo only in a subset of O6-methylguanine-DNA methyltransferase methylated glioblastoma multiforme xenografts. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 212–219. [Google Scholar] [CrossRef]
- Kitange, G.; Carlson, B.; Schroeder, M.; Grogan, P.; Lamont, J.; Decker, P.; Wu, W.; James, C.; Sarkaria, J. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro-oncology 2008, 11, 281–291. [Google Scholar] [CrossRef]
- Brock, C.S.; Newlands, E.S.; Wedge, S.R.; Bower, M.; Evans, H.; Colquhoun, I.; Roddie, M.; Glaser, M.; Brampton, M.H.; Rustin, G.J. Phase I trial of temozolomide using an extended continuous oral schedule. Cancer Res. 1998, 58, 4363–4367. [Google Scholar]
- Arora, P.; Adams, C.H.; Gudelsky, G.; DasGupta, B.; Desai, P.B. Plasma and brain pharmacokinetics of letrozole and drug interaction studies with temozolomide in NOD-scid gamma mice and sprague dawley rats. Cancer Chemother. Pharm. 2019, 83, 81–89. [Google Scholar] [CrossRef]
- Goldwirt, L.; Beccaria, K.; Carpentier, A.; Farinotti, R.; Fernandez, C. Irinotecan and temozolomide brain distribution: A focus on ABCB1. Cancer Chemother. Pharm. 2014, 74, 185–193. [Google Scholar] [CrossRef]
- Goldwirt, L.; Zahr, N.; Farinotti, R.; Fernandez, C. Development of a new UPLC-MSMS method for the determination of temozolomide in mice: Application to plasma pharmacokinetics and brain distribution study. Biomed. Chromatogr. 2013, 27, 889–893. [Google Scholar] [CrossRef]
- League-Pascual, J.C.; Lester-McCully, C.M.; Shandilya, S.; Ronner, L.; Rodgers, L.; Cruz, R.; Peer, C.J.; Figg, W.D.; Warren, K.E. Plasma and cerebrospinal fluid pharmacokinetics of select chemotherapeutic agents following intranasal delivery in a non-human primate model. J. Neurooncol. 2017, 132, 401–407. [Google Scholar] [CrossRef]
- Reyderman, L.; Statkevich, P.; Thonoor, C.M.; Patrick, J.; Batra, V.K.; Wirth, M. Disposition and pharmacokinetics of temozolomide in rat. Xenobiotica 2004, 34, 487–500. [Google Scholar] [CrossRef]
- Zhou, Q.; Guo, P.; Kruh, G.D.; Vicini, P.; Wang, X.; Gallo, J.M. Predicting human tumor drug concentrations from a preclinical pharmacokinetic model of temozolomide brain disposition. Clin. Cancer Res. 2007, 13, 4271–4279. [Google Scholar] [CrossRef]
- Rai, R.; Banerjee, M.; Wong, D.H.; McCullagh, E.; Gupta, A.; Tripathi, S.; Riquelme, E.; Jangir, R.; Yadav, S.; Raja, M.; et al. Temozolomide analogs with improved brain/plasma ratios - Exploring the possibility of enhancing the therapeutic index of temozolomide. Bioorg. Med. Chem. Lett. 2016, 26, 5103–5109. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.F.; Hickman, J.A.; Langdon, S.P.; Chubb, D.; Vickers, L.; Stone, R.; Baig, G.; Goddard, C.; Gibson, N.W.; Slack, J.A. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo5,1-d-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res. 1987, 47, 5846–5852. [Google Scholar] [PubMed]
- Brandes, A.A.; Tosoni, A.; Cavallo, G.; Bertorelle, R.; Gioia, V.; Franceschi, E.; Biscuola, M.; Blatt, V.; Crinò, L.; Ermani, M. Temozolomide 3 weeks on and 1 week off as first-line therapy for recurrent glioblastoma: Phase II study from gruppo italiano cooperativo di neuro-oncologia (GICNO). Br. J. Cancer 2006, 95, 1155–1160. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Gerson, S.L.; Denis, L.; Geyer, C.; Hammond, L.A.; Patnaik, A.; Goetz, A.D.; Schwartz, G.; Edwards, T.; Reyderman, L.; et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br. J. Cancer 2003, 88, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.; Bélanger, K.; Mason, W.P.; Fulton, D.; Kavan, P.; Easaw, J.; Shields, C.; Kirby, S.; Macdonald, D.R.; Eisenstat, D.D.; et al. Phase II trial of continuous dose-intense temozolomide in recurrent malignant glioma: RESCUE study. J. Clin. Oncol. 2010, 28, 2051–2057. [Google Scholar] [CrossRef]
- Wick, A.; Pascher, C.; Wick, W.; Jauch, T.; Weller, M.; Bogdahn, U.; Hau, P. Rechallenge with temozolomide in patients with recurrent gliomas. J. Neurol. 2009, 256, 734–741. [Google Scholar] [CrossRef]
- Wick, W.; Steinbach, J.P.; Küker, W.M.; Dichgans, J.; Bamberg, M.; Weller, M. One week on/one week off: A novel active regimen of temozolomide for recurrent glioblastoma. Neurology 2004, 62, 2113–2115. [Google Scholar] [CrossRef]
- Brobyn, R.D. The Human Toxicology of Dimethyl Sulfoxide. Ann. N. Y. Acad. Sci. 1975, 243, 497–506. [Google Scholar] [CrossRef]
- Santos, N.C.; Figueira-Coelho, J.; Martins-Silva, J.; Saldanha, C. Multidisciplinary utilization of dimethyl sulfoxide: Pharmacological, cellular, and molecular aspects. Biochem. Pharmacol. 2003, 65, 1035–1041. [Google Scholar] [CrossRef]
- Verheijen, M.; Lienhard, M.; Schrooders, Y.; Clayton, O.; Nudischer, R.; Boerno, S.; Timmermann, B.; Selevsek, N.; Schlapbach, R.; Gmuender, H.; et al. DMSO induces drastic changes in human cellular processes and epigenetic landscape in vitro. Sci. Rep. 2019, 9, 4641. [Google Scholar] [CrossRef]
- Galvao, J.; Davis, B.; Tilley, M.; Normando, E.; Duchen, M.R.; Cordeiro, M.F. Unexpected low-dose toxicity of the universal solvent DMSO. FASEB J. 2013, 28, 1317–1330. [Google Scholar] [CrossRef]
- Hanslick, J.L.; Lau, K.; Noguchi, K.K.; Olney, J.W.; Zorumski, C.F.; Mennerick, S.; Farber, N.B. Dimethyl sulfoxide (DMSO) produces widespread apoptosis in the developing central nervous system. Neurobiol. Dis. 2009, 34, 1–10. [Google Scholar] [CrossRef]
- Kaina, B. Temozolomide in Glioblastoma Therapy: Role of Apoptosis, Senescence and Autophagy. Comment on Strobel et al., Temozolomide and Other Alkylating Agents in Glioblastoma Therapy. Biomedicines 2019, 7, 90. [Google Scholar] [CrossRef]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.R.; Parada, L.F. Cell of origin of glioma: Biological and clinical implications. Br. J. Cancer 2016, 115, 1445–1450. [Google Scholar] [CrossRef]
- Zhang, C.; Deng, Y.; Dai, H.; Zhou, W.; Tian, J.; Bing, G.; Zhao, L. Effects of dimethyl sulfoxide on the morphology and viability of primary cultured neurons and astrocytes. Brain Res. Bull. 2017, 128, 34–39. [Google Scholar] [CrossRef]
- Yuan, C.; Gao, J.; Guo, J.; Bai, L.; Marshall, C.; Cai, Z.; Wang, L.; Xiao, M. Dimethyl Sulfoxide Damages Mitochondrial Integrity and Membrane Potential in Cultured Astrocytes. PLoS ONE 2014, 9, e107447. [Google Scholar] [CrossRef]
- Schneider, M.; Ströbele, S.; Nonnenmacher, L.; Siegelin, M.D.; Tepper, M.; Stroh, S.; Hasslacher, S.; Enzenmüller, S.; Strauss, G.; Baumann, B.; et al. A paired comparison between glioblastoma “stem cells” and differentiated cells. Int. J. Cancer 2016, 138, 1709–1718. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef]
- de Witt Hamer, P.C.; van Tilborg, A.A.G.; Eijk, P.P.; Sminia, P.; Troost, D.; van Noorden, C.J.F.; Ylstra, B.; Leenstra, S. The genomic profile of human malignant glioma is altered early in primary cell culture and preserved in spheroids. Oncogene 2008, 27, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother 2015, 15, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Ströbele, S.; Schneider, M.; Schneele, L.; Siegelin, M.D.; Nonnenmacher, L.; Zhou, S.; Karpel-Massle, G.; Westhoff, M.-A.; Halatsch, M.-E.; Debatin, K.-M. A Potential Role for the Inhibition of PI3K Signaling in Glioblastoma Therapy. PLoS ONE 2015, 10, e0131670. [Google Scholar] [CrossRef]
- Jamalzadeh, L.; Ghafoori, H.; Sariri, R.; Rabuti, H.; Nasirzade, J.; Hasani, H.; Aghamaali, M.R. Cytotoxic Effects of Some Common Organic Solvents on MCF-7, RAW-264.7 and Human Umbilical Vein Endothelial Cells. Avicenna J. Med. Biochem. 2016, 4, 10–33453. [Google Scholar] [CrossRef]
- Da Violante, G.; Zerrouk, N.; Richard, I.; Provot, G.; Chaumeil, J.C.; Arnaud, P. Evaluation of the Cytotoxicity Effect of Dimethyl Sulfoxide (DMSO) on Caco2/TC7 Colon Tumor Cell Cultures. Biol. Pharm. Bull. 2002, 25, 1600–1603. [Google Scholar] [CrossRef]
- Singh, M. Effect of dimethyl sulfoxide on in vitro proliferation of skin fibroblast cells. J. Biol. Res. 2017, 8, 78–82. [Google Scholar]
- Hajighasemi, F.; Tajic, S. Assessment of Cytotoxicity of Dimethyl Sulfoxide in Human Hematopoietic Tumor Cell Lines. IJBC 2017, 9, 48–53. [Google Scholar]
- de Abreu Costa, L.; Henrique Fernandes Ottoni, M.; Dos Santos, M.G.; Meireles, A.B.; de Gomes Almeida, V.; de Fátima Pereira, W.; de Alves Avelar-Freitas, B.; Eustáquio Alvim Brito-Melo, G. Dimethyl Sulfoxide (DMSO) Decreases Cell Proliferation and TNF-α, IFN-γ, and IL-2 Cytokines Production in Cultures of Peripheral Blood Lymphocytes. Molecules 2017, 22, 1789. [Google Scholar] [CrossRef]
- Pal, R.; Mamidi, M.K.; Das, A.K.; Bhonde, R. Diverse effects of dimethyl sulfoxide (DMSO) on the differentiation potential of human embryonic stem cells. Arch. Toxicol. 2012, 86, 651–661. [Google Scholar] [CrossRef]
- Cao, X.-G.; Li, X.-X.; Bao, Y.-Z.; Xing, N.-Z.; Chen, Y. Responses of Human Lens Epithelial Cells to Quercetin and DMSO. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3714–3718. [Google Scholar] [CrossRef]
- Yi, X.; Liu, M.; Luo, Q.; Zhuo, H.; Cao, H.; Wang, J.; Han, Y. Toxic effects of dimethyl sulfoxide on red blood cells, platelets, and vascular endothelial cells in vitro. FEBS Open Bio 2017, 7, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Broadwell, R.D.; Salcman, M.; Kaplan, R.S. Morphologic effect of dimethyl sulfoxide on the blood-brain barrier. Science 1982, 217, 164. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kubota, A.; Matsuo, A.; Kawakami, A.; Kamizi, H.; Mochigoe, A.; Hiramachi, T.; Kasaoka, S.; Yoshikawa, H.; Nagata, S. Effect of Absorption Behavior of Solubilizers on Drug Dissolution in the Gastrointestinal Tract: Evaluation Based on In Vivo Luminal Concentration–Time Profile of Cilostazol, a Poorly Soluble Drug, and Solubilizers. J. Pharm. Sci. 2016, 105, 2825–2831. [Google Scholar] [CrossRef][Green Version]
- Huszthy, P.C.; Daphu, I.; Niclou, S.P.; Stieber, D.; Nigro, J.M.; Sakariassen, P.Ø.; Miletic, H.; Thorsen, F.; Bjerkvig, R. In vivo models of primary brain tumors: Pitfalls and perspectives. Neuro-Oncology 2012, 14, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Lenting, K.; Verhaak, R.; Ter Laan, M.; Wesseling, P.; Leenders, W. Glioma: Experimental models and reality. Acta Neuropathol. 2017, 133, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Vogel, T.W.; Zhuang, Z.; Li, J.; Okamoto, H.; Furuta, M.; Lee, Y.-S.; Zeng, W.; Oldfield, E.H.; Vortmeyer, A.O.; Weil, R.J. Proteins and protein pattern differences between glioma cell lines and glioblastoma multiforme. Clin. Cancer Res. 2005, 11, 3624–3632. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Walling, J.; Kotliarov, Y.; Center, A.; Steed, M.E.; Ahn, S.J.; Rosenblum, M.; Mikkelsen, T.; Zenklusen, J.C.; Fine, H.A. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol. Cancer Res. 2008, 6, 21–30. [Google Scholar] [CrossRef]
- Ledur, P.F.; Onzi, G.R.; Zong, H.; Lenz, G. Culture conditions defining glioblastoma cells behavior: What is the impact for novel discoveries? Oncotarget 2017, 8, 69185–69197. [Google Scholar] [CrossRef]
- Jacobs, V.L.; Valdes, P.A.; Hickey, W.F.; de Leo, J.A. Current review of in vivo GBM rodent models: Emphasis on the CNS-1 tumour model. ASN Neuro 2011, 3, e00063. [Google Scholar] [CrossRef] [PubMed]
- Radaelli, E.; Ceruti, R.; Patton, V.; Russo, M.; Degrassi, A.; Croci, V.; Caprera, F.; Stortini, G.; Scanziani, E.; Pesenti, E.; et al. Immunohistopathological and neuroimaging characterization of murine orthotopic xenograft models of glioblastoma multiforme recapitulating the most salient features of human disease. Histol. Histopathol. 2009, 24, 879–891. [Google Scholar] [CrossRef]
- Allen, M.; Bjerke, M.; Edlund, H.; Nelander, S.; Westermark, B. Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 2016, 8, 354re3. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; van Meir, E.G. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.; Camby, I.; Salmon, I.; van Ham, P.; Brotchi, J.; Pasteels, J.L. Relationship between DNA ploidy level and tumor sociology behavior in 12 nervous cell lines. Cytometry 1995, 20, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Diserens, A.C.; de Tribolet, N.; Martin-Achard, A.; Gaide, A.C.; Schnegg, J.F.; Carrel, S. Characterization of an established human malignant glioma cell line: LN-18. Acta Neuropathol. 1981, 53, 21–28. [Google Scholar] [CrossRef]
- Berte, N.; Piée-Staffa, A.; Piecha, N.; Wang, M.; Borgmann, K.; Kaina, B.; Nikolova, T. Targeting Homologous Recombination by Pharmacological Inhibitors Enhances the Killing Response of Glioblastoma Cells Treated with Alkylating Drugs. Mol. Cancer. 2016, 15, 2665–2678. [Google Scholar] [CrossRef]
- Hermisson, M.; Klumpp, A.; Wick, W.; Wischhusen, J.; Nagel, G.; Roos, W.; Kaina, B.; Weller, M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. J. Neurochem. 2006, 96, 766–776. [Google Scholar] [CrossRef]
- Chahal, M.; Abdulkarim, B.; Xu, Y.; Guiot, M.-C.; Easaw, J.C.; Stifani, N.; Sabri, S. O6-Methylguanine-DNA methyltransferase is a novel negative effector of invasion in glioblastoma multiforme. Mol. Cancer. 2012, 11, 2440–2450. [Google Scholar] [CrossRef]
- Happold, C.; Roth, P.; Wick, W.; Schmidt, N.; Florea, A.-M.; Silginer, M.; Reifenberger, G.; Weller, M. Distinct molecular mechanisms of acquired resistance to temozolomide in glioblastoma cells. J. Neurochem. 2012, 122, 444–455. [Google Scholar] [CrossRef]
- Fukai, J.; Koizumi, F.; Nakao, N. Enhanced anti-tumor effect of zoledronic acid combined with temozolomide against human malignant glioma cell expressing O6-methylguanine DNA methyltransferase. PLoS ONE 2014, 9, e104538. [Google Scholar] [CrossRef]
- Gaspar, N.; Marshall, L.; Perryman, L.; Bax, D.A.; Little, S.E.; Viana-Pereira, M.; Sharp, S.Y.; Vassal, G.; Pearson, A.D.J.; Reis, R.M.; et al. MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res. 2010, 70, 9243–9252. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Mattei, C.; Vitale, F.; Marampon, F.; Colapietro, A.; Rossi, G.; Ventura, L.; Vetuschi, A.; Di Cesare, E.; et al. Enhancement of radiosensitivity by the novel anticancer quinolone derivative vosaroxin in preclinical glioblastoma models. Oncotarget 2017, 8, 29865–29886. [Google Scholar] [CrossRef] [PubMed]
- Nie, E.; Jin, X.; Wu, W.; Yu, T.; Zhou, X.; Zhi, T.; Shi, Z.; Zhang, J.; Liu, N.; You, Y. BACH1 Promotes Temozolomide Resistance in Glioblastoma through Antagonizing the Function of p53. Sci. Rep. 2016, 6, 39743. [Google Scholar] [CrossRef] [PubMed]
- Aasland, D.; Reich, T.R.; Tomicic, M.T.; Switzeny, O.J.; Kaina, B.; Christmann, M. Repair gene O6 -methylguanine-DNA methyltransferase is controlled by SP1 and up-regulated by glucocorticoids, but not by temozolomide and radiation. J. Neurochem. 2018, 144, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, S.; Wang, L.; Yachi, K.; Mahabir, R.; Narita, T.; Itoh, T.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol. Cancer. 2012, 11, 1289–1299. [Google Scholar] [CrossRef]
- Wang, H.-H.; Chang, T.-Y.; Lin, W.-C.; Wei, K.-C.; Shin, J.-W. GADD45A plays a protective role against temozolomide treatment in glioblastoma cells. Sci. Rep. 2017, 7, 8814. [Google Scholar] [CrossRef]
- Atif, F.; Patel, N.R.; Yousuf, S.; Stein, D.G. The Synergistic Effect of Combination Progesterone and Temozolomide on Human Glioblastoma Cells. PLoS ONE 2015, 10, e0131441. [Google Scholar] [CrossRef] [PubMed]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.-S.; Zhong, W.-Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010, 70, 3228–3238. [Google Scholar] [CrossRef]
- Li, Y.; Dowbenko, D.; Lasky, L.A. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 2002, 277, 11352–11361. [Google Scholar] [CrossRef]
- Anderson, C.W.; Joan Allalunis-Turner, M. Human TP53 from the Malignant Glioma-Derived Cell Lines M059J and M059K Has a Cancer-Associated Mutation in Exon 8. Radiat. Res. 2000, 154, 473–476. [Google Scholar] [CrossRef]
- Agnihotri, S.; Gajadhar, A.S.; Ternamian, C.; Gorlia, T.; Diefes, K.L.; Mischel, P.S.; Kelly, J.; McGown, G.; Thorncroft, M.; Carlson, B.L.; et al. Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients. J. Clin. Investig. 2012, 122, 253–266. [Google Scholar] [CrossRef]
- Belot, N.; Rorive, S.; Doyen, I.; Lefranc, F.; Bruyneel, E.; Dedecker, R.; Micik, S.; Brotchi, J.; Decaestecker, C.; Salmon, I.; et al. Molecular characterization of cell substratum attachments in human glial tumors relates to prognostic features. Glia 2001, 36, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Hlavaty, J.; Jandl, G.; Liszt, M.; Petznek, H.; König-Schuster, M.; Sedlak, J.; Egerbacher, M.; Weissenberger, J.; Salmons, B.; Günzburg, W.H.; et al. Comparative evaluation of preclinical in vivo models for the assessment of replicating retroviral vectors for the treatment of glioblastoma. J. Neurooncol. 2011, 102, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Huang, G.; Ho, B.; Yemma, M.; Morrison, C.D.; Lee, J.; Eliceiri, B.P.; Cance, W.G. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol. Cancer. 2013, 12, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Lundy, D.J.; Lee, K.-J.; Peng, I.-C.; Hsu, C.-H.; Lin, J.-H.; Chen, K.-H.; Tien, Y.-W.; Hsieh, P.C.H. Inducing a Transient Increase in Blood-Brain Barrier Permeability for Improved Liposomal Drug Therapy of Glioblastoma Multiforme. ACS Nano 2019, 13, 97–113. [Google Scholar] [CrossRef]
- George, J.; Banik, N.L.; Ray, S.K. Combination of hTERT knockdown and IFN-gamma treatment inhibited angiogenesis and tumor progression in glioblastoma. Clin. Cancer Res. 2009, 15, 7186–7195. [Google Scholar] [CrossRef]
- Burden-Gulley, S.M.; Qutaish, M.Q.; Sullivant, K.E.; Lu, H.; Wang, J.; Craig, S.E.L.; Basilion, J.P.; Wilson, D.L.; Brady-Kalnay, S.M. Novel cryo-imaging of the glioma tumor microenvironment reveals migration and dispersal pathways in vivid three-dimensional detail. Cancer Res. 2011, 71, 5932–5940. [Google Scholar] [CrossRef]
- Papachristodoulou, A.; Silginer, M.; Weller, M.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P. Therapeutic Targeting of TGFβ Ligands in Glioblastoma Using Novel Antisense Oligonucleotides Reduces the Growth of Experimental Gliomas. Clin. Cancer Res. 2019, 25, 7189–7201. [Google Scholar] [CrossRef]
- Szabo, E.; Schneider, H.; Seystahl, K.; Rushing, E.J.; Herting, F.; Weidner, K.M.; Weller, M. Autocrine VEGFR1 and VEGFR2 signaling promotes survival in human glioblastoma models in vitro and in vivo. Neuro-Oncology 2016, 18, 1242–1252. [Google Scholar] [CrossRef]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.-P.; Glackin, C.A.; Wakimoto, H.; González-Herrero, I.; et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 2010, 9, 194. [Google Scholar] [CrossRef]
- Kim, S.-S.; Rait, A.; Kim, E.; Pirollo, K.F.; Nishida, M.; Farkas, N.; Dagata, J.A.; Chang, E.H. A nanoparticle carrying the p53 gene targets tumors including cancer stem cells, sensitizes glioblastoma to chemotherapy and improves survival. ACS Nano 2014, 8, 5494–5514. [Google Scholar] [CrossRef]
- Camphausen, K.; Purow, B.; Sproull, M.; Scott, T.; Ozawa, T.; Deen, D.F.; Tofilon, P.J. Influence of in vivo growth on human glioma cell line gene expression: Convergent profiles under orthotopic conditions. Proc. Natl. Acad. Sci. USA 2005, 102, 8287–8292. [Google Scholar] [CrossRef] [PubMed]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462. [Google Scholar] [CrossRef] [PubMed]
- Busek, P.; Sedo, A. Dipeptidyl Peptidase-IV and Related Proteases in Brain Tumors. In Evolution of the Molecular Biology of Brain Tumors and the Therapeutic Implications; Lichtor, T., Ed.; IntechOpen: London, UK, 2013; pp. 235–269. [Google Scholar] [CrossRef][Green Version]
- Plowman, J.; Waud, W.R.; Koutsoukos, A.D.; Rubinstein, L.V.; Moore, T.D.; Grever, M.R. Preclinical antitumor activity of temozolomide in mice: Efficacy against human brain tumor xenografts and synergism with 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 1994, 54, 3793–3799. [Google Scholar] [PubMed]
- Mathieu, V.; de Nève, N.; Le Mercier, M.; Dewelle, J.; Gaussin, J.-F.; Dehoux, M.; Kiss, R.; Lefranc, F. Combining bevacizumab with temozolomide increases the antitumor efficacy of temozolomide in a human glioblastoma orthotopic xenograft model. Neoplasia 2008, 10, 1383–1392. [Google Scholar] [CrossRef]
- Wakimoto, H.; Mohapatra, G.; Kanai, R.; Curry, W.T.; Yip, S.; Nitta, M.; Patel, A.P.; Barnard, Z.R.; Stemmer-Rachamimov, A.O.; Louis, D.N.; et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro-Oncology 2012, 14, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Teng, J.; da Hora, C.C.; Kantar, R.S.; Nakano, I.; Wakimoto, H.; Batchelor, T.T.; Chiocca, E.A.; Badr, C.E.; Tannous, B.A. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro-Oncology 2017, 19, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Kesari, S.; Farrell, C.J.; Curry, W.T.; Zaupa, C.; Aghi, M.; Kuroda, T.; Stemmer-Rachamimov, A.; Shah, K.; Liu, T.-C.; et al. Human glioblastoma-derived cancer stem cells: Establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009, 69, 3472–3481. [Google Scholar] [CrossRef]
- Joseph, J.V.; van Roosmalen, I.A.M.; Busschers, E.; Tomar, T.; Conroy, S.; Eggens-Meijer, E.; Peñaranda Fajardo, N.; Pore, M.M.; Balasubramanyian, V.; Wagemakers, M.; et al. Serum-Induced Differentiation of Glioblastoma Neurospheres Leads to Enhanced Migration/Invasion Capacity That Is Associated with Increased MMP9. PLoS ONE 2015, 10, e0145393. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; de Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- D’Alessandris, Q.G.; Biffoni, M.; Martini, M.; Runci, D.; Buccarelli, M.; Cenci, T.; Signore, M.; Stancato, L.; Olivi, A.; de Maria, R.; et al. The clinical value of patient-derived glioblastoma tumorspheres in predicting treatment response. Neuro-Oncology 2017, 19, 1097–1108. [Google Scholar] [CrossRef]
- Laks, D.R.; Masterman-Smith, M.; Visnyei, K.; Angenieux, B.; Orozco, N.M.; Foran, I.; Yong, W.H.; Vinters, H.V.; Liau, L.M.; Lazareff, J.A.; et al. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells 2009, 27, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Pallini, R.; Ricci-Vitiani, L.; Banna, G.L.; Signore, M.; Lombardi, D.; Todaro, M.; Stassi, G.; Martini, M.; Maira, G.; Larocca, L.M.; et al. Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 8205–8212. [Google Scholar] [CrossRef]
- Xie, Y.; Bergström, T.; Jiang, Y.; Johansson, P.; Marinescu, V.D.; Lindberg, N.; Segerman, A.; Wicher, G.; Niklasson, M.; Baskaran, S.; et al. The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtypes. EBioMedicine 2015, 2, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Panosyan, E.H.; Laks, D.R.; Masterman-Smith, M.; Mottahedeh, J.; Yong, W.H.; Cloughesy, T.F.; Lazareff, J.A.; Mischel, P.S.; Moore, T.B.; Kornblum, H.I. Clinical outcome in pediatric glial and embryonal brain tumors correlates with in vitro multi-passageable neurosphere formation. Pediatr. Blood Cancer 2010, 55, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Joo, K.M.; Kim, J.; Jin, J.; Kim, M.; Seol, H.J.; Muradov, J.; Yang, H.; Choi, Y.-L.; Park, W.-Y.; Kong, D.-S.; et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep. 2013, 3, 260–273. [Google Scholar] [CrossRef]
- Ernst, A.; Hofmann, S.; Ahmadi, R.; Becker, N.; Korshunov, A.; Engel, F.; Hartmann, C.; Felsberg, J.; Sabel, M.; Peterziel, H.; et al. Genomic and expression profiling of glioblastoma stem cell-like spheroid cultures identifies novel tumor-relevant genes associated with survival. Clin. Cancer Res. 2009, 15, 6541–6550. [Google Scholar] [CrossRef]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells--much more complex than expected. Mol. Cancer 2011, 10, 128. [Google Scholar] [CrossRef]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; de Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Ghods, A.J.; Irvin, D.; Liu, G.; Yuan, X.; Abdulkadir, I.R.; Tunici, P.; Konda, B.; Wachsmann-Hogiu, S.; Black, K.L.; Yu, J.S. Spheres isolated from 9L gliosarcoma rat cell line possess chemoresistant and aggressive cancer stem-like cells. Stem Cells 2007, 25, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef]
- Welker, A.M.; Jaros, B.D.; An, M.; Beattie, C.E. Changes in tumor cell heterogeneity after chemotherapy treatment in a xenograft model of glioblastoma. Neuroscience 2017, 356, 35–43. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Kang, M.-K.; Kang, S.-K. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. 2007, 16, 837–847. [Google Scholar] [CrossRef]
- de Vleeschouwer, S.; Bergers, G. Glioblastoma: To Target the Tumor Cell or the Microenvironment. In Glioblastoma; de Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef]
- Goers, L.; Freemont, P.; Polizzi, K.M. Co-culture systems and technologies: Taking synthetic biology to the next level. J. R. Soc. Interface 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Lovett, M.L.; Nieland, T.J.F.; Dingle, Y.-T.L.; Kaplan, D.L. Innovations in 3D Tissue Models of Human Brain Physiology and Diseases. Adv. Funct. Mater. 2020, 1909146. [Google Scholar] [CrossRef]
- Heinrich, M.A.; Bansal, R.; Lammers, T.; Zhang, Y.S.; Michel Schiffelers, R.; Prakash, J. 3D-Bioprinted Mini-Brain: A Glioblastoma Model to Study Cellular Interactions and Therapeutics. Adv. Mater. Weinh. 2019, 31, e1806590. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Liu, Z.; Ling, F.; Xu, G. Astrocytes protect glioma cells from chemotherapy and upregulate survival genes via gap junctional communication. Mol. Med. Rep. 2016, 13, 1329–1335. [Google Scholar] [CrossRef]
- Yang, N.; Yan, T.; Zhu, H.; Liang, X.; Leiss, L.; Sakariassen, P.Ø.; Skaftnesmo, K.O.; Huang, B.; Costea, D.E.; Enger, P.Ø.; et al. A co-culture model with brain tumor-specific bioluminescence demonstrates astrocyte-induced drug resistance in glioblastoma. J. Transl. Med. 2014, 12, 278. [Google Scholar] [CrossRef]
- Chen, W.; Wang, D.; Du, X.; He, Y.; Chen, S.; Shao, Q.; Ma, C.; Huang, B.; Chen, A.; Zhao, P.; et al. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med. Oncol. 2015, 32, 43. [Google Scholar] [CrossRef] [PubMed]
- Munoz, J.L.; Rodriguez-Cruz, V.; Greco, S.J.; Ramkissoon, S.H.; Ligon, K.L.; Rameshwar, P. Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis. 2014, 5, e1145. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.Q.; Toh, X.Y.; Ho, I.A.W.; Sia, K.C.; Newman, J.P.; Yulyana, Y.; Ng, W.-H.; Lai, S.H.; Ho, M.M.F.; Dinesh, N.; et al. Combined treatment of Nimotuzumab and rapamycin is effective against temozolomide-resistant human gliomas regardless of the EGFR mutation status. BMC Cancer 2015, 15, 255. [Google Scholar] [CrossRef]
- Sato, Y.; Kurose, A.; Ogawa, A.; Ogasawara, K.; Traganos, F.; Darzynkiewicz, Z.; Sawai, T. Diversity of DNA damage response of astrocytes and glioblastoma cell lines with various p53 status to treatment with etoposide and temozolomide. Cancer Biol. 2009, 8, 452–457. [Google Scholar] [CrossRef]
- Vairano, M.; Graziani, G.; Tentori, L.; Tringali, G.; Navarra, P.; Dello Russo, C. Primary cultures of microglial cells for testing toxicity of anticancer drugs. Toxicol. Lett. 2004, 148, 91–94. [Google Scholar] [CrossRef]
- Virrey, J.J.; Dong, D.; Stiles, C.; Patterson, J.B.; Pen, L.; Ni, M.; Schönthal, A.H.; Chen, T.C.; Hofman, F.M.; Lee, A.S. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol. Cancer Res. 2008, 6, 1268–1275. [Google Scholar] [CrossRef]
- Leite, D.M.; Zvar Baskovic, B.; Civita, P.; Neto, C.; Gumbleton, M.; Pilkington, G.J. A human co-culture cell model incorporating microglia supports glioblastoma growth and migration, and confers resistance to cytotoxics. FASEB J. 2020, 34, 1710–1727. [Google Scholar] [CrossRef]
- Persano, L.; Rampazzo, E.; Basso, G.; Viola, G. Glioblastoma cancer stem cells: Role of the microenvironment and therapeutic targeting. Biochem. Pharmacol. 2013, 85, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Ngo, M.T.; Harley, B.A.C. Perivascular signals alter global gene expression profile of glioblastoma and response to temozolomide in a gelatin hydrogel. Biomaterials 2019, 198, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Nair, J.; Dutt, S. Chapter 2 Preclinical Orthotopic Murine Xenograft Models in Glioblastoma. Available online: https://avidscience.com/book/brain-tumor/ (accessed on 29 April 2020).
- Miyai, M.; Tomita, H.; Soeda, A.; Yano, H.; Iwama, T.; Hara, A. Current trends in mouse models of glioblastoma. J. Neurooncol. 2017, 135, 423–432. [Google Scholar] [CrossRef]
- Gargiulo, G. Next-Generation in vivo Modeling of Human Cancers. Front. Oncol. 2018, 8, 429. [Google Scholar] [CrossRef]
- O’Reilly, S.M.; Newlands, E.S.; Brampton, M.; Glaser, M.G.; Rice-Edwards, J.M.; Illingworth, R.D.; Richards, P.G.; Kennard, C.; Colquhoun, I.R.; Lewis, P.; et al. Temozolomide: A new oral cytotoxic chemotherapeutic agent with promising activity against primary brain tumours. Eur. J. Cancer 1993, 29, 940–942. [Google Scholar] [CrossRef]
- Newlands, E.S.; O’Reilly, S.M.; Glaser, M.G.; Bower, M.; Evans, H.; Brock, C.; Brampton, M.H.; Colquhoun, I.; Lewis, P.; Rice-Edwards, J.M.; et al. The charing cross hospital experience with temozolomide in patients with gliomas. Eur. J. Cancer 1996, 32, 2236–2241. [Google Scholar] [CrossRef]
- Hirst, T.C.; Vesterinen, H.M.; Sena, E.S.; Egan, K.J.; Macleod, M.R.; Whittle, I.R. Systematic review and meta-analysis of temozolomide in animal models of glioma: Was clinical efficacy predicted? Br. J. Cancer 2013, 108, 64–71. [Google Scholar] [CrossRef]
- Uhl, E.W.; Warner, N.J. Mouse Models as Predictors of Human Responses: Evolutionary Medicine. Curr. Pathobiol. Rep. 2015, 3, 219–223. [Google Scholar] [CrossRef]
- Chu, X.; Bleasby, K.; Evers, R. Species differences in drug transporters and implications for translating preclinical findings to humans. Expert Opin. Drug Metab. Toxicol. 2013, 9, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Aday, S.; Cecchelli, R.; Hallier-Vanuxeem, D.; Dehouck, M.P.; Ferreira, L. Stem Cell-Based Human Blood-Brain Barrier Models for Drug Discovery and Delivery. Trends Biotechnol. 2016, 34, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Wu, J.; Xie, Y.; Kim, S.; Michelhaugh, S.; Jiang, J.; Mittal, S.; Sanai, N.; Li, J. Protein Expression and Functional Relevance of Efflux and Uptake Drug Transporters at the Blood-Brain Barrier of Human Brain and Glioblastoma. Clin. Pharm. 2020, 107, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Friedman, H.S.; Dolan, M.E.; Pegg, A.E.; Marcelli, S.; Keir, S.; Catino, J.J.; Bigner, D.D.; Schold, S.C. Activity of temozolomide in the treatment of central nervous system tumor xenografts. Cancer Res. 1995, 55, 2853–2857. [Google Scholar] [PubMed]
- Le Calvé, B.; Rynkowski, M.; Le Mercier, M.; Bruyère, C.; Lonez, C.; Gras, T.; Haibe-Kains, B.; Bontempi, G.; Decaestecker, C.; Ruysschaert, J.-M.; et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia 2010, 12, 727–739. [Google Scholar] [CrossRef]
- Martignoni, M.; Groothuis, G.M.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Therapeutics 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Tsang, L.L.H.; Quarterman, C.P.; Gescher, A.; Slack, J.A. Comparison of the cytotoxicity in vitro of temozolomide and dacarbazine, prodrugs of 3-methyl-(triazen-1-yl)imidazole-4-carboxamide. Cancer Chemother. Pharm. 1991, 27, 342–346. [Google Scholar] [CrossRef]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.; Tsang, L.L.; Slack, J.A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef]
- Aasland, D.; Götzinger, L.; Hauck, L.; Berte, N.; Meyer, J.; Effenberger, M.; Schneider, S.; Reuber, E.E.; Roos, W.P.; Tomicic, M.T.; et al. Temozolomide Induces Senescence and Repression of DNA Repair Pathways in Glioblastoma Cells via Activation of ATR-CHK1, p21, and NF-κB. Cancer Res. 2019, 79, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.; Fakurnejad, S.; Sayegh, E.T.; Clark, A.J.; Ivan, M.E.; Sun, M.Z.; Safaee, M.; Bloch, O.; James, C.D.; Parsa, A.T. Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med. 2014, 12, 107. [Google Scholar] [CrossRef]
- Barth, R.F.; Kaur, B. Rat brain tumor models in experimental neuro-oncology: The C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J. Neurooncol. 2009, 94, 299–312. [Google Scholar] [CrossRef]
- Ménétrier-Caux, C.; Ray-Coquard, I.; Blay, J.-Y.; Caux, C. Lymphopenia in Cancer Patients and its Effects on Response to Immunotherapy: An opportunity for combination with Cytokines? J. Immunother. Cancer 2019, 7, 85. [Google Scholar] [CrossRef]
- Fadul, C.E.; Fisher, J.L.; Gui, J.; Hampton, T.H.; Côté, A.L.; Ernstoff, M.S. Immune modulation effects of concomitant temozolomide and radiation therapy on peripheral blood mononuclear cells in patients with glioblastoma multiforme. Neuro-Oncology 2011, 13, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Kizilarslanoglu, M.C.; Aksoy, S.; Yildirim, N.O.; Ararat, E.; Sahin, I.; Altundag, K. Temozolomide-related infections: Review of the literature. J. BUON 2011, 16, 547–550. [Google Scholar] [PubMed]
- Dutoit, V.; Philippin, G.; Widmer, V.; Marinari, E.; Vuilleumier, A.; Migliorini, D.; Schaller, K.; Dietrich, P.-Y. Impact of Radiochemotherapy on Immune Cell Subtypes in High-Grade Glioma Patients. Front. Oncol. 2020, 10, 89. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines (Basel) 2016, 4, 28. [Google Scholar] [CrossRef]
- Kim, T.-G.; Kim, C.-H.; Park, J.-S.; Park, S.-D.; Kim, C.K.; Chung, D.-S.; Hong, Y.-K. Immunological factors relating to the antitumor effect of temozolomide chemoimmunotherapy in a murine glioma model. Clin. Vaccine Immunol. 2010, 17, 143–153. [Google Scholar] [CrossRef]
- Campian, J.L.; Piotrowski, A.F.; Ye, X.; Hakim, F.T.; Rose, J.; Yan, X.-Y.; Lu, Y.; Gress, R.; Grossman, S.A. Serial changes in lymphocyte subsets in patients with newly diagnosed high grade astrocytomas treated with standard radiation and temozolomide. J. Neurooncol. 2017, 135, 343–351. [Google Scholar] [CrossRef]
- Chiba, Y.; Hashimoto, N.; Tsuboi, A.; Oka, Y.; Murao, A.; Kinoshita, M.; Kagawa, N.; Oji, Y.; Hosen, N.; Nishida, S.; et al. Effects of concomitant temozolomide and radiation therapies on WT1-specific T-cells in malignant glioma. Jpn. J. Clin. Oncol. 2010, 40, 395–403. [Google Scholar] [CrossRef][Green Version]
- Koumarianou, A.; Christodoulou, M.-I.; Patapis, P.; Papadopoulos, I.; Liakata, E.; Giagini, A.; Stavropoulou, A.; Poulakaki, N.; Tountas, N.; Xiros, N.; et al. The effect of metronomic versus standard chemotherapy on the regulatory to effector T-cell equilibrium in cancer patients. Exp. Hematol. Oncol. 2014, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Karachi, A.; Dastmalchi, F.; Mitchell, D.A.; Rahman, M. Temozolomide for immunomodulation in the treatment of glioblastoma. Neuro-Oncology 2018, 20, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 2017, 97, 498–518. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Won, W.-J.; Deshane, J.S.; Leavenworth, J.W.; Oliva, C.R.; Griguer, C.E. Metabolic and functional reprogramming of myeloid-derived suppressor cells and their therapeutic control in glioblastoma. Cell Stress 2019, 3, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Ding, A.S.; Routkevitch, D.; Jackson, C.; Lim, M. Targeting Myeloid Cells in Combination Treatments for Glioma and Other Tumors. Front. Immunol. 2019, 10, 1715. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188-204.e22. [Google Scholar] [CrossRef] [PubMed]
- Billerbeck, E.; Barry, W.T.; Mu, K.; Dorner, M.; Rice, C.M.; Ploss, A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rγ(null) humanized mice. Blood 2011, 117, 3076–3086. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef]
- Khoshnevis, M.; Carozzo, C.; Bonnefont-Rebeix, C.; Belluco, S.; Leveneur, O.; Chuzel, T.; Pillet-Michelland, E.; Dreyfus, M.; Roger, T.; Berger, F.; et al. Development of induced glioblastoma by implantation of a human xenograft in Yucatan minipig as a large animal model. J. Neurosci. Methods 2017, 282, 61–68. [Google Scholar] [CrossRef]
- Koehler, J.W.; Miller, A.D.; Miller, C.R.; Porter, B.; Aldape, K.; Beck, J.; Brat, D.; Cornax, I.; Corps, K.; Frank, C.; et al. A Revised Diagnostic Classification of Canine Glioma: Towards Validation of the Canine Glioma Patient as a Naturally Occurring Preclinical Model for Human Glioma. J. Neuropathol. Exp. Neurol. 2018, 77, 1039–1054. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Delord, J.-P.; Gonçalves, A.; Gavoille, C.; Dubot, C.; Isambert, N.; Campone, M.; Trédan, O.; Massiani, M.-A.; Mauborgne, C.; et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol. 2015, 16, 1324–1334. [Google Scholar] [CrossRef]
- Tannock, I.F.; Hickman, J.A. Limits to Personalized Cancer Medicine. N. Engl. J. Med. 2016, 375, 1289–1294. [Google Scholar] [CrossRef]
- McGranahan, T.; Therkelsen, K.E.; Ahmad, S.; Nagpal, S. Current State of Immunotherapy for Treatment of Glioblastoma. Curr. Treat. Opt. Oncol. 2019, 20, 24. [Google Scholar] [CrossRef]
- Burton, D.; Stolzing, A. Cellular senescence: Immunosurveillance and future immunotherapy. Ageing Res. Rev. 2018, 43. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Vendor | Solubility in DMSO [mg/mL] | Stock Concentration [mM] | Online Reference |
|---|---|---|---|
| BioCrick | 9.7 | 50 | https://www.biocrick.com/Temozolomide-BCC4386.html |
| TargetMol | 9.7 | 50 | https://www.targetmol.com/compound/Temozolomide |
| Tocris | 9.7 | 50 | https://www.tocris.com/products/temozolomide_2706 |
| Abmole | >20 | >100 | http://www.abmole.com/products/temozolomide.html |
| Merck/Sigma Aldrich | >20 | >100 | https://www.sigmaaldrich.com/catalog/product/sigma/t2577?lang=de®ion=DE |
| AbaChem Scene | 20.83 | 107.29 | https://www.chemscene.com/85622-93-1.html |
| MedChem Express | 20.83 | 107.29 | https://www.medchemexpress.com/Temozolomide.html |
| Apex Biotechnology | >29.61 | >152.6 | http://www.apexbt.com/temozolomide.html |
| Selleckchem | 38 | 195.72 | https://www.selleckchem.com/products/Methazolastone.html |
| Adooq | 39 | 200.87 | https://www.adooq.com/methazolastone.html |
| Santa Cruz Biotechnology | 39 | 200.87 | https://www.scbt.com/scbt/product/temozolomide-85622-93-1 |
| DMSO Concen-Tration Used (v/v) | Cellular System | Reported Effects | Additional Comments | Reference |
|---|---|---|---|---|
| 0.1, 0.5, 1, 1.5, 2, 3 and 5% | Human umbilical vein endothelial cells, RAW264.7 mouse macrophage cell line, MCF-7 human breast cancer cell line | Growth inhibition at concentration > 0.5% | Only looked at cell viability | [75] |
| 2, 4, 10, 20, 50 and 100% | Caco-2 cell line (enterocyte-like) | Lactate dehydrogenase release and Neutral Red uptake significantly altered at concentrations > 10% | Looked at alterations in apical membrane permeability or on cell-to-cell tight junctional complexes | [76] |
| 0.001, 0.01, 0.1, 1, 3, 5, 7 and 9% | GSF3.2 goat skin fibroblast cell line | 0.01–0.001% enhance proliferation, 0.5–3% retarded proliferation, > 3% no cell survival | Only looked at cell viability | [77] |
| 0.1, 0.2, 0.5, 1, 2 and 5% | Molt-4, Jurkat, U937 and THP1 leukaemic cell lines | Cytotoxic effects at concentrations > 1% | [78] | |
| 0.5, 1, 2, 2.5, 5, 10 and 20% | Peripheral blood lymphocytes | 1% and 2% reduce relative proliferation, 5% and 10% reduce percentage total lymphocytes and cytokine producers | [79] | |
| 0.01, 0.1 and 1% | Embryonic stem cells | All tested concentrations lead to reduced viability, alter morphology and adhesion and lead to abnormal differentiation | [80] | |
| 1% | Human lens epithelial cells | Decreased cell viability, increased cellular apoptosis, and upregulated Bax in these cells | [81] | |
| 0.2, 0.4 and 0.6% | EAhy926 umbilical vascular endothelial cell line, red blood cells | Concentrations of > 0.2% consistently cause haemolysis in red blood cells, concentrations of >0.2% consistently cause cell cycle arrest and all concentration increase apoptosis in EAhy926 | The authors conclude that DMSO is toxic to the haematologic system | [82] |
| 0.25, 0.5, 1, 2, 3, 4, 5 and 6% | Platelets | Aggregation induced by various factors was consistently inhibited by concentrations of > 1% |
| Cell Line | Age [yr] | Sex | Cell Culture Condition | Karyotype | MGMT Status (Promotor Methylation) | TP53 Status * | PTEN Status * | CDKN2A (p14ARF/p16INK4a) Status * | Orthotopic Xenograft Model |
|---|---|---|---|---|---|---|---|---|---|
| A172 | 53 A,B | M A,B | Adherent, Serum 10% A,B | Diploid to Hyper-triploid (n ~ 80) B,D | Negativ (methylated) G,H,I,J,K,L | wt C,R | Homozygous Deletion A,C,R | Homozygous Deletion A,C,R | T U,V NT W |
| DBTRG-05MG | 59 A,B | F A,B | Hypotetraploid (n ~ 87–91) A,B | Positive (unmethylated) M vs. Negative J | wt R,S | Homozygous Deletion R | Homozygous Deletion R | T X,Y | |
| LN18 | 65 A | M A | Adherent, Serum 5–10% A,C | n ~ 70–80, modal: 78 E | Positive (un-methylated) G,H,I,J,L,N | Homozygous * vs. Heterozygous C,R Missense Mutation (Cys→Ser) A | Wt A,C,R | Homozygous Deletion A,C,R | T W,Z |
| LN229 | 60 A | F A | Hypertriploid (n ~ 82) F | Negative (methylated) F,G,I,J,K,L | Homozygous * vs. Heterozygous C,R Missense Mutation (Pro→Leu) A | wtA,C,F,R | Homozygous Deletion A,C,R | T W,Aa | |
| LN308 (LNZ308) | 65 C | M C | Hypertriploid (n ~ 75–81) F | Negative (methylated) F,G,I,O | null/nullC | Splice-site (Deletion exon6) C,F | Wt C | T Bb,Cc | |
| M059K/J | 33 A | M A | Adherent, Serum 10% A,B | M059K: n ~ 65–79, modal: 75 A | Positive N vs. Negative J | Homozygous Missense Mutation (Glu→Lys) T | Heterozygous Frameshift Deletion R | M059J: wt R | na |
| T98G | 61 A,B | M A,B | Hyperpentaploid (n ~ 128–132) A,D | Positive (unmehtylated + methylated) G,H,I,J,L,M,N,O,P | Homozygous Misssense Mutation (Met→Ile) C,R | Homozygous Missense Mutation (Leu→Arg) C,R | Homozygous Deletion C,R | T V,Dd,Ee NTW | |
| U87 ** | 44 B,C | F B,C | Hypodiploid (n ~ 44, 48%) A,D,F | Negative (methylated) F, G, H, K, L, M, O, P, Q | wt C,R | Homozygous Splice Site (c.209+1G>T) A,C,F,R | Homozygous Deletion A,C,R | T V,W,Ee, Ff, Gg | |
| U118MG ** | 50 A | M A | Cytogenetically similar to U138 A,D | Positive (un-methylated) K,L,N,Q | Homozygous Missense Mutation (Arg→Gln) C | Homozygous Splice Site (c. 1026+1G>T) A,C | Homozygous Deletion A,C | T V,Hh NT W | |
| U138MG ** | 47 A | M A | Adherent, Serum 10% A,B | Hyperdiploid to Pentaploid A,D | Positive (un-mehtylated) H,K,L.M,O vs. Negative G | Homozygous Missense Mutation (Arg→Gln) C,R | Homozygous Splice Site C,R | Homozygous Deletion C,R | T Ii NT V |
| U251 ** | 75 C | M C | Diploid (n ~ 46) B | Negative (methylated) G,H,J,L,M | Homozygous Missense Mutation (Arg→His) C | Heterozygous Frame-shift Insertion (c.722_723dup) C | Homozygous Deletion C | T Ff,Jj | |
| U373 ** | 61 C | M C | Diploid (n ~ 46) B,D | Low positiveH vs. Negative (methylated) G,L,P | Homozygous Missense Mutation (Arg→His) C | Frameshift Mutation C | wt vs. p14 ARF Deletion Variant C | T V,Ee,Kk |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbener, V.J.; Burster, T.; Goreth, A.; Pruss, M.; von Bandemer, H.; Baisch, T.; Fitzel, R.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.-M.; et al. Considering the Experimental Use of Temozolomide in Glioblastoma Research. Biomedicines 2020, 8, 151. https://doi.org/10.3390/biomedicines8060151
Herbener VJ, Burster T, Goreth A, Pruss M, von Bandemer H, Baisch T, Fitzel R, Siegelin MD, Karpel-Massler G, Debatin K-M, et al. Considering the Experimental Use of Temozolomide in Glioblastoma Research. Biomedicines. 2020; 8(6):151. https://doi.org/10.3390/biomedicines8060151
Chicago/Turabian StyleHerbener, Verena J., Timo Burster, Alicia Goreth, Maximilian Pruss, Hélène von Bandemer, Tim Baisch, Rahel Fitzel, Markus D. Siegelin, Georg Karpel-Massler, Klaus-Michael Debatin, and et al. 2020. "Considering the Experimental Use of Temozolomide in Glioblastoma Research" Biomedicines 8, no. 6: 151. https://doi.org/10.3390/biomedicines8060151
APA StyleHerbener, V. J., Burster, T., Goreth, A., Pruss, M., von Bandemer, H., Baisch, T., Fitzel, R., Siegelin, M. D., Karpel-Massler, G., Debatin, K.-M., Westhoff, M.-A., & Strobel, H. (2020). Considering the Experimental Use of Temozolomide in Glioblastoma Research. Biomedicines, 8(6), 151. https://doi.org/10.3390/biomedicines8060151