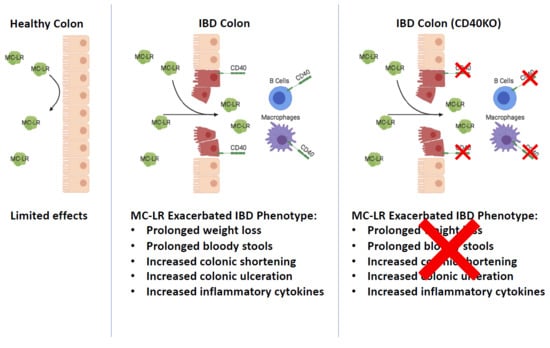
CD40 Receptor Knockout Protects against Microcystin-LR (MC-LR) Prolongation and Exacerbation of Dextran Sulfate Sodium (DSS)-Induced Colitis

 , ,
, , 
 and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
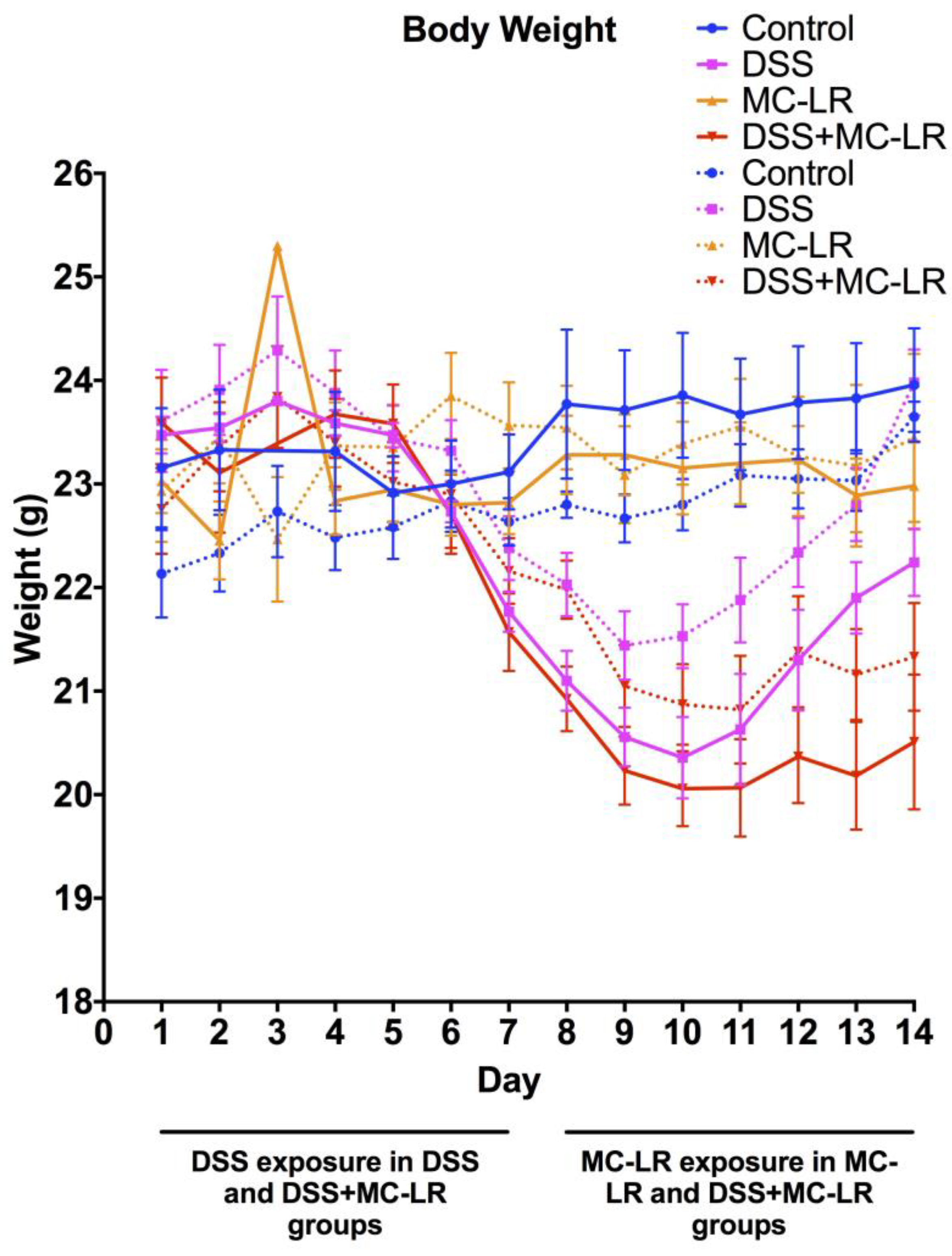
2.1. Body Weight and Survival
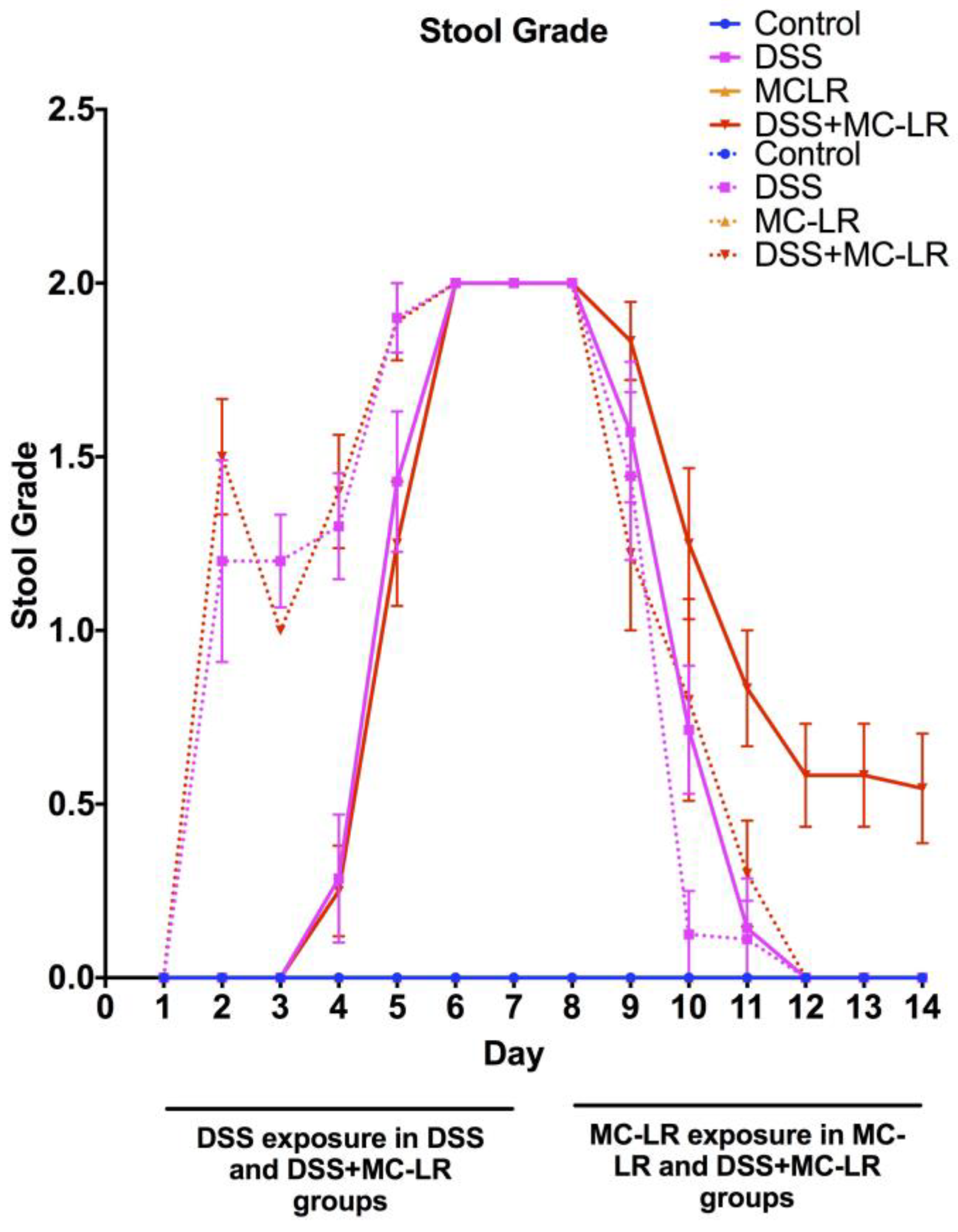
2.2. Stool Grading
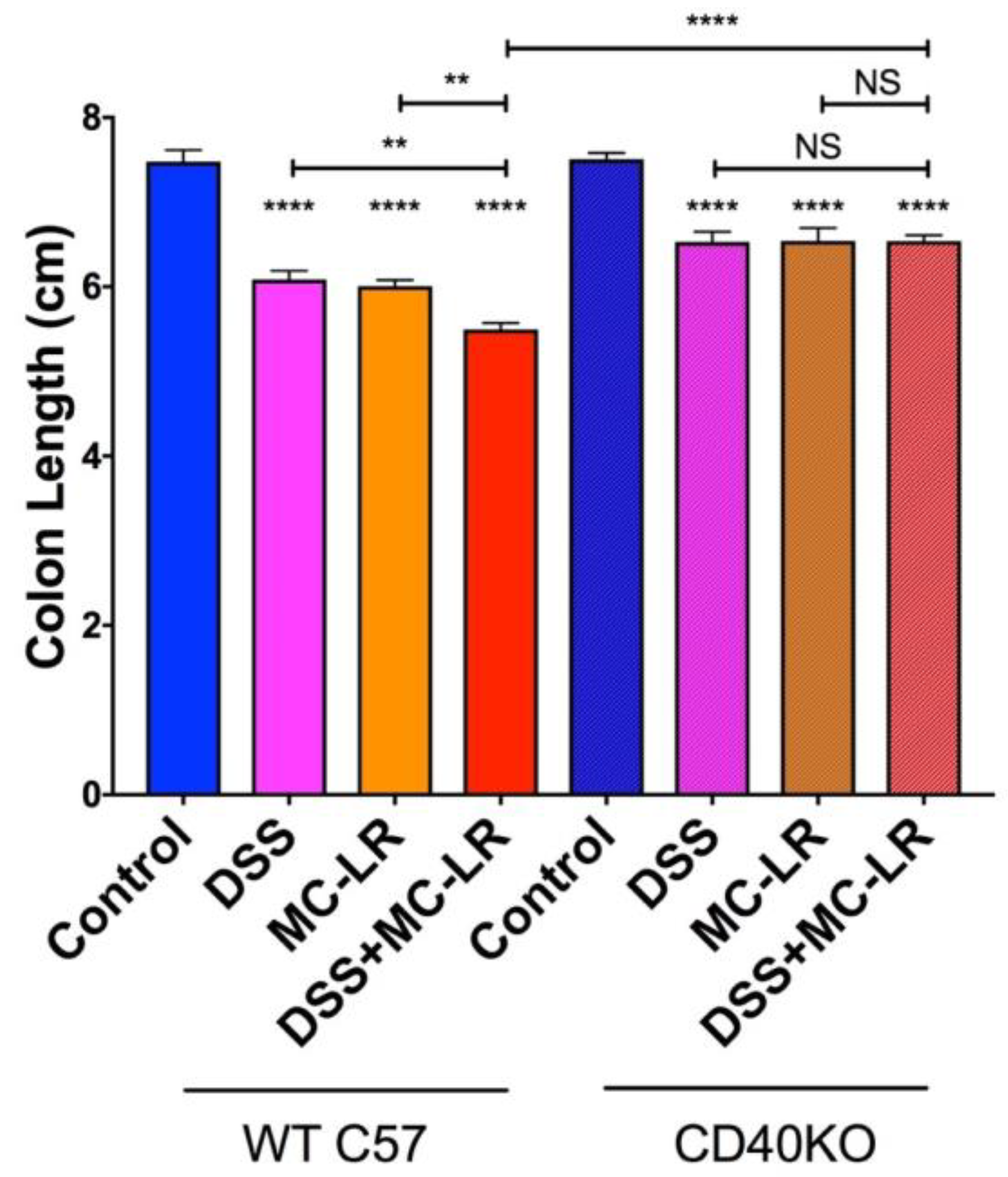
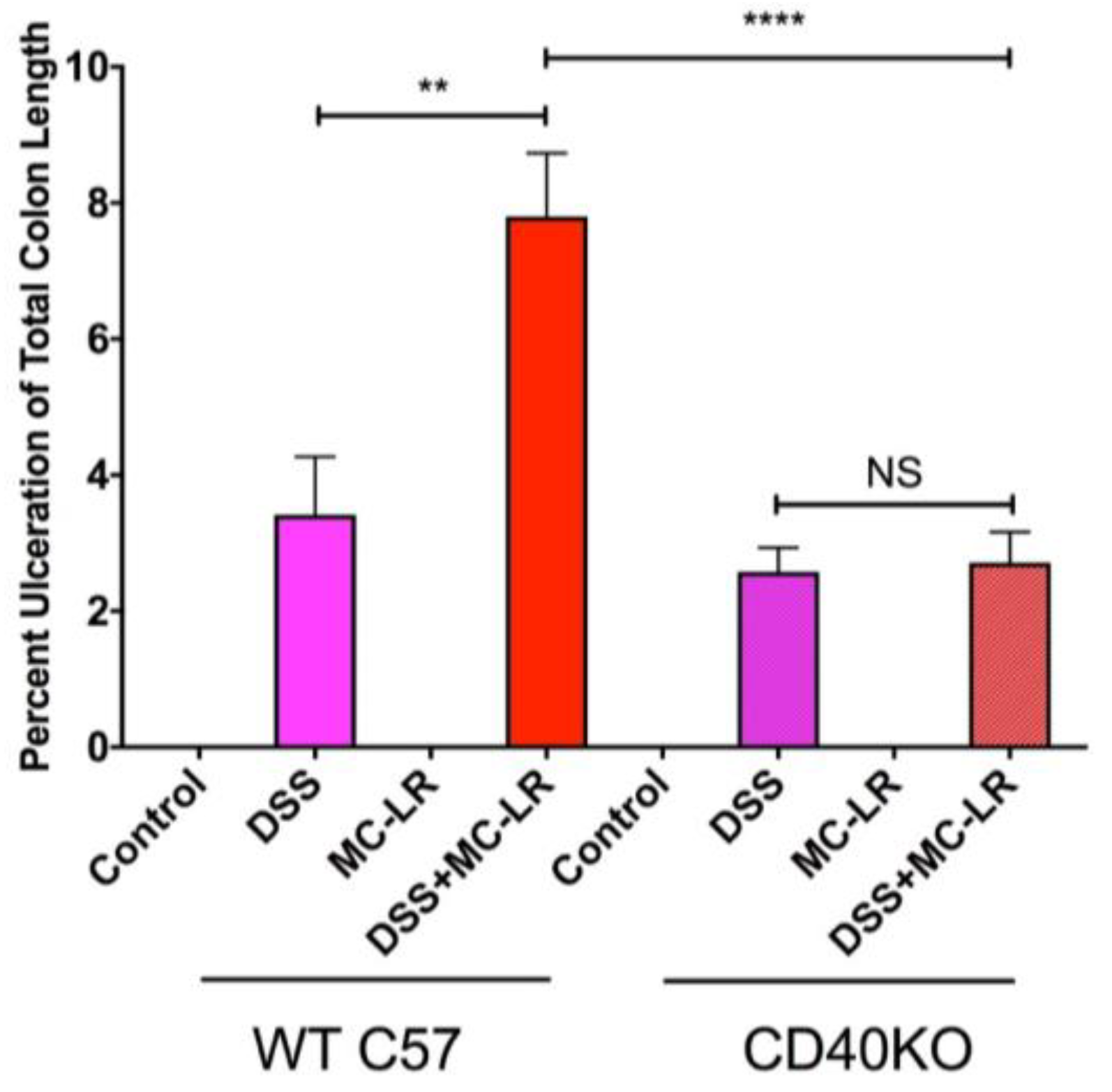
2.3. Colon Length
2.4. Histopathology
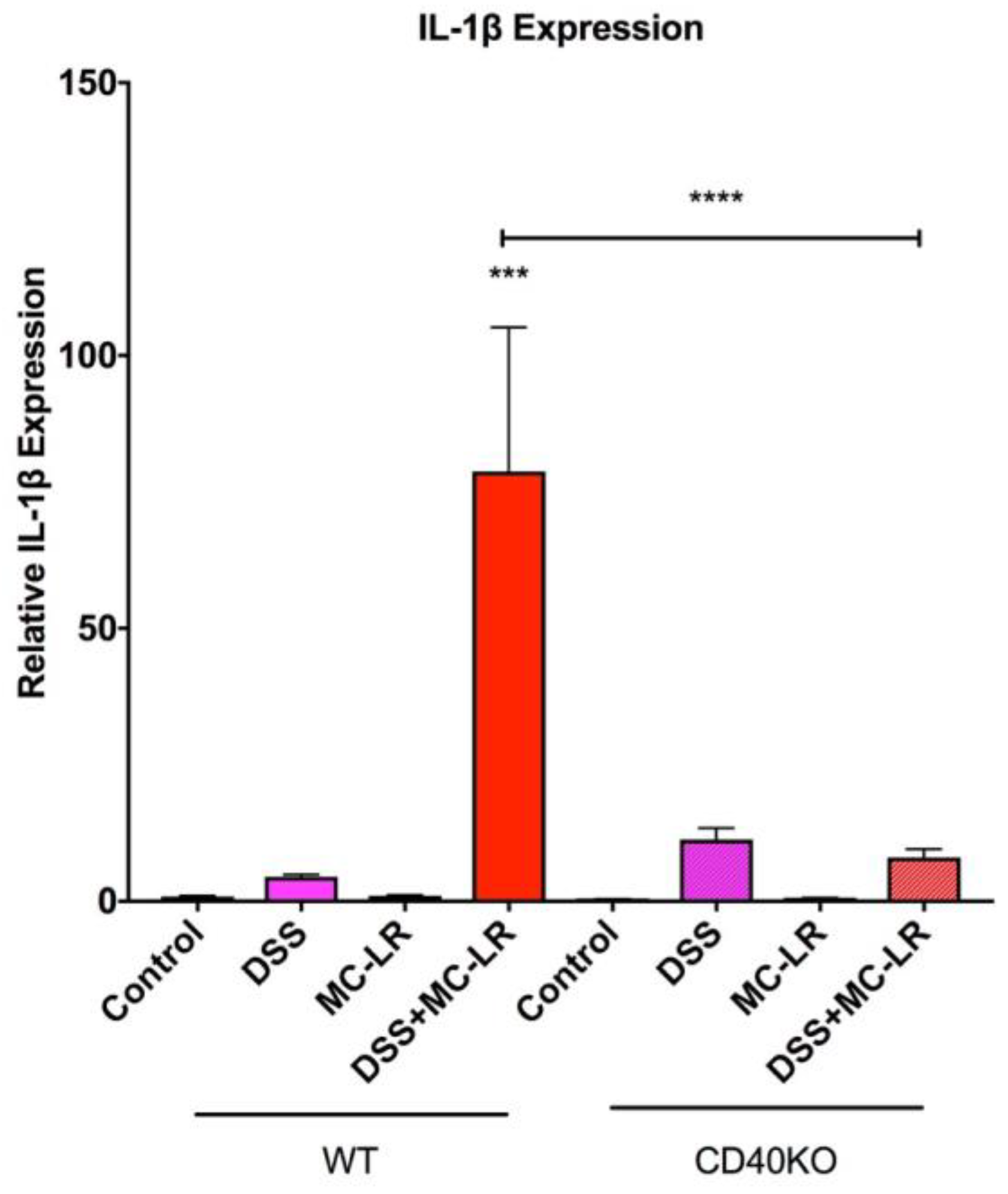
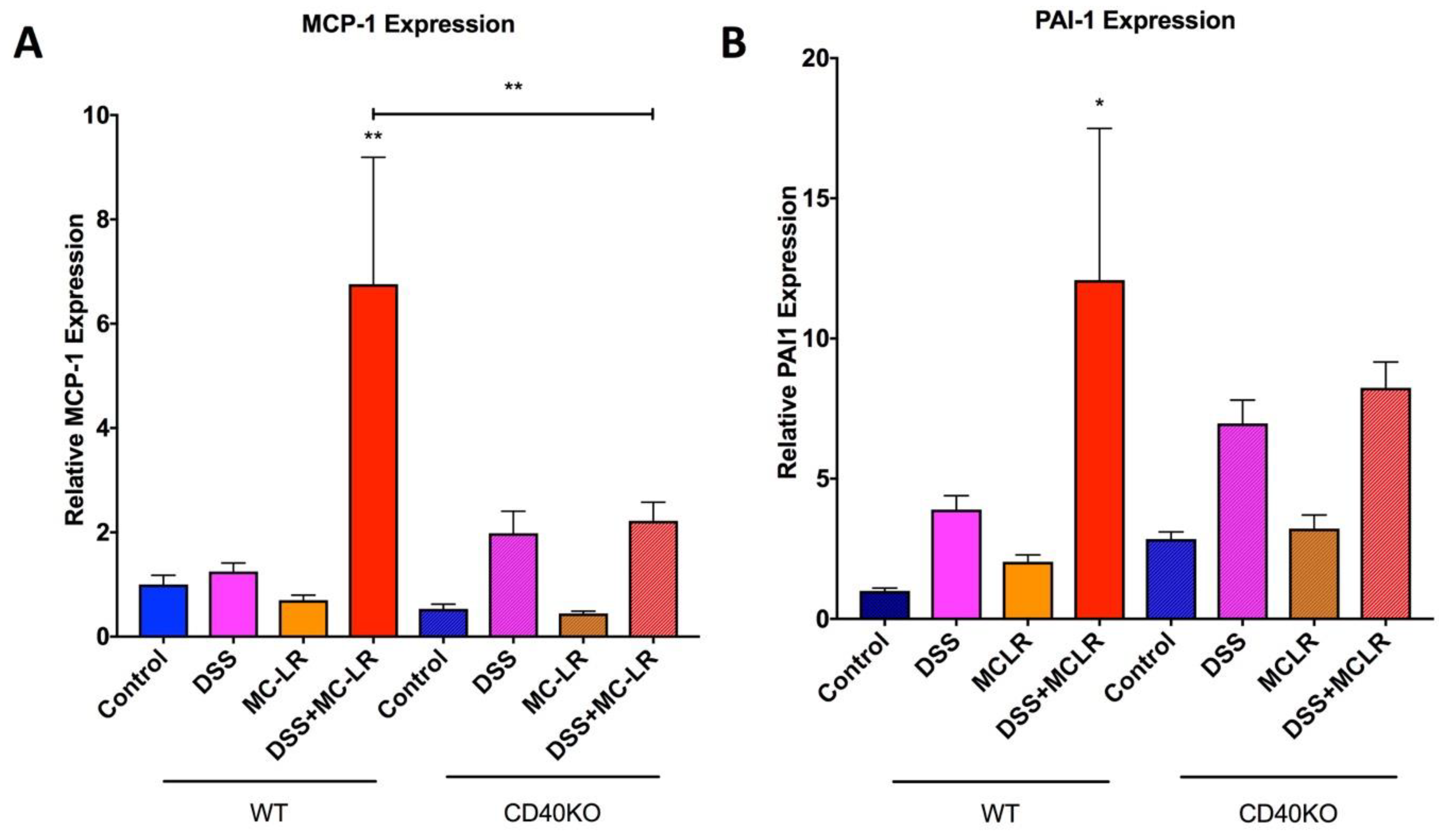
2.5. Gene Expression in the Colon
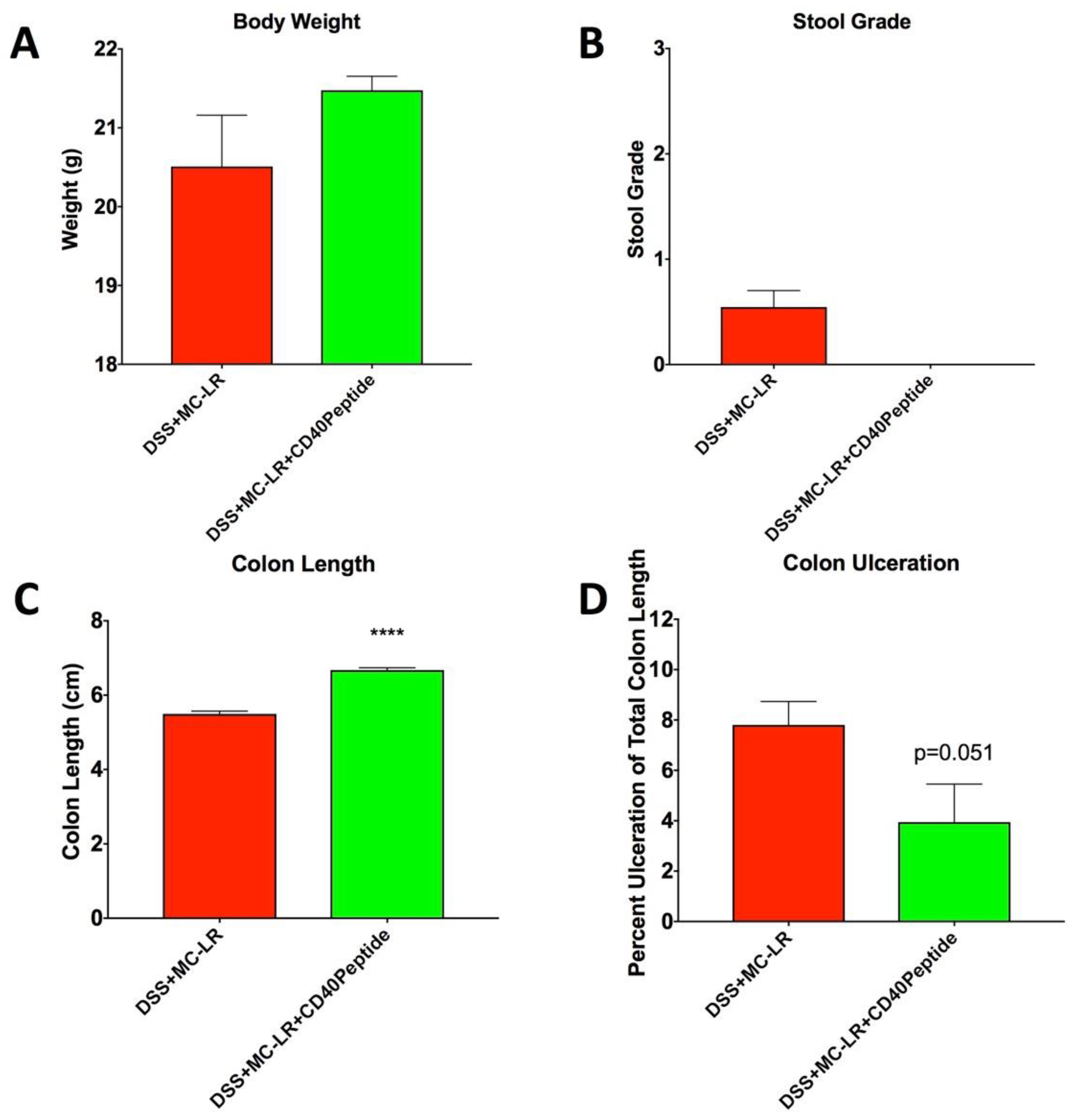
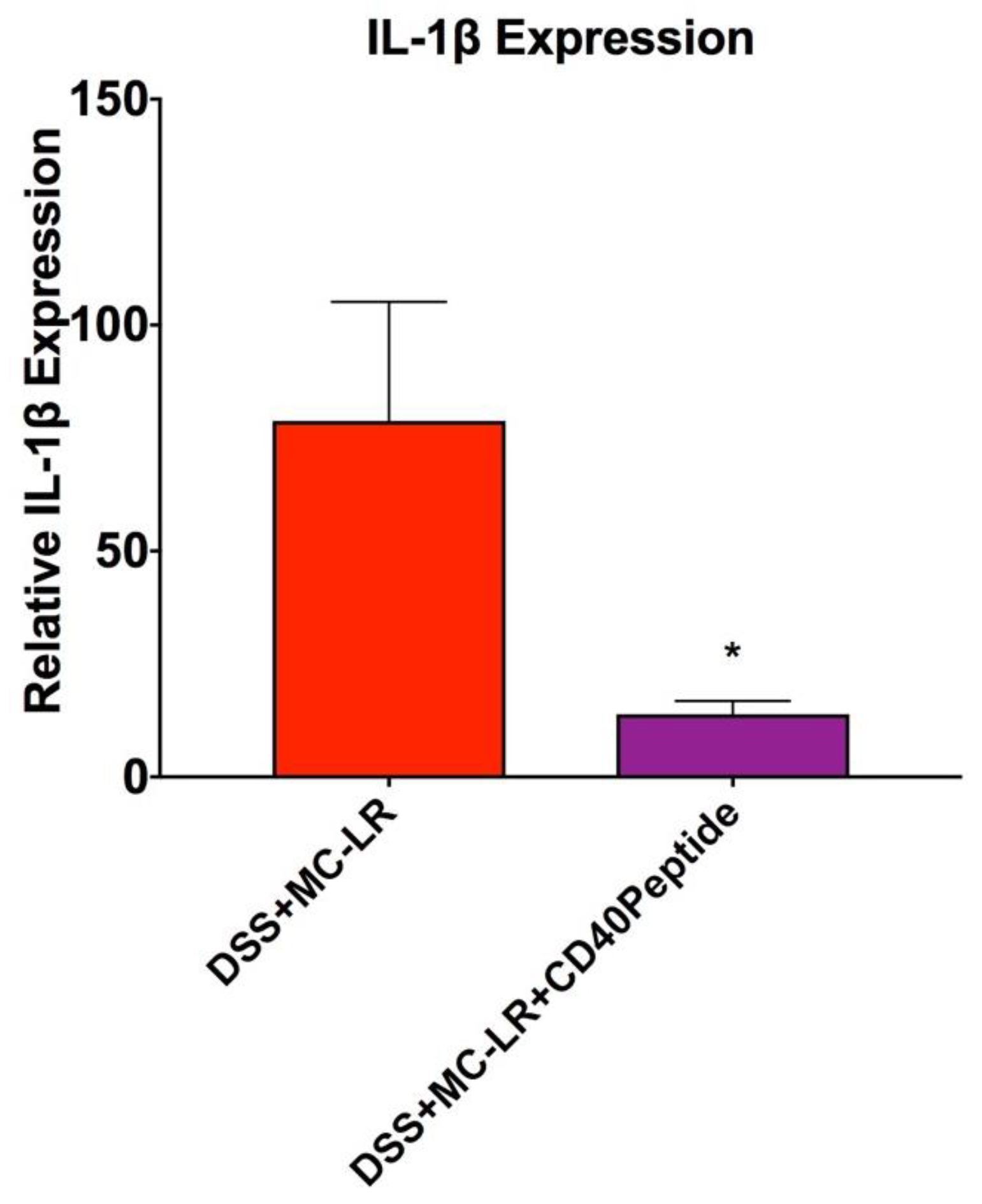
2.6. Proof-of-Concept Study with CD40 Receptor Blocking Peptide
3. Discussion
4. Materials and Methods
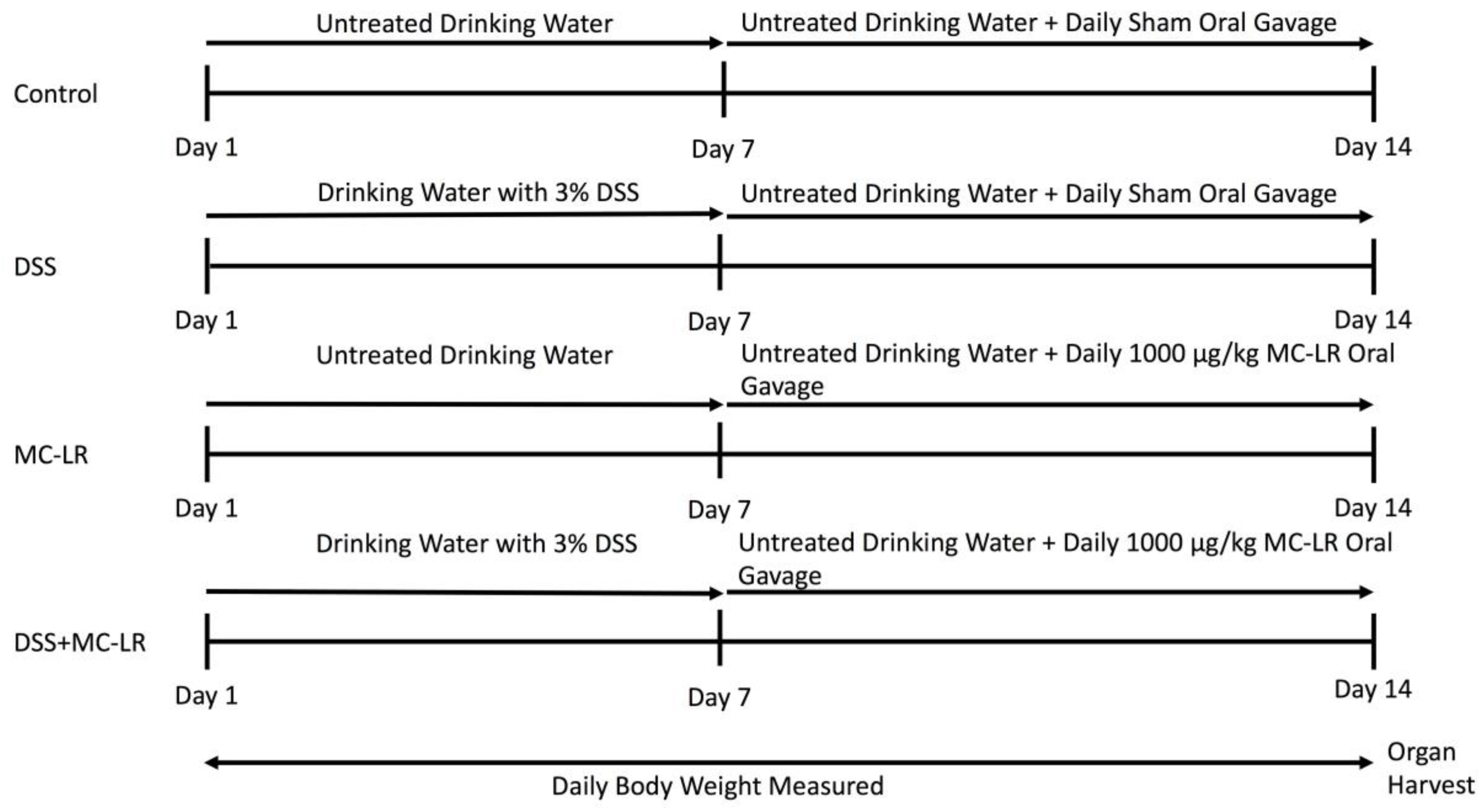
4.1. Mice and Experimental Design
4.2. Histology
4.3. RNA Extraction and RT-qPCR Method
4.4. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Jeengar, M.K.; Thummuri, D.; Magnusson, M.; Naidu, V.G.M.; Uppugunduri, S. Uridine Ameliorates Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Sci. Rep. 2017, 7, 3924. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Dahlhamer, J.M.; Zammitti, E.P.; Wheaton, A.G.; Croft, J.B. Prevalence of Inflammatory Bowel Disease Among Adults Aged ≥18 Years—United States, 2015. Morb. Mortal. Wkly. Rep. 2016, 65, 1166–1169. [Google Scholar]
- Molodecky, N.A.; Kaplan, G.G. Environmental Risk Factors for Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2010, 6, 339–346. [Google Scholar]
- Campos, A.; Vasconcelos, V. Molecular Mechanisms of Microcystin Toxicity in Animal Cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar] [CrossRef]
- Greer, B.; Meneely, J.P.; Elliott, C.T. Uptake and accumulation of Microcystin-LR based on exposure through drinking water: An animal model assessing the human health risk. Sci. Rep. 2018, 8, 4913. [Google Scholar] [CrossRef]
- Chorus, I. Introduction: Cyanotoxins—Research for Environmental Safety and Human Health. In Cyanotoxins: Occurrence, Causes, Consequences; Chorus, I., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; pp. 1–4. [Google Scholar]
- Lone, Y.; Koiri, R.K.; Bhide, M. An overview of the toxic effect of potential human carcinogen Microcystin-LR on testis. Toxicol. Rep. 2015, 2, 289–296. [Google Scholar] [CrossRef]
- Sedan, D.; Laguens, M.; Copparoni, G.; Aranda, J.O.; Leda, G.; Marra, C.A.; Andrinolo, D. Hepatic and intestine alterations in mice after prolonged exposure to low oral doses of Microcystin-LR. Toxicon 2015, 104, 26–33. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Williams, C.D.; Ni, H.; Kumer, S.C.; Schmitt, T.; Kane, B.; Jaeschke, H. Microcystin-LR induced liver injury in mice and in primary human hepatocytes is caused by oncotic necrosis. Toxicon 2017, 125, 99–109. [Google Scholar] [CrossRef]
- Mrdjen, I.; Morse, M.A.; Ruch, R.J.; Knobloch, T.J.; Choudhary, S.; Weghorst, C.; Lee, J. Impact of Microcystin-LR on Liver Function Varies by Dose and Sex in Mice. Toxins 2018, 10, 435. [Google Scholar] [CrossRef]
- Milutinović, A.; Zorc-Pleskovic, R.; Petrovič, D.; Zorc, M.; Suput, D. Microcystin-LR induces alterations in heart muscle. Folia Boil. 2006, 52, 116–118. [Google Scholar]
- La-Salete, R.; Oliveira, M.; Palmeira, C.; Almeida, J.; Peixoto, F. Mitochondria a key role in microcystin-LR kidney intoxication. J. Appl. Toxicol. 2007, 28, 55–62. [Google Scholar] [CrossRef]
- Yi, X.; Xu, S.; Huang, F.; Wen, C.; Zheng, S.; Feng, H.; Guo, J.; Chen, J.; Feng, X.; Yang, F.; et al. Effects of Chronic Exposure to Microcystin-LR on Kidney in Mice. Int. J. Environ. Res. Public Health 2019, 16, 5030. [Google Scholar] [CrossRef]
- Carmichael, N.G.; Enzmann, H.; Pate, I.; Waechter, F. The significance of mouse liver tumor formation for carcinogenic risk assessment: Results and conclusions from a survey of ten years of testing by the agrochemical industry. Environ. Health Perspect. 1997, 105, 1196–1203. [Google Scholar] [CrossRef]
- Svircev, Z.; Drobac, D.; Tokodi, N.; Mijović, B.; Codd, G.A.; Meriluoto, J. Toxicology of microcystins with reference to cases of human intoxications and epidemiological investigations of exposures to cyanobacteria and cyanotoxins. Arch. Toxicol. 2017, 91, 621–650. [Google Scholar] [CrossRef]
- Jochimsen, E.M.; Carmichael, W.; An, J.; Cardo, D.M.; Cookson, S.T.; Holmes, C.E.; Antunes, M.B.; Filho, D.A.D.M.; Lyra, T.M.; Barreto, V.S.T.; et al. Liver Failure and Death after Exposure to Microcystins at a Hemodialysis Center in Brazil. N. Engl. J. Med. 1998, 338, 873–878. [Google Scholar] [CrossRef]
- Carmichael, W.W.; Azevedo, S.M.; An, J.S.; Molica, R.J.; Jochimsen, E.M.; Lau, S.; Rinehart, K.L.; Shaw, G.R.; Eaglesham, G.K. Human fatalities from cyanobacteria: Chemical and biological evidence for cyanotoxins. Environ. Health Perspect. 2001, 109, 663–668. [Google Scholar] [CrossRef]
- Azevedo, S.M.F.D.O.E.; Carmichael, W.W.; Jochimsen, E.M.; Rinehart, K.L.; Lau, S.; Shaw, G.R.; Eaglesham, G.K. Human intoxication by microcystins during renal dialysis treatment in Caruaru—Brazil. Toxicology 2002, 181, 441–446. [Google Scholar] [CrossRef]
- Lad, A.; Su, R.C.; Breidenbach, J.D.; Stemmer, P.M.; Carruthers, N.; Sanchez, N.K.; Khalaf, F.K.; Zhang, S.; Kleinhenz, A.L.; Dube, P.; et al. Chronic Low Dose Oral Exposure to Microcystin-LR Exacerbates Hepatic Injury in a Murine Model of Non-Alcoholic Fatty Liver Disease. Toxins 2019, 11, 486. [Google Scholar] [CrossRef]
- Su, R.C.; Lad, A.; Breidenbach, J.D.; Blomquist, T.M.; Gunning, W.T.; Dube, P.; Kleinhenz, A.L.; Malhotra, D.; Haller, S.T.; Kennedy, D.J. Hyperglycemia induces key genetic and phenotypic changes in human liver epithelial HepG2 cells which parallel the Leprdb/J mouse model of non-alcoholic fatty liver disease (NAFLD). PLoS ONE 2019, 14, e0225604. [Google Scholar] [CrossRef]
- Su, R.C.; Blomquist, T.M.; Kleinhenz, A.L.; Khalaf, F.K.; Dube, P.; Lad, A.; Breidenbach, J.D.; Mohammed, C.J.; Zhang, S.; Baum, C.E.; et al. Exposure to the Harmful Algal Bloom (HAB) Toxin Microcystin-LR (MC-LR) Prolongs and Increases Severity of Dextran Sulfate Sodium (DSS)-Induced Colitis. Toxins 2019, 11, 371. [Google Scholar] [CrossRef]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; Fiocchi, C. The CD40/CD40L costimulatory pathway in inflammatory bowel disease. Gut 2004, 53, 1035–1043. [Google Scholar] [CrossRef]
- Girardi, B.; Principi, M.; Pricci, M.; Giorgio, F.; Iannone, A.; Losurdo, G.; Ierardi, E.; Di Leo, A.; Barone, M. Chemoprevention of inflammation-related colorectal cancer by silymarin-, acetyl-11-keto-beta-boswellic acid-, curcumin- and maltodextrin-enriched dietetic formulation in animal model. Carcinogenesis 2018, 39, 1274–1282. [Google Scholar] [CrossRef]
- Dieleman, L.A.; Palmen, M.J.H.J.; Akol, H.; Bloemena, E.; Peña, A.S.; Meuwissen, S.G.M.; Van Rees, E.P. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol. 1998, 114, 385–391. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol. 2014, 104, 15.25.1–15.25.14. [Google Scholar] [CrossRef]
- Smith, C.A.; Farrah, T.; Goodwin, R.G. The TNF receptor superfamily of cellular and viral proteins: Activation, costimulation, and death. Cell 1994, 76, 959–962. [Google Scholar] [CrossRef]
- Schonbeck, U.; Libby, P. The CD40/CD154 receptor/ligand dyad. Cell. Mol. Life Sci. 2001, 58, 4–43. [Google Scholar]
- Pietravalle, F.; Lecoanet-Henchoz, S.; Blasey, H.; Aubry, J.-P.; Elson, G.; Edgerton, M.D.; Bonnefoy, J.-Y.; Gauchat, J.-F. Human Native Soluble CD40L Is a Biologically Active Trimer, Processed Inside Microsomes. J. Boil. Chem. 1996, 271, 5965–5967. [Google Scholar] [CrossRef]
- Grammer, A.; Lipsky, P.E. CD40-mediated regulation of immune responses by TRAF-dependent and TRAF-independent signaling mechanisms. Adv. Immunol. 2000, 76, 61–178. [Google Scholar] [PubMed]
- Ngaotepprutaram, T.; Kaplan, B.L.; Kaminski, N.E. Impaired NFAT and NFkappaB activation are involved in suppression of CD40 ligand expression by Delta(9)-tetrahydrocannabinol in human CD4(+) T cells. Toxicol. Appl. Pharmacol. 2013, 273, 209–218. [Google Scholar] [CrossRef]
- Afford, S.; Ahmed-Choudhury, J.; Randhawa, S.; Russell, C.; Youster, J.; Crosby, H.A.; Eliopoulos, A.; Hubscher, S.G.; Young, L.S.; Adams, D.H. CD40 activation-induced, Fas-dependent apoptosis and NF-κB/AP-1 signaling in human intrahepatic biliary epithelial cells. FASEB J. 2001, 15, 2345–2354. [Google Scholar] [CrossRef]
- Aversa, G.; Punnonen, J.; Carballido, J.M.; Cocks, B.G.; De Vries, J.E. CD40 ligand-CD40 interaction in Ig isotype switching in mature and immature human B cells. Semin. Immunol. 1994, 6, 295–301. [Google Scholar] [CrossRef]
- Zan, H.; Cerutti, A.; Dramitinos, P.; Schaffer, A.; Casali, P. CD40 engagement triggers switching to IgA1 and IgA2 in human B cells through induction of endogenous TGF-beta: Evidence for TGF-beta but not IL-10-dependent direct S mu-->S alpha and sequential S mu-->S gamma, S gamma-->S alpha DNA recombination. J. Immunol. 1998, 161, 5217–5225. [Google Scholar]
- Nonoyama, S.; Hollenbaugh, D.; Aruffo, A.; Ledbetter, J.A.; Ochs, H.D. B cell activation via CD40 is required for specific antibody production by antigen-stimulated human B cells. J. Exp. Med. 1993, 178, 1097–1102. [Google Scholar] [CrossRef]
- Callard, R.E.; Armitage, R.J.; Fanslow, W.C.; Spriggs, M.K. CD40 ligand and its role in X-linked hyper-IgM syndrome. Immunol. Today 1993, 14, 559–564. [Google Scholar] [CrossRef]
- Aruffo, A.; Farrington, M.; Hollenbaugh, D.; Li, X.; Milatovich, A.; Nonoyama, S.; Bajorath, J.; Grosmaire, L.S.; Stenkamp, R.; Neubauer, M.; et al. The CD40 ligand, gp39, is defective in activated T cells from patients with X-linked hyper-IgM syndrome. Cell 1993, 72, 291–300. [Google Scholar] [CrossRef]
- Suttles, J.; Milhorn, D.M.; Miller, R.W.; Poe, J.C.; Wahl, L.M.; Stout, R.D. CD40 signaling of monocyte inflammatory cytokine synthesis through an ERK1/2-dependent pathway. A target of interleukin (il)-4 and il-10 anti-inflammatory action. J. Biol. Chem. 1999, 274, 5835–5842. [Google Scholar] [CrossRef]
- Burger, D.; Molnarfi, N.; Gruaz, L.; Dayer, J.-M. Differential induction of IL-1beta and TNF by CD40 ligand or cellular contact with stimulated T cells depends on the maturation stage of human monocytes. J. Immunol. 2004, 173, 1292–1297. [Google Scholar] [CrossRef]
- Pontrelli, P.; Ursi, M.; Ranieri, E.; Capobianco, C.; Schena, F.; Gesualdo, L.; Grandaliano, G. CD40L Proinflammatory and Profibrotic Effects on Proximal Tubular Epithelial Cells: Role of NF-κB and Lyn. J. Am. Soc. Nephrol. 2006, 17, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.T.; Kumarasamy, S.; Folt, D.A.; Wuescher, L.M.; Stepkowski, S.; Karamchandani, M.; Waghulde, H.; Mell, B.; Chaudhry, M.; Maxwell, K.; et al. Targeted disruption of Cd40 in a genetically hypertensive rat model attenuates renal fibrosis and proteinuria, independent of blood pressure. Kidney Int. 2017, 91, 365–374. [Google Scholar] [CrossRef]
- Li, H.; Nord, E.P. CD40 ligation stimulates MCP-1 and IL-8 production, TRAF6 recruitment, and MAPK activation in proximal tubule cells. Am. J. Physiol. Physiol. 2002, 282, F1020–F1033. [Google Scholar] [CrossRef] [PubMed]
- Laxmanan, S.; Datta, D.; Geehan, C.; Briscoe, D.; Pal, S. CD40: A Mediator of Pro- and Anti-Inflammatory Signals in Renal Tubular Epithelial Cells. J. Am. Soc. Nephrol. 2005, 16, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.T.; Kalra, P.A.; Ritchie, J.P.; Chrysochou, C.; Brewster, P.; He, W.; Yu, H.; Shapiro, J.I.; Cooper, C.J. Effect of CD40 and sCD40L on renal function and survival in patients with renal artery stenosis. Hypertension 2013, 61, 894–900. [Google Scholar] [CrossRef][Green Version]
- Xie, J.X.; Alderson, H.; Ritchie, J.; Kalra, P.A.; Xie, Y.; Ren, K.; Nguyen, H.; Chen, T.; Brewster, P.; Gupta, R.; et al. Circulating CD40 and sCD40L Predict Changes in Renal Function in Subjects with Chronic Kidney Disease. Sci. Rep. 2017, 7, 7942. [Google Scholar] [CrossRef]
- Xie, J.X.; Zhang, S.; Cui, X.; Zhang, J.; Yu, H.; Khalaf, F.K.; Malhotra, D.; Kennedy, D.J.; Shapiro, J.I.; Tian, J.; et al. Na/K-ATPase/src complex mediates regulation of CD40 in renal parenchyma. Nephrol. Dial. Transplant. 2017, 33, 1138–1149. [Google Scholar] [CrossRef]
- Zhang, S.; Breidenbach, J.D.; Khalaf, F.K.; Dube, P.R.; Mohammed, C.J.; Lad, A.; Stepkowski, S.; Hinds, T.D.; Kumarasamy, S.; Kleinhenz, A.; et al. Renal Fibrosis Is Significantly Attenuated Following Targeted Disruption of Cd40 in Experimental Renal Ischemia. J. Am. Hear. Assoc. 2020, 9, e014072. [Google Scholar] [CrossRef]
- Battaglia, E.; Biancone, L.; Resegotti, A.; Emanuelli, G.; Fronda, G.R.; Camussi, G. Expression of CD40 and its ligand, CD40L, in intestinal lesions of Crohn’s disease. Am. J. Gastroenterol. 1999, 94, 3279–3284. [Google Scholar] [CrossRef]
- Polese, L.; Angriman, I.; Cecchetto, A.; Norberto, L.; Scarpa, M.; Ruffolo, C.; Barollo, M.; Sommariva, A.; D’Amico, D.F. The role of CD40 in ulcerative colitis: Histochemical analysis and clinical correlation. Eur. J. Gastroenterol. Hepatol. 2002, 14, 237–241. [Google Scholar] [CrossRef]
- Gelbmann, C.M.; Leeb, S.N.; Vogl, D.; Maendel, M.; Herfarth, H.; Schölmerich, J.; Falk, W.; Rogler, G. Inducible CD40 expression mediates NFkappaB activation and cytokine secretion in human colonic fibroblasts. Gut 2003, 52, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.D.; West, G.A.; Danese, S.; De La Motte, C.; Phillips, M.H.; Strong, S.A.; Willis, J.; Fiocchi, C. CD40-mediated immune-nonimmune cell interactions induce mucosal fibroblast chemokines leading to T-Cell transmigration. Gastroenterology 2004, 126, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Colpaert, S.; D’Haens, G.R.; Kasran, A.; De Boer, M.; Rutgeerts, P.; Geboes, K.; Ceuppens, J. Hyperexpression of CD40 ligand (CD154) in inflammatory bowel disease and its contribution to pathogenic cytokine production. J. Immunol. 1999, 163, 4049–4057. [Google Scholar] [PubMed]
- Borcherding, F.; Nitschke, M.; Hundorfean, G.; Rupp, J.; Von Smolinski, R.; Bieber, K.; Van Kooten, C.; Lehnert, H.; Fellermann, K.; Büning, J. The CD40-CD40L Pathway Contributes to the Proinflammatory Function of Intestinal Epithelial Cells in Inflammatory Bowel Disease. Am. J. Pathol. 2010, 176, 1816–1827. [Google Scholar] [CrossRef]
- Danese, S.; De La Motte, C.; Sturm, A.; Vogel, J.D.; West, G.A.; Strong, S.A.; Katz, J.A.; Fiocchi, C. Platelets trigger a CD40-dependent inflammatory response in the microvasculature of inflammatory bowel disease patients. Gastroenterology 2003, 124, 1249–1264. [Google Scholar] [CrossRef]
- Vaitaitis, G.; Olmstead, M.H.; Waid, D.M.; Carter, J.R.; Wagner, D.H.J. A CD40-targeted peptide controls and reverses type 1 diabetes in NOD mice. Diabetologia 2014, 57, 2366–2373. [Google Scholar] [CrossRef]
- Viennois, E.; Tahsin, A.; Merlin, D. Purification of Total RNA from DSS-treated Murine Tissue via Lithium Chloride Precipitation. BioProtocol 2018, 8, e2829. [Google Scholar] [CrossRef]
- Kennedy, D.J.; Khalaf, F.K.; Sheehy, B.; Weber, M.E.; Agatisa-Boyle, B.; Conic, J.; Hauser, K.; Medert, C.M.; Westfall, K.; Bucur, P.; et al. Telocinobufagin, a Novel Cardiotonic Steroid, Promotes Renal Fibrosis via Na(+)/K(+)-ATPase Profibrotic Signaling Pathways. Int. J. Mol. Sci. 2018, 19, 2566. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, R.C.; Warner, E.A.; Breidenbach, J.D.; Lad, A.; Blomquist, T.M.; Kleinhenz, A.L.; Modyanov, N.; Malhotra, D.; Kennedy, D.J.; Haller, S.T. CD40 Receptor Knockout Protects against Microcystin-LR (MC-LR) Prolongation and Exacerbation of Dextran Sulfate Sodium (DSS)-Induced Colitis. Biomedicines 2020, 8, 149. https://doi.org/10.3390/biomedicines8060149
Su RC, Warner EA, Breidenbach JD, Lad A, Blomquist TM, Kleinhenz AL, Modyanov N, Malhotra D, Kennedy DJ, Haller ST. CD40 Receptor Knockout Protects against Microcystin-LR (MC-LR) Prolongation and Exacerbation of Dextran Sulfate Sodium (DSS)-Induced Colitis. Biomedicines. 2020; 8(6):149. https://doi.org/10.3390/biomedicines8060149
Chicago/Turabian StyleSu, Robin C., Emily A. Warner, Joshua D. Breidenbach, Apurva Lad, Thomas M. Blomquist, Andrew L. Kleinhenz, Nikolai Modyanov, Deepak Malhotra, David J. Kennedy, and Steven T. Haller. 2020. "CD40 Receptor Knockout Protects against Microcystin-LR (MC-LR) Prolongation and Exacerbation of Dextran Sulfate Sodium (DSS)-Induced Colitis" Biomedicines 8, no. 6: 149. https://doi.org/10.3390/biomedicines8060149
APA StyleSu, R. C., Warner, E. A., Breidenbach, J. D., Lad, A., Blomquist, T. M., Kleinhenz, A. L., Modyanov, N., Malhotra, D., Kennedy, D. J., & Haller, S. T. (2020). CD40 Receptor Knockout Protects against Microcystin-LR (MC-LR) Prolongation and Exacerbation of Dextran Sulfate Sodium (DSS)-Induced Colitis. Biomedicines, 8(6), 149. https://doi.org/10.3390/biomedicines8060149