New Insights into Mechanisms of Long-term Protective Anti-tumor Immunity Induced by Cancer Vaccines Modified by Virus Infection


Abstract
1. Introduction
2. Strategy of Designing a Tumor Vaccine Modified by Virus Infection
3. Target Structures in the Virus-modified Vaccines ATV-NDV and IO-VACR
3.1. Peptides from Tumor Neoantigens
3.2. Viral PAMPs
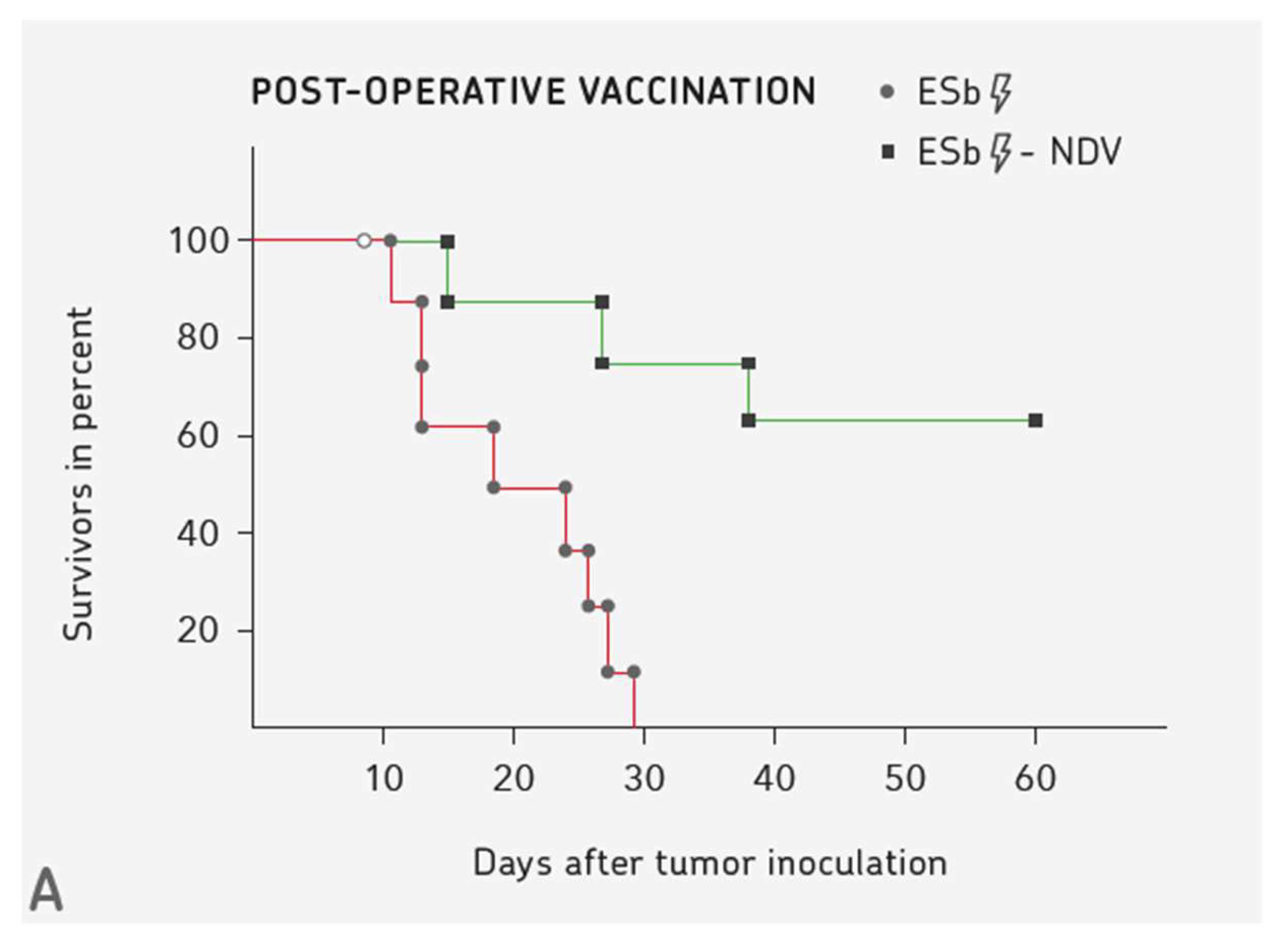
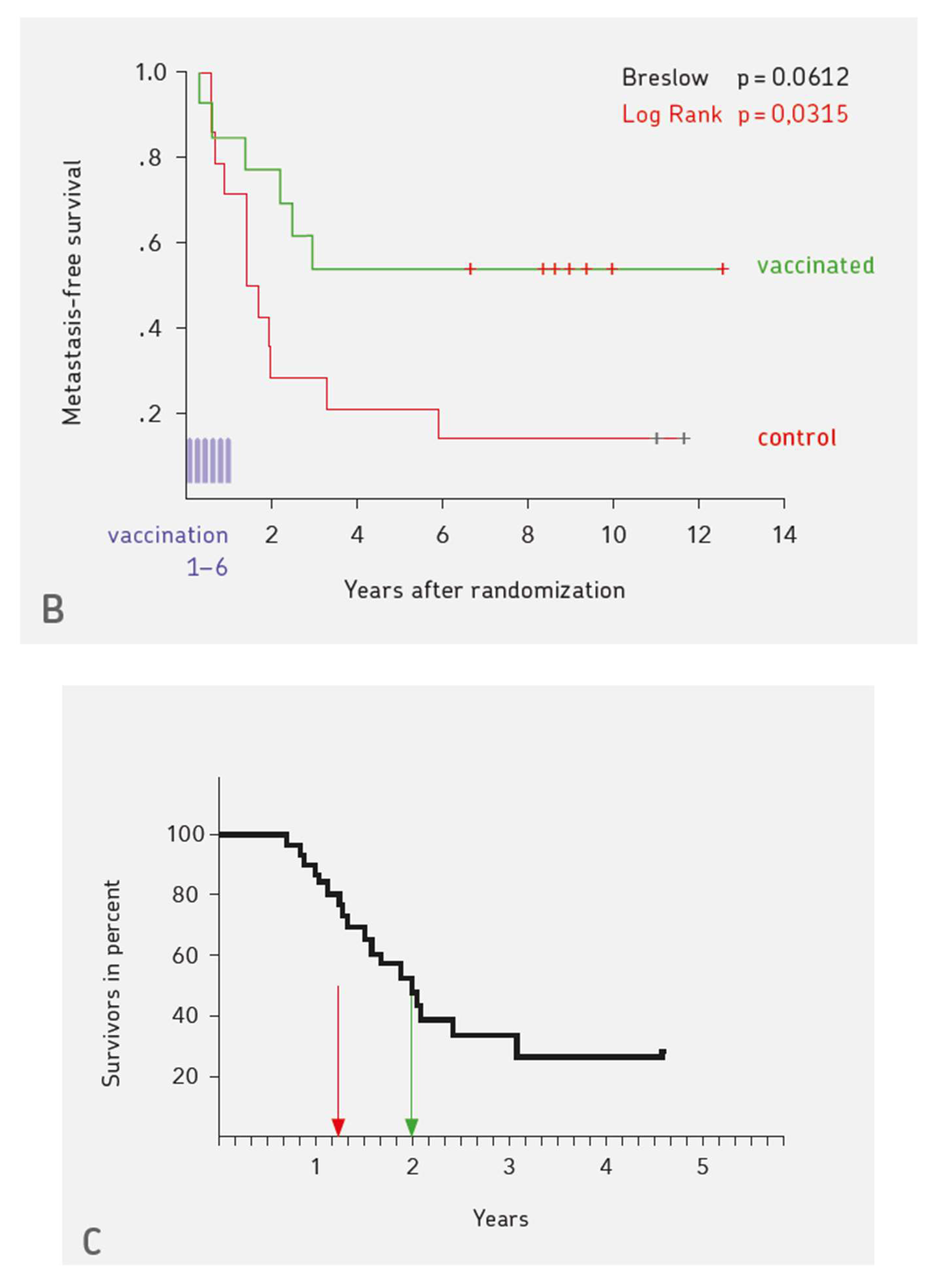
4. Three Examples for Vaccination-induced Long-term Protective Anti-cancer Immunity
5. 50 Years of Clinical Application of NDV
6. Mechanisms of TA Transport
6.1. TA Uptake and Transport via the Lymphatic System
6.2. TA Uptake and Transport via the Blood to Spleen and Bone Marrow
7. Bone Marrow and its Importance for T Cell Mediated Immune Responses to Blood-borne Antigens
7.1. Effect of Transient Dietary Restriction
7.2. Antigen Specific Cognate T-DC Interactions in the BM
- Naïve T cells (CD62Lhigh, CD44low,CD69neg) expressing a transgenic ovalbumin (OVA)-specific TCR were shown to home to BM parenchyma upon the transfer to naïve (B6) wild-type or splenectomized alymphoplastic mutant (Map3k14aly/aly) C57BL/6 mice. Upon OVA challenge (0,45 mg/mouse i.v.), BM DCs took up the blood-borne antigen and processed it via MHC I and II pathways. The transferred CD4+ or CD8+ T cells formed multicellular clusters with the BM-resident DCs (CD11c+) as APCs, became activated, proliferated, and differentiated into effector T cells and MTCs.
- BM responses could be generated in mice without lymph nodes and spleen. Thus, BM is autonomous in generating primary CD4+ and CD8+ T cell responses.
- In the absence of administered adjuvant, the BM responses were not tolerogenic and they resulted in generation of CTL activity, protective anti-tumor immunity, and immunological memory.
7.3. Antigen Processing and Presentation, Scanning of APCs by T Cells, Synapse Formation, APC-T Cluster Formation, CD4-CD8 T-T Interactions
7.4. Therapeutic Potential of BM derived MTCs
7.5. TA-specific Treg Cells from BM Exert Peripheral Tumor Immune Suppression
8. Mechanisms of Maintenance of Long-term T cell Memory
8.1. Dynamics and Longevity of Memory
8.2. Bone Marrow Niches for Maintenance of Memory T cells
- CD4+ MTCs helping antibody producing B cells were studied in the BM. In a secondary immune response to systemic antigen, antigen-specific helper T cells of the BM were found to aggregate together with MHC class II-expressing B cells. After 10 days, the immune clusters disappeared again. 30 days later, the expanded CD4+ MTCs returned to their BM niches and they were maintained there as resting cells [72].
- CXCR4 was found to be crucial for the entry of CD8+ T cells into the BM. This chemokine receptor also controls subsequent CD8+ T cell localization via attraction by CXCL12 (SDF-1α/β) toward BM niches, which support their survival [73].
- A hypothesis, recently proposed, suggests the existence of two niches in the BM to explain life-long T cell memory, one for T cell cycling and the other for T cell quiescence [74].
- A deuterium labelling study in mice supports a dynamic model for the maintenance of MTCs in the BM. This provides support for specialized BM niches. These are organized in such a way that MTCs can continuously self-renew and recirculate between the blood, BM, spleen, and lymph nodes [75].
8.3. Tissue-resident Memory T cells (T MTCs)
8.4. Stem Cell-like Memory T Cells (S MTCs)
9. Recruitment and Re-activation of MTCs from BM by Virus-modified Tumor Vaccine
10. Discussion
11. Summary
12. Conclusions
- (i)
- to release the tumor induced brakes on T cells by targeted checkpoint inhibitory antibodies,
- (ii)
- to boost instruction of the immune system via TA containing vaccines,
- (iii)
- to boost instruction by DC vaccines, thus bypassing the process of in situ antigen processing,
- (iv)
- to boost recognition bypassing instruction, the field of adoptive T-cell therapy, and
- (v)
- to boost recognition bypassing instruction and TA pMHC presentation, as exemplified by chimeric antigen receptor (CAR) T cells, bispecific T cell engagers (BITEs), or superantigens (SAGs).
Funding
Acknowledgments
Conflicts of Interest
References
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef]
- Kurd, N.; Robey, E.A. T-cell selection in the thymus: A spatial and temporal perspective. Immunol. Rev. 2016, 271, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z.; et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer 2019, 7, 1–21. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Speiser, D.E.; Knuth, A.; Bachmann, M.F. Virus-like particles for vaccination against cancer. WIREs Nanomed. Nanobiotechnol. 2020, 12, 1579. [Google Scholar] [CrossRef]
- Gubin, M.M.; Artyomov, M.N.; Mardis, E.R.; Schreiber, R.D. Tumor neoantigens: Building a framework for personalized cancer immunotherapy. J. Cin. Investig. 2015, 125, 3413–3421. [Google Scholar] [CrossRef] [PubMed]
- Rajewsky, K.; Schirrmacher, V.; Nase, S.; Jerne, N.K. The reqirement of more than one antigenic determinant for immunogenicity. J. Exp. Med. 1969, 129, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Baxter, A.G.; Hodgkin, P.D. Activation rules: The two-signal theory of immune activation. Nat. Rev. Immunol. 2002, 2, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Szabo, S.J.; Sullivan, B.M.; Peng, S.L.; Glimcher, L.H. Molecular mechanisms regulating Th1 immune responses. Annu. Rev. Immunol. 2003, 21, 713–758. [Google Scholar] [CrossRef]
- Sinkovics, J. Isolation of Newcastle Disease Virus from human oculoglandular illness. Kisérletes Orv. 1949, 1, 34–43. [Google Scholar]
- Cassel, W.A.; Garrett, R.E. Newcastle disease virus as an antineoplastic agent. Cancer 1965, 18, 863–868. [Google Scholar] [CrossRef]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Durai, V.; Bagadia, P.; Granja, J.M.; Satpathy, A.T.; Kulkarni, D.H.; Davidson, J.T., 4th; Wu, R.; Patel, S.J.; Iwata, A.; Liu, T.T.; et al. Cryptic activation of an Irf8 enhancer governs cDC1 fate specification. Nat. Immunol. 2019, 20, 1161–1173. [Google Scholar] [CrossRef]
- Mildner, A.; Jung, S. Development and function of dendritic cell subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, E.S.; Mellman, L. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 2005, 23, 975–1028. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F. Heterogeneity of human CD4+ T cells against microbes. Annu. Rev. Immunol. 2016, 34, 317–334. [Google Scholar] [CrossRef]
- Bevan, M.J. Helping the CD8+ T cell response. Nat. Rev. Immunol. 2004, 4, 595–602. [Google Scholar] [CrossRef]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef]
- Teijeira, A.; Garasa, S.; Etxeberria, I.; Gato-Canas, M.; Melero, I.; Delgoffe, G.M. Metabolic consequences of T-cell costimulation in anticancer immunity. Cancer Immunol. Res. 2019, 7, 1564–1569. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Von Hoegen, P.; Heicappell, R.; Griesbach, A.; Altevogt, P.; Schirrmacher, V. Prevention of metastatic spread by post-operative immunotherapy with virally modified autologous tumor cells. III. Postoperative activation of tumor-specific CTLP from mice with metastases requires stimulation with the specific antigen plus additional signals. Invasion Metastasis 1989, 9, 117–133. [Google Scholar] [PubMed]
- Schirrmacher, V.; Haas, C.; Bonifer, R.; Ahlert, T.; Gerhards, R.; Ertel, C. Human tumor cell modification by virus infection: An efficient and safe way to produce cancer vaccine with pleiotropic immune stimulatory properties when using Newcastle disease virus. Gene Ther. 1999, 6, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Lorenzen, D.; Van Gool, S.W.; Stuecker, W. A new strategy of cancer immunotherapy combining hyperthermia/oncolytic virus pretreatment with specific autologous anti-tumor vaccination—A Review. Austin Oncol. Case Rep. 2017, 2, 1006. [Google Scholar]
- Schirrmacher, V.; Heicappell, R. Prevention of metastatic spread by postoperative immunotherapy with virally modified autologous tumor cells. II. Establishment of specific systemic anti-tumor immunity. Clin. Exp. Metastasis 1987, 5, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Schirrmacher, V.; Fournier, P. The hemagglutinin-neuraminidase gene of Newcastle Disease Virus: A powerful molecular adjuvant for DNA anti-tumor vaccination. Vaccine 2010, 28, 6891–6900. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.; Wilden, H.; Schirrmacher, V. Importance of retinoic acid-inducible gene I and of receptor for type I interferon for cellular resistance to infection by Newcastle disease virus. Int. J. Oncol. 2012, 40, 287–298. [Google Scholar] [CrossRef]
- Zeng, J.; Fournier, P.; Schirrmacher, V. Induction of interferon-alpha and tumor-necrosis factor-related apoptosis-inducing ligand in human blood mononuclear cells by hemagglutinin-neuraminidase but not F protein of Newcastle disease virus. Virology 2002, 297, 19–30. [Google Scholar] [CrossRef]
- Koks, C.A.; Garg, A.D.; Ehrhardt, M.; Riva, M.; Vandenberk, L.; Boon, L.; De Vleeschouwer, S.; Agostinis, P.; Graf, N.; Van Gool, S.W. Newcastle disease virotherapy induces long-term survival and tumor-specific immune memory in orthotopic glioma through induction of immunogenic cell death. Int. J. Cancer 2015, 136, 313–325. [Google Scholar] [CrossRef]
- Harris, J.; Sharp, F.A.; Lavelle, E.C. The role of inflammasomes in the immunostimulatory effects of particulate vaccine adjuvants. Eur. J. Immunol. 2010, 40, 634–638. [Google Scholar] [CrossRef]
- Schirrmacher, V.; van Gool, S.; Stuecker, W. Breaking therapy resistance: An update on oncolytic Newcasle disease virus for improvements of cancer therapy. Biomedicines 2019, 7, 66. [Google Scholar] [CrossRef]
- Heicappell, R.; Schirrmacher, V.; von Hoegen, P.; Ahlert, T.; Appelhans, B. Prevention of metastatic spread by postoperative immunotherapy with virally modified autologous tumor cells. I. Parameters for optimal therapeutic effects. Int. J. Cancer 1986, 37, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Schulze, T.; Kemmner, W.; Weitz, J.; Wernecke, K.D.; Schirrmacher, V.; Schlag, P.M. Efficiency of adjuvant active specific immunization with Newcastle disease virus modified tumor cells in colorectal cancer patients following resection of liver metastases: Results of a prospective randomized trial. Caner Immunol. Immunother. 2009, 58, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Fournier, P.; Schlag, P. Autologous tumor cell vaccines for post-operative active-specific immunotherapy of colorectal carcinoma: Long-term patient survival and mechanism of function. Expert Rev. Vaccines 2014, 13, 117–130. [Google Scholar] [CrossRef]
- Song, H.; Zhong, L.; He, J.; Huand, Y.; Zhao, Y. Application of Newcastle disease virus in the treatment of colorectal cancer. World J. Clin. Cases 2019, 7, 2143–2154. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Feyen, O.; Prix, L.; Schirrmacher, V.; Stuecker, W. The induction of immunogenic cell death (ICD) during maintenance chemotherapy and susequent multimodal immunotherapy for glioblastoma (GBM). Austin Oncol. Case Rep. 2018, 3, 1010. [Google Scholar]
- Schirrmacher, V. Fifty years of clinical application of Newcastle disease virus: Time to celebrate! Biomedicines 2016, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Pabst, R. The bone marrow is not only a primary lymphoid organ: The critical role for T lymphocyte migration and housing of long-term memory plasma cells. Eur. J. Immunol. 2018, 48, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, L.C.; Carneiro, J. Blutbildung. In Histologie, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 326–342. [Google Scholar]
- Feuerer, M.; Beckhove, P.; Garbi, N.; Mahnke, Y.; Limmer, A.; Hommel, M.; Hämmerling, G.J.; Kyewski, B.; Hamann, A.; Umansky, V.; et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nat. Med. 2003, 9, 1151–1157. [Google Scholar] [CrossRef]
- Siracusa, F.; McGrath, M.A.; Maschmeyer, P.; Bardua, M.; Lehmann, K.; Heinz, G.; Durek, P.; Heinrich, F.F.; Mashreghi, M.F.; Chang, H.D.; et al. Nonfollicular reactivation of bone marrow resident memory CD4 T cells in immune clusters of the bone marrow. Proc. Natl. Acad. Sci. USA 2018, 115, 1334–1339. [Google Scholar] [CrossRef]
- Schirrmacher, V. Cancer-reactive memory T cells from bone marrow: Spontaneous induction and therapeutic potential (Review). Int. J. Oncol. 2015, 47, 2005–2016. [Google Scholar] [CrossRef]
- Li, M.; Davey, G.M.; Sutherland, R.M.; Kurts, C.; Lew, A.M.; Hirst, C.; Carbone, F.R.; Heath, W.R. Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J. Immunol. 2001, 166, 6099–6103. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.; Han, S.J.; Enamorado, M.; Link, V.M.; Huang, B.; Moseman, E.A.; Kishton, R.J.; Shannon, J.P.; Dixit, D.; Schwab, S.R. The bone marrow protects and optimizes immunological memory during dietary restriction. Cell 2019, 178, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Miggitsch, C.; Meryk, A.; Naismith, E.; Pangrazzi, L.; Ejaz, A.; Jenewein, B.; Wagner, S.; Nägele, F.; Fenkart, G.; Trieb, K.; et al. Human bone marrow adipocytes display distinct immune regulatory properties. EBioMedicine 2019, 46, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Apcher, S.; Millot, G.; Daskalogianni, C.; Scherl, A.; Manoury, B.; Fahraeus, R. Translation of pre-spliced RNAs in the nuclear compartment generates peptides for MHC class I pathway. Proc. Natl. Acad. Sci. USA 2013, 110, 17951–17956. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W.; Dersh, D.; Fahraeus, R. Peptide channeling: The key to MHC class I immunosurveillance? Trends Cell Biol. 2019, 29, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.P.; Malbert-Colas, L.; Lista, M.J.; Daskalogianni, C.; Apcher, S.; Pla, M.; Findakly, S.; Blondel, M.; Fahraeus, R. Nuclear processing of nascent transcripts determines synthesis of full-length proteins and antigenic peptides. Nucleic Acids Res. 2019, 47, 3086–3100. [Google Scholar] [CrossRef]
- Zaghouani, H.; Kuzu, Y.; Kuzu, H.; Brumeanu, T.D.; Swiggard, W.J.; Steinman, R.M.; Bona, C.A. Contrasting efficacy of presentation by major histocompatibility complex class I and class II products when peptides are administered within a common protein carrier, self immunoglobulin. Eur. J. Immunol. 1993, 23, 2746–2750. [Google Scholar] [CrossRef]
- Manz, B.N.; Jackson, B.L.; Petit, R.S.; Dustion, M.L.; Groves, J. T-cell triggering thresholds are modulated by the number of antigen within individual T-cell clusters. Proc. Natl. Acad. Sci. USA 2011, 108, 9089–9094. [Google Scholar] [CrossRef]
- Lanzavecchia, A.; Sallusto, F. From synapses to immunological memory: The role of sustained T cell stimulation. Curr. Opin. Immunol. 2000, 12, 92–98. [Google Scholar] [CrossRef]
- Fooksman, D.R.; Vardhana, S.; Vasiliver-Shamis, G.; Liese, J.; Blair, D.A.; Waite, J.; Sacristán, C.; Victora, G.D.; Zanin-Zhorov, A.; Dustin, M.L. Functional anatomy of T cell activation and synapse formation. Annu. Rev. Immunol. 2010, 28, 79–105. [Google Scholar] [CrossRef]
- Bai, L.; Beckhove, F.; Feuerer, M.; Umansky, V.; Choi, C.; Solomayer, F.S.; Diel, I.J.; Schirrmacher, V. Cognate interactions between memory T cells and tumor antigen-presenting dendritic cells from bone marrow of breast cancer patients: Bidirectional cell stimulation, survival and antitumor activity in vivo. Int. J. Cancer 2003, 103, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Hommel, M.; Kyewski, B. Dynamic changes during the immune response in T cell-antigen-presenting cell clusters isolated from lymph nodes. J. Exp. Med. 2003, 197, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Beckhove, P.; Mahnke, Y.; Hommel, M.; Kyewsky, B.; Hamann, A.; Umansky, V.; Schirrmacher, V. Bone marrow microenvironment facilitating dendritic cell: CD4 T cell interactions and maintenance of CD4 memory. Int. J. Oncol. 2004, 25, 867–876. [Google Scholar] [PubMed]
- Müerköster, S.; Wacholski, O.; Zerban, H.; Schirrmacher, V.; Umansly, V.; Rocha, M. Graft-versus-leukemia reactivity involves cluster formation between superantigen-reactive donor T lymphocytes and host macrophages. Clin. Cancer Res. 1998, 4, 3095–3106. [Google Scholar] [PubMed]
- Beckhove, P.; Feuerer, M.; Dolenc, M.; Schuetz, F.; Choi, C.; Sommerfeldt, N.; Schwendemann, J.; Ehlert, K.; Altevogt, P.; Bastert, G.; et al. Specifically activated memory T cell subsets from cancer patients recognize and reject xenotransplanted autologous tumors. J. Clin. Investig. 2004, 114, 67–76. [Google Scholar] [CrossRef]
- Williama, M.A.; Tyznik, A.J.; Bevan, M.J. Interleukin-2 signals during priming are required for secondary expansion of CD8 memory T cells. Nature 2006, 441, 890–893. [Google Scholar] [CrossRef]
- Fournier, P.; Aigner, M.; Schirrmacher, V. Transcriptome analysis and cytokine profiling of naive T cells stimulated by a tumor vaccine via CD3 and CD25. Int. J. Oncol. 2010, 37, 1439–1452. [Google Scholar]
- Bourgeois, C.; Rocha, B.; Tanchot, C. A role for CD40 expression on CD8 T cells in the generation of CD8 T cell memory. Science 2002, 297, 2060–2063. [Google Scholar] [CrossRef]
- Bennett, S.R.; Carbone, F.R.; Karamalis, F.; Flavell, R.A.; Miller, J.F.; Heath, W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998, 393, 478–480. [Google Scholar] [CrossRef]
- Schoenberger, S.P.; Toes, R.E.; van der Voort, E.I.; Offringa, R.; Melief, C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef]
- Feuerer, M.; Beckhove, P.; Bai, L.; Solomayer, E.; Bastert, G.; Diel, I.J.; Pedain, C.; Oberniedermayer, M.; Schirrmacher, V.; Umansky, V. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat. Med. 2001, 7, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.A.; Jenkins, M.R.; Rudd-Schmidt, J.A.; Brennan, A.J.; Danne, J.C.; Mannering, S.I.; Trapani, J.A.; Voskoboinik, I. Rapid and unidirectional perforin pore delivery at the cytotoxic immune synapse. J. Immunol. 2013, 191, 2328–2334. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.F.; Bzeih, H.; Schirra, C.; Chitirala, P.; Halimani, M.; Cordat, E.; Krause, E.; Rettig, J.; Pattu, V. Endocytosis of cytotoxic granules is essential for multiple killing of target cells by T lymphocytes. J. Immunol. 2016, 197, 2473–2484. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.N.; Gebhardt, T.; Carbone, F.R.; Heath, W.R. Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol. 2013, 31, 137–161. [Google Scholar] [CrossRef]
- Ge, Y.; Böhm, H.H.; Rathinasamy, A.; Xydia, M.; Hu, X.; Pincha, M.; Umansky, L.; Breyer, C.; Hillier, M.; Bonertz, A.; et al. Tumor-specific regulatory T cells from bone marrow orchestrate antitumor immunity in breast cancer. Cancer Immunol. Immunother. 2019, 7, 1998–2012. [Google Scholar] [CrossRef]
- Khazaie, K.; Prifti, S.; Beckhove, P.; Griesbach, A.; Russell, S.; Collins, M.; Schirrmacher, V. Persistence of dormant tumor cells in the bone marrow of tumor cell-vaccinated mice correlates with long-term immunological protection. Proc. Natl. Acad. Sci. USA 1994, 91, 7430–7434. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Schirrmacher, V. Characteristics of a potent tumor vaccine-induced secondary anti-tumor T cell response. Int. J. Oncol. 2004, 24, 1427–1434. [Google Scholar] [PubMed]
- Mahnke, Y.D.; Schirrmacher, V. A novel tumour model system for the study of long-term protective immunity and immune T cell memory. Cell. Immunol. 2003, 221, 89–99. [Google Scholar] [CrossRef]
- Sercan Alp, Ö.; Durlanik, S.; Schulz, D.; McGrath, M.; Grün, J.R.; Bardua, M.; Ikuta, K.; Sgouroudis, E.; Riedel, R.; Zehentmeier, S.; et al. Memory CD8+ T cells colocalize with IL-7+ stromal cells in bone marrow and rest in terms of proliferation and transcription. Eur. J. Immunol. 2015, 45, 975–987. [Google Scholar] [CrossRef]
- Herndler-Brandstetter, D.; Landgraf, K.; Jenewein, B.; Tzankow, A.; Brunauer, R.; Brunner, S.; Parson, W.; Kloss, F.; Gassner, R.; Lepperdinger, G.; et al. Human bone marrow hosts polyfunctional memory CD4+ and CD8+ T cells with close contact to IL-15 producing cells. J. Immunol. 2011, 186, 6965–6971. [Google Scholar] [CrossRef]
- Chang, H.D.; Tokoyoda, K.; Radbruch, A. Immunological memories of the bone marrow. Immuno. Rev. 2018, 283, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, M.; Gessel, S.; van der Voort, R.; Slot, E.; Lucas, B.; Gielen, E.; Hoogenboezem, M.; Rademakers, T.; Geerman, S.; van Buul, J.D.; et al. CXCR4, but not CXCR3, drives CD8+ T-cell entry into and migration through the murine bone marrow. Eur. J. Immunol. 2019, 49, 576–589. [Google Scholar] [CrossRef] [PubMed]
- DiRosa, F. Two niches in the bone marrow: A hypothesis on life-long T cell memory. Trends Immunol. 2016, 37, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Baliu-Piqué, M.; Otto, S.A.; Borghans, J.A.M.; Tesselaar, K. In vivo deuterium labelling in mice supports a dynamic model for memory T-cell maintenance in the bone marrow. Immunol. Lett. 2019, 210, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Zens, K.; Münz, C. Tissue resident T cell memory or how the magnificent seven are chilling in the bone. Eur. J. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Pascutti, M.F.; Geerman, S.; Collins, N.; Brasser, G.; Nota, B.; Stark, R.; Behr, F.; Oja, A.; Slot, E.; Panagioti, E.; et al. Peripheral and systemic antigens elicit an expandable pool of resident memory CD8+ T cells in the bone marrow. Eur. J. Immunol. 2019. [Google Scholar] [CrossRef]
- Mami-Chouaib, F.; Blanc, C.; Corgnac, S.; Hans, S.; Malenica, I.; Granier, C.; Tihy, I.; Tartour, E. Resident memory T cells, critical components in tumor immunology. J. Immunother. 2018, 6, 87. [Google Scholar] [CrossRef]
- Menares, E.; Gálvez-Cancino, F.; Cáceres-Morgado, P.; Ghorani, E.; López, E.; Diaz, X.; Saavedra-Almaraz, J.; Figueroa, D.A.; Roa, E.; Quezada, S.A.; et al. Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat. Commun. 2019, 10, 4401. [Google Scholar] [CrossRef]
- Knight, F.C.; Gilchuk, P.; Kumar, A.; Becker, K.W.; Sevimli, S.; Jacobson, M.E.; Suryadevara, N.; Wang-Bishop, L.; Boyd, K.L.; Crowe, J.E., Jr.; et al. Mucosal immunization with a pH-responsive nanoparticle vaccine induces protective CD8+ lung-resident memory T cells. ACS Nano 2019. [Google Scholar] [CrossRef]
- Bartolomé-Casado, R.; Landsverk, O.J.B.; Chauhan, S.K.; Richter, L.; Phung, D.; Greiff, V.; Risnes, L.F.; Yao, Y.; Neumann, R.S.; Yaqub, S.; et al. Resident memory CD8 T cells persist for years in human small intestine. J. Exp. Med. 2019, 216, 2412–2426. [Google Scholar] [CrossRef]
- Kudernatsch, R.F.; Letsch, A.; Guerreiro, M.; Löbel, M.; Bauer, S.; Volk, H.D.; Scheibenbogen, C. Human bone marrow contains a subset of quiescent early memory CD8+ T cells characterized by high CD127 expression and efflux capacity. Eur. J. Immunol. 2014, 44, 3532–3542. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Li, Y.; Zhang, S.; Zhou, N.; Liu, B.; Pan, T.; Zhang, X.; Luo, H.; Huang, Z.; Li, X.; et al. Preferential homing of tumor-specific and functional CD8+ stem cell-like memory T cells to the bone marrow. J. Immunother. 2019. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, F.; Alp, Ö.S.; Maschmeyer, P.; McGrath, M.; Mashreghi, M.F.; Hoiyo, S.; Chang, H.D.; Tokoyada, K.; Radbruch, A. Maintenance of CD8+ memory T lymphocytes in the spleen but not in the bone marrow is dependent on proliferation. Eur. J. Immunol. 2017, 47, 1900–1905. [Google Scholar] [CrossRef]
- Abdelsamed, H.A.; Moustaki, A.; Fan, Y.; Dogra, P.; Ghoneim, H.E.; Zebley, C.C.; Triplett, B.M.; Sekaly, R.P.; Youngblood, B. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J. Exp. Med. 2017, 214, 1593–1606. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef]
- Kondo, T.; Imura, Y.; Ando, M.; Chikuma, S.; Yoshimura, A. In vitro conversion of activated T cells into stem cell memory-like T cells. Methods Mol. Biol. 2019, 2048, 41–51. [Google Scholar] [CrossRef]
- Cieri, N.; Camissa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Koopman, J.; Fiola, C.; Fournier, P.; Schirrmacher, V. Dendritic cells pulsed with viral oncolysates potently stimulate autologous T cells from cancer patients. Int. J. Oncol. 2002, 21, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Schild, H.; von Hoegen, P.; Schirrmacher, V. Modification of tumor cells by a low dose of Newcastle disease virus. II. Augmented tumor-specific T cell responses as a result of CD4+ and CD8+ immune T cell cooperation. Cancer Immunol. Immunother. 1989, 28, 22–28. [Google Scholar] [CrossRef]
- Von Hoegen, P.; Zawatzky, R.; Schirrmacher, V. Modification of tumor cells by a low dose of Newcastle disease virus. III. Potentiation of tumor-specific cytolytic T cell activity via induction of interferon-alpha/beta. Cell. Immunol. 1990, 126, 80–90. [Google Scholar] [CrossRef]
- Raeber, M.E.; Zurbuchen, Y.; Impellizzieri, D.; Boyman, O. The role of cytokines in T-cell memory in health and disease. Immunol. Rev. 2018, 283, 176–193. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Gerner, M.Y.; Lins, D.C.; Mescher, M.F. Signal 3 availability limits the CD8 T cell response to a solid tumor. J. Immunol. 2007, 178, 6752–6760. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [PubMed]
- Boissonnas, A.; Fetler, L.; Zeelenberg, I.S.; Hugues, S.; Amigorena, S. In vivo imaging of cytotoxic T cell. infiltration and elimination of a solid tumor. J. Exp. Med. 2007, 204, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Sommerfeld, N.; Schütz, F.; Sohn, C.; Förster, J.; Schirrmacher, V.; Beckhove, P. The shaping of a polyvalent and highly individual T-cell repertoire in the bone marrow of breast cancer patients. Cancer Res. 2006, 66, 8258–8265. [Google Scholar] [CrossRef]
- Jiang, X.; Muthusamy, V.; Fedorova, O.; Kong, Y.; Kim, D.J.; Bosenberg, M.; Pyle, A.M.; Iwasaki, A. Intratumoral delivery of RIG-I agonist SLR14 induces robust antitumor responses. J. Exp. Med. 2019. [Google Scholar] [CrossRef]
- Jarahian, M.; Watzl, C.; Fournier, P.; Arnold, A.; Djandji, D.; Zahedi, S.; Cerwenka, A.; Paschen, A.; Schirrmacher, V.; Momburg, F. Activation of natural killer cells by Newcastle disease virus hemagglutinin-neuraminidase. J. Virol. 2009, 83, 8108–8121. [Google Scholar] [CrossRef]
- Umansky, V.; Shatrov, V.A.; Lehmann, V.; Schirrmacher, V. Induction of NO synthesis in macrophages by Newcastle disease virus is associated with activation of nuclear factor-kappa B. Int. Immunol. 1996, 8, 491–498. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Bai, L.; Umansky, V.; Yu, L.; Xing, Y.; Qian, Z. Newcastle disease virus activates macrophages for anti-tumor activity. Int. J. Oncol. 2000, 16, 363–373. [Google Scholar] [CrossRef]
- Zaslawsky, E.; Hershberg, U.; Seto, J.; Pham, A.M.; Marques, S.; Duke, J.L.; Wetmur, J.G.; Tenoever, B.R.; Sealfon, S.C.; Kleinstein, S.H. Antiviral response dictated by choreographed cascade of transcription factors. J. Immunol. 2010, 184, 2908–2917. [Google Scholar] [CrossRef]
- Washburn, B.; Weigand, M.A.; Grosse-Wilde, A.; Janke, M.; Stahl, H.; Rieser, E.; Sprick, M.R.; Schirrmacher, V.; Walczak, H. TNF-related apoptosis-inducing ligand mediates tumoricidal activity of human monocytes stimulated by Newcastle disease virus. J. Immunol. 2003, 170, 1814–1821. [Google Scholar] [CrossRef]
- Brownlie, R.J.; Zamoyska, R. T cell receptor signalling networks: Branched, diversified and bounded. Nat. Rev. Immunol. 2013, 13, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Sikora, A.G.; Jaffarzad, N.; Hailemichael, Y.; Gelbard, A.; Stonier, S.W.; Schluns, K.S.; Frasca, L.; Lou, Y.; Liu, C.; Andersson, H.A.; et al. IFN-alpha enhances peptide vaccine-induced CD8+ T cell numbers, effector function, and antitumor activity. J. Immunol. 2009, 182, 7398–7407. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. Oncolytic Newcastle disease virus as a prospective anti-cancer therapy. A biologic agent with potential to break therapy resistance. Expert Opin. Biol. Ther. 2015, 15, 1757–1771. [Google Scholar] [CrossRef] [PubMed]
- Niraula, S.; Seruga, B.; Ocana, A.; Shao, T.; Goldstein, R.; Tannock, I.F.; Amir, E. The price we pay for progress: A meta-analysis of harms of newly. approved anticancer drugs. J. Clin. Oncol. 2012, 30, 3012–3019. [Google Scholar] [CrossRef]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Heidegger, S.; Wintges, A.; Stritzke, F.; Bek, S.; Steiger, K.; Koenig, P.A.; Göttert, S.; Engleitner, T.; Öllinger, R.; Nedelko, T.; et al. RIG-I activation is critical for responsiveness to checkpoint blockade. Sci. Immunol. 2019, 4, 8943. [Google Scholar] [CrossRef]
- Hirbod-Mobarakeh, A.; Gordan, H.A.; Zahiri, Z.; Mirshahvalad, M.; Hosseinverdi, S.; Rini, B.I.; Rezaei, N. Specific immunotherapy in renal cancer: A systematic review. Ther. Adv. Urol. 2017, 9, 45–58. [Google Scholar] [CrossRef]
- Schirrmacher, V. Quo Vadis Cancer Therapy? LAP Lambert Academic Publishing: Saarbrücken, Germany, 2017; pp. 1–353. [Google Scholar]



{kind=link}
{kind=link}
| Feature | pDC | cDC | iDC |
|---|---|---|---|
| Surface CD | CD123 | CD11c | CD11c |
| TF | TCF4 | IRF4 | IRF8 |
| Cytokine | IFNα,β | IL-12 | IL-2 |
| APC function for | viruses | bacteria | virus-infected cells |
| Routes | infection | extra- or intracellular 1 | cross-presentation 2 |
| Loaded MHC | class I | class II | class I |
| Cognate T cell | CD8 | CD4 | CD8 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schirrmacher, V. New Insights into Mechanisms of Long-term Protective Anti-tumor Immunity Induced by Cancer Vaccines Modified by Virus Infection. Biomedicines 2020, 8, 55. https://doi.org/10.3390/biomedicines8030055
Schirrmacher V. New Insights into Mechanisms of Long-term Protective Anti-tumor Immunity Induced by Cancer Vaccines Modified by Virus Infection. Biomedicines. 2020; 8(3):55. https://doi.org/10.3390/biomedicines8030055
Chicago/Turabian StyleSchirrmacher, Volker. 2020. "New Insights into Mechanisms of Long-term Protective Anti-tumor Immunity Induced by Cancer Vaccines Modified by Virus Infection" Biomedicines 8, no. 3: 55. https://doi.org/10.3390/biomedicines8030055
APA StyleSchirrmacher, V. (2020). New Insights into Mechanisms of Long-term Protective Anti-tumor Immunity Induced by Cancer Vaccines Modified by Virus Infection. Biomedicines, 8(3), 55. https://doi.org/10.3390/biomedicines8030055