
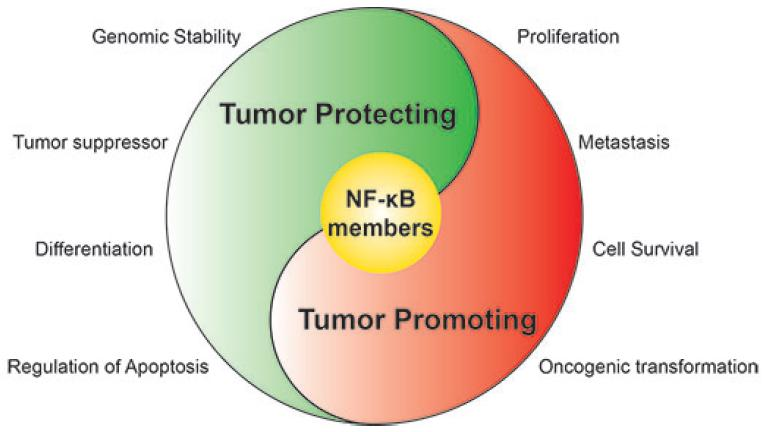
NF-κB Members Left Home: NF-κB-Independent Roles in Cancer


Abstract
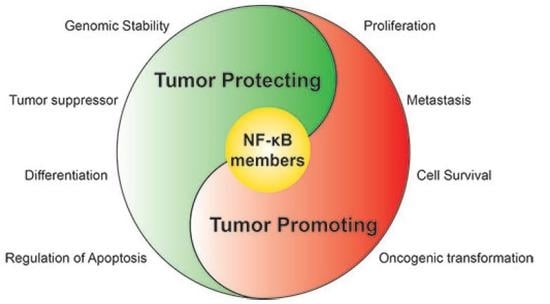
:
1. Introduction
2. Breast Cancer
3. Prostate Cancer
4. Colorectal Cancer
5. Skin Cancer
6. Liver Cancer
7. Renal Cancer
8. Lung Cancer
9. Conclusions
Conflicts of Interest
References
- Sen, R.; Baltimore, D. Inducibility of κ immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell 1986, 47, 921–928. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-β links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.D.; Goodlad, J.R.; Limb, G.A.; Powell, J.J.; Thompson, R.P.; Punchard, N.A. Activation of nuclear factor κB in Crohn’s disease. Inflamm. Res. 1998, 47, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; Clauson, C.L.; Niedernhofer, L.J.; Robbins, P.D. NF-κB in aging and disease. Aging Dis. 2011, 2, 449–465. [Google Scholar] [PubMed]
- Siebenlist, U.; Franzoso, G.; Brown, K. Structure, regulation and function of NF-κB. Annu Rev. Cell Biol. 1994, 10, 405–455. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S. The NF-κB and IκB proteins: New discoveries and insights. Annu. Rev. Immunol. 1996, 14, 649–683. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-κB and rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y. Regulatory functions of ubiquitination in the immune system. Nat. Immunol. 2002, 3, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krähn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef] [PubMed]
- Rayet, B.; Gélinas, C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene 1999, 18, 6938–6947. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Aggarwal, B.B. Nuclear transcription factor-κB as a target for cancer drug development. Leukemia 2002, 16, 1053–1068. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.F.; Kuo, H.P.; Chen, C.T.; Hsu, J.M.; Chou, C.K.; Wei, Y.; Sun, H.L.; Li, L.Y.; Ping, B.; Huang, W.C.; et al. IKKβ suppression of tsc1 links inflammation and tumor angiogenesis via the mtor pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Illario, M.; Finelli, R.; Iaccarino, G. To NFκB or not to NFκB: The dilemma on how to inhibit a cancer cell fate regulator. Transl. Med. UniSa 2012, 4, 73–85. [Google Scholar] [PubMed]
- Park, K.J.; Krishnan, V.; O'Malley, B.W.; Yamamoto, Y.; Gaynor, R.B. Formation of an IKKα-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol. Cell 2005, 18, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Rizzo, P.; Osipo, C.; Pannuti, A.; Wyatt, D.; Cheung, L.W.; Sonenshein, G.; Osborne, B.A.; Miele, L. Notch-1 activates estrogen receptor-α-dependent transcription via IKKα in breast cancer cells. Oncogene 2010, 29, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Prajapati, S.; Park, K.J.; Kelly, N.J.; Yamamoto, Y.; Gaynor, R.B. IKKα regulates estrogen-induced cell cycle progression by modulating E2F1 expression. J. Biol. Chem. 2006, 281, 6699–6706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tan, W.; Wu, X.; Poustovoitov, M.; Strasner, A.; Li, W.; Borcherding, N.; Ghassemian, M.; Karin, M. A NIK-IKKα module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell 2013, 23, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Lee, D.F.; Xia, W.; Golfman, L.S.; Ou-Yang, F.; Yang, J.Y.; Zou, Y.; Bao, S.; Hanada, N.; Saso, H.; et al. IκB kinase promotes tumorigenesis through inhibition of forkhead foxo3a. Cell 2004, 117, 225–237. [Google Scholar] [CrossRef]
- Dan, H.C.; Adli, M.; Baldwin, A.S. Regulation of mammalian target of rapamycin activity in PTEN-inactive prostate cancer cells by IκB kinase α. Cancer Res. 2007, 67, 6263–6269. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Ebbs, A.; Pasparakis, M.; Van Dyke, T.; Basseres, D.S.; Baldwin, A.S. Akt-dependent activation of mtorc1 complex involves phosphorylation of mtor (mammalian target of rapamycin) by IκB kinase α (IKKα). J. Biol. Chem. 2014, 289, 25227–25240. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.C.; Antonia, R.J.; Baldwin, A.S. PI3K/Akt promotes feedforward mTORC2 activation through IKKα. Oncotarget 2016, 7, 21064–21075. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Sellers, W.R. Akt-regulated pathways in prostate cancer. Oncogene 2005, 24, 7465–7474. [Google Scholar] [CrossRef] [PubMed]
- Hoberg, J.E.; Yeung, F.; Mayo, M.W. Smrt derepression by the IκB kinase α: A prerequisite to NF-κB transcription and survival. Mol. Cell 2004, 16, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.L.; Tan, W.; Ricono, J.M.; Korchynskyi, O.; Zhang, M.; Gonias, S.L.; Cheresh, D.A.; Karin, M. Nuclear cytokine-activated IKKα controls prostate cancer metastasis by repressing maspin. Nature 2007, 446, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Majada, V.; Aguilera, C.; Villanueva, A.; Vilardell, F.; Robert-Moreno, A.; Aytés, A.; Real, F.X.; Capella, G.; Mayo, M.W.; Espinosa, L.; et al. Nuclear ikk activity leads to dysregulated notch-dependent gene expression in colorectal cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Majada, V.; Pujadas, J.; Vilardell, F.; Capella, G.; Mayo, M.W.; Bigas, A.; Espinosa, L. Aberrant cytoplasmic localization of N-CoR in colorectal tumors. Cell Cycle 2007, 6, 1748–1752. [Google Scholar] [CrossRef] [PubMed]
- Margalef, P.; Fernández-Majada, V.; Villanueva, A.; Garcia-Carbonell, R.; Iglesias, M.; López, L.; Martínez-Iniesta, M.; Villà-Freixa, J.; Mulero, M.C.; Andreu, M.; et al. A truncated form of IKKα is responsible for specific nuclear IKK activity in colorectal cancer. Cell Rep. 2012, 2, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Margalef, P.; Colomer, C.; Villanueva, A.; Montagut, C.; Iglesias, M.; Bellosillo, B.; Salazar, R.; Martínez-Iniesta, M.; Bigas, A.; Espinosa, L. Braf-induced tumorigenesis is IKKα-dependent but NF-κB-independent. Sci. Signal. 2015, 8, ra38. [Google Scholar] [CrossRef] [PubMed]
- Göktuna, S.I.; Canli, O.; Bollrath, J.; Fingerle, A.A.; Horst, D.; Diamanti, M.A.; Pallangyo, C.; Bennecke, M.; Nebelsiek, T.; Mankan, A.K.; et al. IKKα promotes intestinal tumorigenesis by limiting recruitment of M1-like polarized myeloid cells. Cell Rep. 2014, 7, 1914–1925. [Google Scholar]
- Hu, Y.; Baud, V.; Delhase, M.; Zhang, P.; Deerinck, T.; Ellisman, M.; Johnson, R.; Karin, M. Abnormal morphogenesis but intact IKK activation in mice lacking the ikkalpha subunit of IκB kinase. Science 1999, 284, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Baud, V.; Oga, T.; Kim, K.I.; Yoshida, K.; Karin, M. IKKα controls formation of the epidermis independently of NF-κB. Nature 2001, 410, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Descargues, P.; Sil, A.K.; Karin, M. IKKα, a critical regulator of epidermal differentiation and a suppressor of skin cancer. EMBO J. 2008, 27, 2639–2647. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Xia, X.; Zhu, F.; Park, E.; Carbajal, S.; Kiguchi, K.; DiGiovanni, J.; Fischer, S.M.; Hu, Y. IKKα is required to maintain skin homeostasis and prevent skin cancer. Cancer Cell 2008, 14, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Xia, X.; Liu, B.; Shen, J.; Hu, Y.; Person, M. IKKα shields 14–3-3σ, a G2/M cell cycle checkpoint gene, from hypermethylation, preventing its silencing. Mol. Cell 2007, 27, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Marinari, B.; Moretti, F.; Botti, E.; Giustizieri, M.L.; Descargues, P.; Giunta, A.; Stolfi, C.; Ballaro, C.; Papoutsaki, M.; Alemà, S.; et al. The tumor suppressor activity of IKKα in stratified epithelia is exerted in part via the TGF-β antiproliferative pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 17091–17096. [Google Scholar] [CrossRef] [PubMed]
- Descargues, P.; Sil, A.K.; Sano, Y.; Korchynskyi, O.; Han, G.; Owens, P.; Wang, X.J.; Karin, M. IKKα is a critical coregulator of a smad4-independent TGFβ-smad2/3 signaling pathway that controls keratinocyte differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 2487–2492. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Shi, Y.; Yan, B.; Xiao, D.; Lai, W.; Pan, Y.; Jiang, Y.; Chen, L.; Mao, C.; Zhou, J.; et al. Lgr5 expression is controled by IKKα in basal cell carcinoma through activating stat3 signaling pathway. Oncotarget 2016, 7, 27280–27294. [Google Scholar] [CrossRef] [PubMed]
- Mulero, M.C.; Ferres-Marco, D.; Islam, A.; Margalef, P.; Pecoraro, M.; Toll, A.; Drechsel, N.; Charneco, C.; Davis, S.; Bellora, N.; et al. Chromatin-bound IκBα regulates a subset of polycomb target genes in differentiation and cancer. Cancer Cell 2013, 24, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Alameda, J.P.; Gaspar, M.; Ramírez, Á.; Navarro, M.; Page, A.; Suárez-Cabrera, C.; Fernández, M.G.; Mérida, J.R.; Paramio, J.M.; García-Fernández, R.A.; et al. Deciphering the role of nuclear and cytoplasmic IKKα in skin cancer. Oncotarget 2016, 7, 29531–29547. [Google Scholar] [PubMed]
- Toll, A.; Margalef, P.; Masferrer, E.; Ferrándiz-Pulido, C.; Gimeno, J.; Pujol, R.M.; Bigas, A.; Espinosa, L. Active nuclear IKK correlates with metastatic risk in cutaneous squamous cell carcinoma. Arch. Dermatol. Res. 2015, 307, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, C.; Hoya-Arias, R.; Haegeman, G.; Espinosa, L.; Bigas, A. Recruitment of IκBα to the hes1 promoter is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2004, 101, 16537–16542. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the rela component of NF-κB. Nature 1995, 376, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Klement, J.F.; Rice, N.R.; Car, B.D.; Abbondanzo, S.J.; Powers, G.D.; Bhatt, P.H.; Chen, C.H.; Rosen, C.A.; Stewart, C.L. IκBα deficiency results in a sustained NF-κB response and severe widespread dermatitis in mice. Mol. Cell. Biol. 1996, 16, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- Rebholz, B.; Haase, I.; Eckelt, B.; Paxian, S.; Flaig, M.J.; Ghoreschi, K.; Nedospasov, S.A.; Mailhammer, R.; Debey-Pascher, S.; Schultze, J.L.; et al. Crosstalk between keratinocytes and adaptive immune cells in an IκBα protein-mediated inflammatory disease of the skin. Immunity 2007, 27, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Van Hogerlinden, M.; Rozell, B.L.; Ahrlund-Richter, L.; Toftgård, R. Squamous cell carcinomas and increased apoptosis in skin with inhibited rel/nuclear factor-κB signaling. Cancer Res. 1999, 59, 3299–3303. [Google Scholar] [PubMed]
- Seitz, C.S.; Lin, Q.; Deng, H.; Khavari, P.A. Alterations in NF-κB function in transgenic epithelial tissue demonstrate a growth inhibitory role for NF-κB. Proc. Natl. Acad. Sci. USA 1998, 95, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Dajee, M.; Lazarov, M.; Zhang, J.Y.; Cai, T.; Green, C.L.; Russell, A.J.; Marinkovich, M.P.; Tao, S.; Lin, Q.; Kubo, Y.; et al. NF-κB blockade and oncogenic ras trigger invasive human epidermal neoplasia. Nature 2003, 421, 639–643. [Google Scholar] [CrossRef] [PubMed]
- van Hogerlinden, M.; Rozell, B.L.; Toftgård, R.; Sundberg, J.P. Characterization of the progressive skin disease and inflammatory cell infiltrate in mice with inhibited NF-κB signaling. J. Investig. Dermatol. 2004, 123, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Motola-Kuba, D.; Zamora-Valdés, D.; Uribe, M.; Méndez-Sánchez, N. Hepatocellular carcinoma. An overview. Ann. Hepatol. 2006, 5, 16–24. [Google Scholar] [PubMed]
- Okuda, K. Hepatocellular carcinoma. J. Hepatol. 2000, 32, 225–237. [Google Scholar] [CrossRef]
- Vainer, G.W.; Pikarsky, E.; Ben-Neriah, Y. Contradictory functions of NF-κB in liver physiology and cancer. Cancer Lett. 2008, 267, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Koppe, C.; Verheugd, P.; Gautheron, J.; Reisinger, F.; Kreggenwinkel, K.; Roderburg, C.; Quagliata, L.; Terracciano, L.; Gassler, N.; Tolba, R.H.; et al. IκB kinaseα/β control biliary homeostasis and hepatocarcinogenesis in mice by phosphorylating the cell-death mediator receptor-interacting protein kinase 1. Hepatology 2016, 64, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Maeda, S.; Chang, L.; Karin, M. Loss of hepatic NF-κB activity enhances chemical hepatocarcinogenesis through sustained c-jun n-terminal kinase 1 activation. Proc. Natl. Acad. Sci. USA 2006, 103, 10544–10551. [Google Scholar] [CrossRef] [PubMed]
- Bettermann, K.; Vucur, M.; Haybaeck, J.; Koppe, C.; Janssen, J.; Heymann, F.; Weber, A.; Weiskirchen, R.; Liedtke, C.; Gassler, N.; et al. Tak1 suppresses a nemo-dependent but NF-κB-independent pathway to liver cancer. Cancer Cell 2010, 17, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of nemo/IKKγ in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Kondylis, V.; Polykratis, A.; Ehlken, H.; Ochoa-Callejero, L.; Straub, B.K.; Krishna-Subramanian, S.; Van, T.M.; Curth, H.M.; Heise, N.; Weih, F.; et al. Nemo prevents steatohepatitis and hepatocellular carcinoma by inhibiting ripk1 kinase activity-mediated hepatocyte apoptosis. Cancer Cell 2015, 28, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Ehlken, H.; Krishna-Subramanian, S.; Ochoa-Callejero, L.; Kondylis, V.; Nadi, N.E.; Straub, B.K.; Schirmacher, P.; Walczak, H.; Kollias, G.; Pasparakis, M. Death receptor-independent fadd signalling triggers hepatitis and hepatocellular carcinoma in mice with liver parenchymal cell-specific nemo knockout. Cell Death Differ. 2014, 21, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Aigelsreiter, A.; Haybaeck, J.; Schauer, S.; Kiesslich, T.; Bettermann, K.; Griessbacher, A.; Stojakovic, T.; Bauernhofer, T.; Samonigg, H.; Kornprat, P.; et al. Nemo expression in human hepatocellular carcinoma and its association with clinical outcome. Hum. Pathol. 2012, 43, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zheng, M.Q.; Lu, J.W.; Jiang, Q.; Wang, T.H.; Huang, X.E. Cxcl12-cxcr4 promotes proliferation and invasion of pancreatic cancer cells. Asian Pac. J. Cancer Prev. 2013, 14, 5403–5408. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, A.M.; Häuselmann, I.; Borsig, L.; Bolduan, S.; Schindler, M.; Schraml, P.; Heikenwalder, M.; Moch, H. A novel PVHl-independent but nemo-driven pathway in renal cancer promotes hif stabilization. Oncogene 2016, 35, 3125–3138. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Whitelaw, M.L.; Peet, D.J. Activity of hypoxia-inducible factor 2α is regulated by association with the NF-κB essential modulator. J. Biol. Chem. 2005, 280, 14240–14251. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Ju, T.K.; Hung, M.C.; Chen, C.C. Phosphorylation of CBP by IKKα promotes cell growth by switching the binding preference of cbp from p53 to NF-κB. Mol. Cell 2007, 26, 75–87. [Google Scholar] [CrossRef] [PubMed]
 Activation/Regulation/Phosphorylation;
Activation/Regulation/Phosphorylation;  Migration;
Migration;  Inactivation.
Activation/Regulation/Phosphorylation; Migration; Inactivation.
Inactivation.
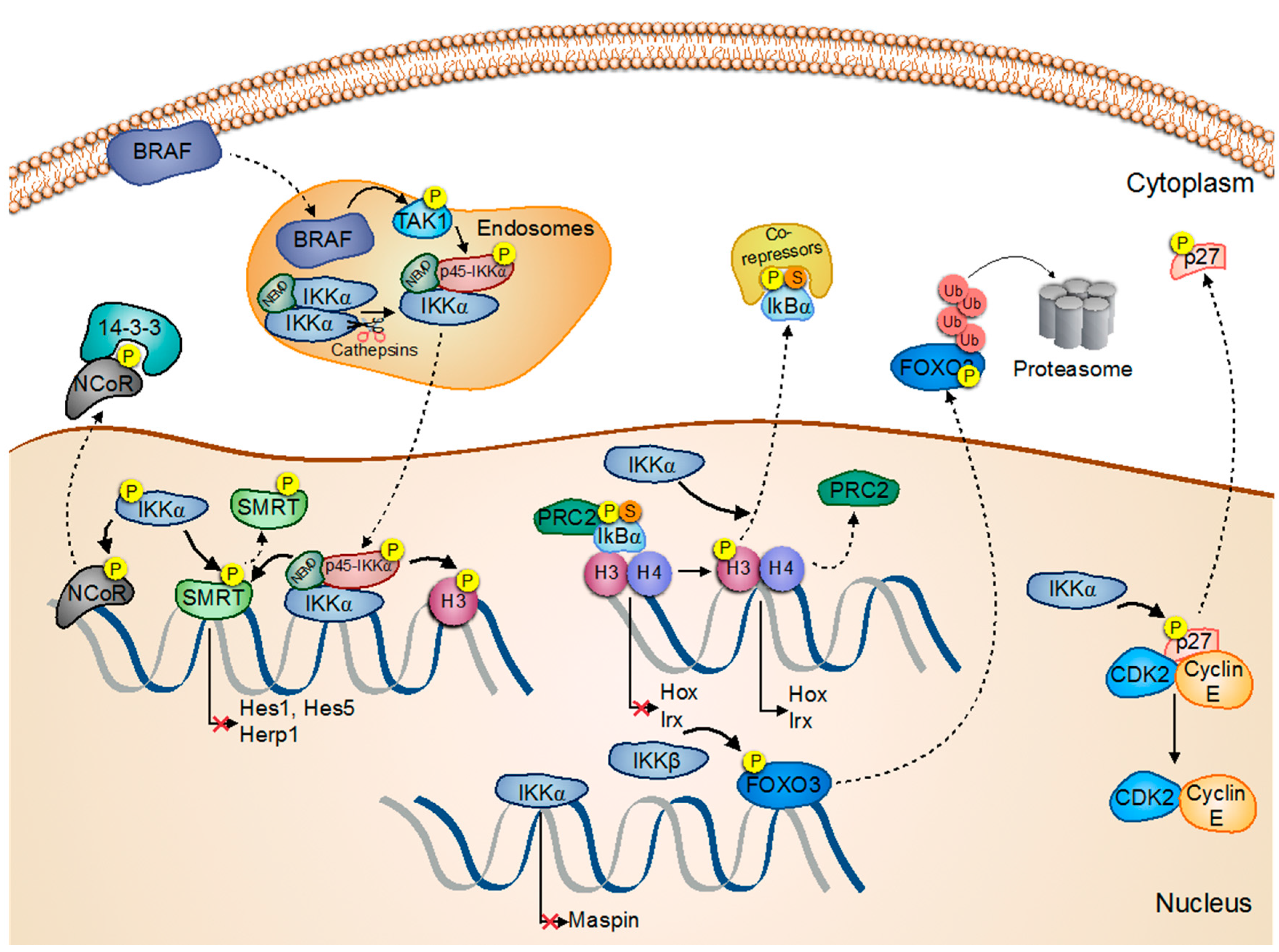
Activation/Regulation/Phosphorylation; Migration; Inactivation.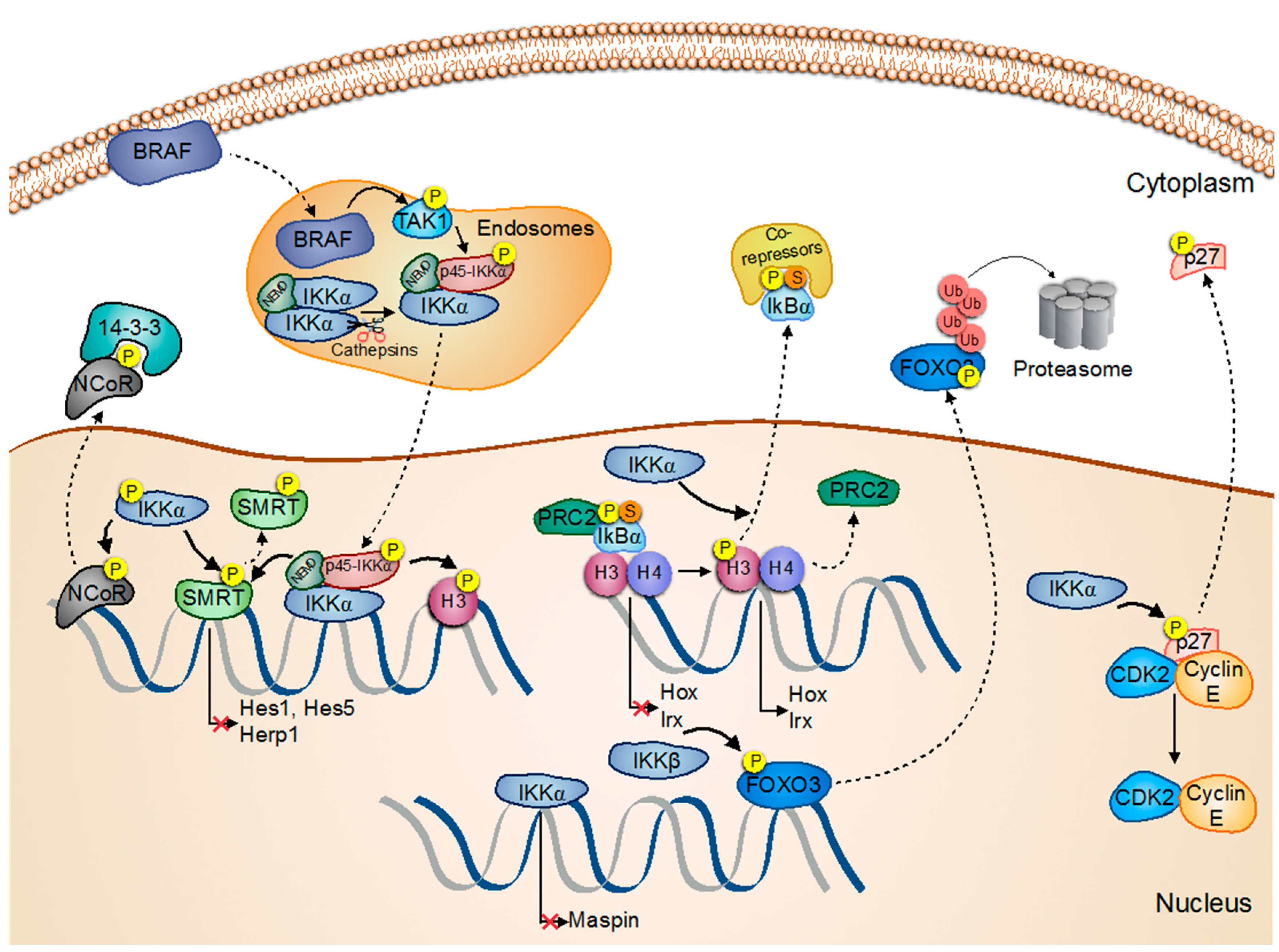 Activation/Regulation/Phosphorylation; Inactivation;
Activation/Regulation/Phosphorylation; Inactivation;  Inhibition
Activation/Regulation/Phosphorylation; Inactivation; Inhibition
Inhibition
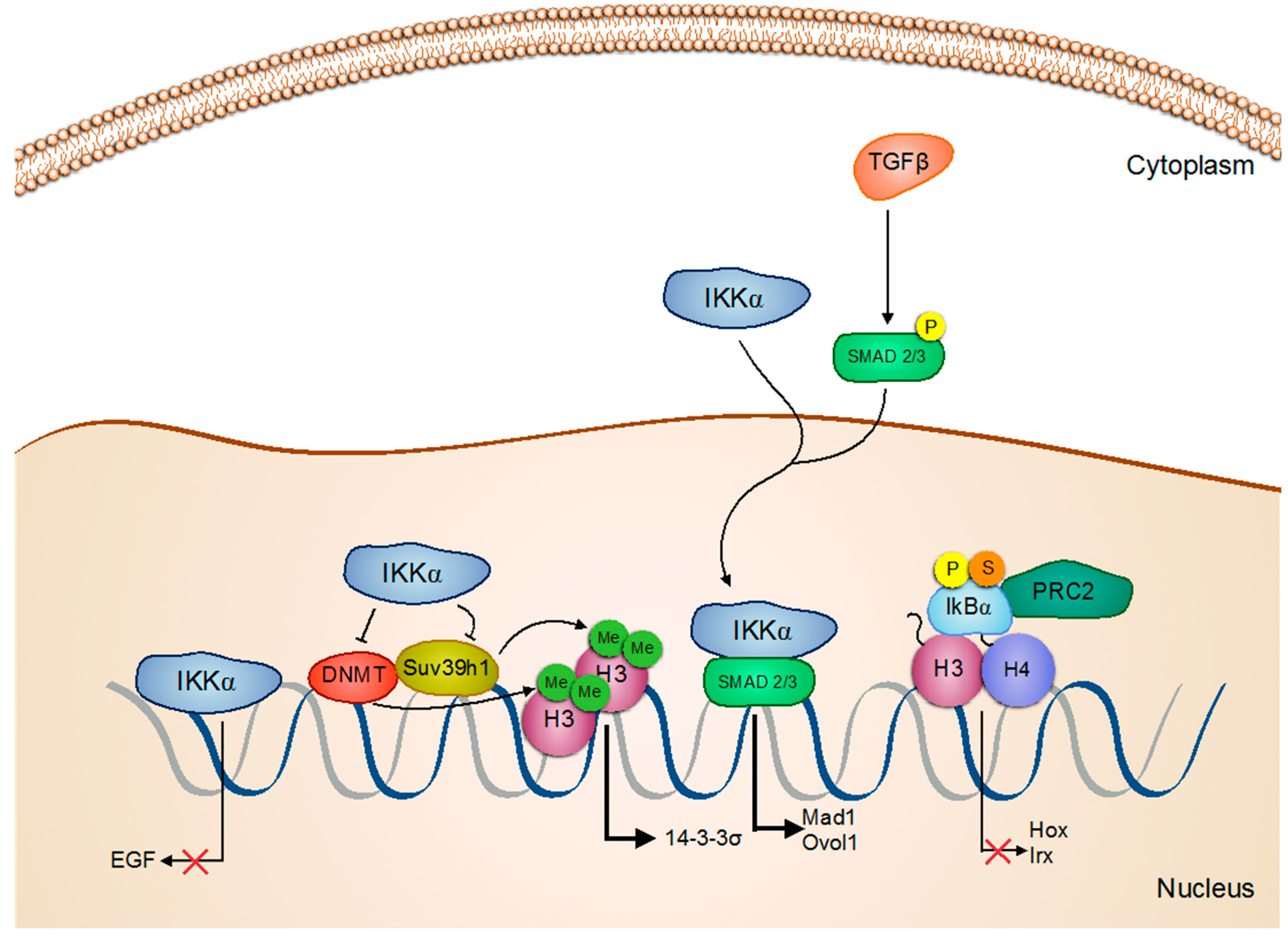
Activation/Regulation/Phosphorylation; Inactivation; Inhibition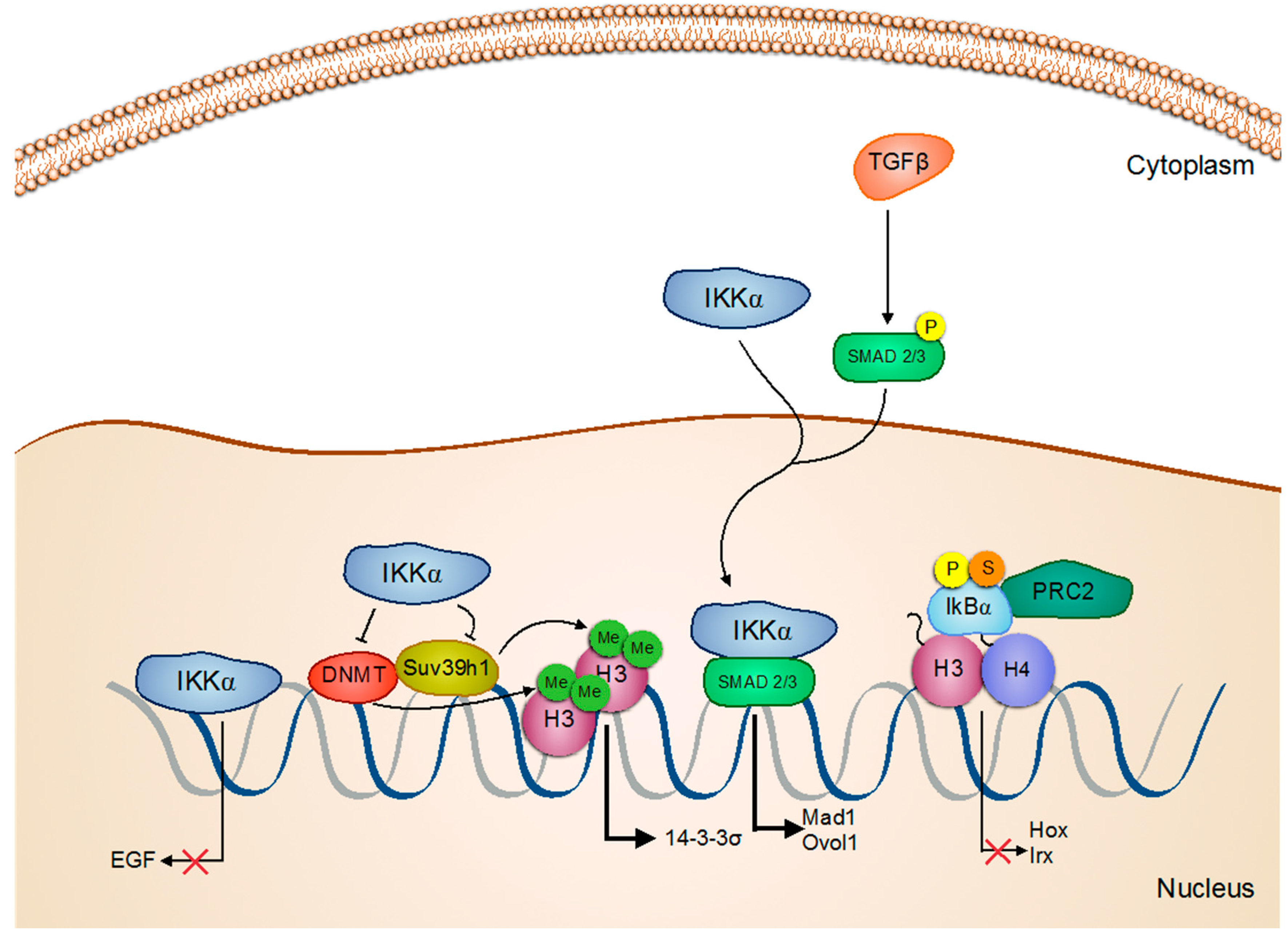

{kind=link}
{kind=link}
{kind=link}
| Protein | Substrate | Effect | Cancer Type | References |
|---|---|---|---|---|
| IKKα | Phosphorylation of ERα and SRC-3 | Estrogen-dependent gene transcription | Breast Cancer | [20] |
| Cooperation with Notch1 to activate transcription of ERα-dependent genes | Cell proliferation | Breast Cancer | [21] | |
| E2F1 transcription | Cell cycle progression | Breast Cancer | [22] | |
| Phosphorylation of p27 | Expansion of tumour-initiating cells | Breast Cancer | [23] | |
| Phosphorylation of mTORC | Cell proliferation | Prostate Cancer | [25,26] | |
| Activation of mTORC2 | Akt activation | Prostate Cancer | [27] | |
| Phosphorylation of SMRT | Increased cell survival Regulation of Notch-dependent gene transcription: Tumour growth | Prostate Cancer CRC | [29] [31] | |
| Maspin gene repression | Metastasis induction | Prostate Cancer SCC | [30] [46] | |
| Phosphorylation of NCoR | Increased gene transcription | CRC | [32] | |
| Regulation of IFNγ-expressing M1-like myeloid cells recruitment | Enhanced tumorigenesis | CRC | [35] | |
| Repression of EGF transcription | Prevention of SCC | SCC | [39] | |
| Prevents hypermethylation of 14-3-3sigma through Suv39h1 | Maintenance of genomic stability in keratinocytes | Skin Cancer | [40] | |
| Myc inhibition | Tumour-suppressive activity | SCC | [41] | |
| Myc inhibition | Keratinocyte proliferation and differentiation | Skin Cancer | [42] | |
| LGR5 expression | Oncogenic transformation | BCC | [43] | |
| Chromatin release of PS-IκBα | Oncogenic transformation | Skin Cancer | [44] | |
| N: c-Myc, Maspin and Integrin-α6 expression: Cyt: Increases EGFR, MMP-9 and VEGF-A activity | Cancer progression | NMSC | [45] | |
| Phosphorylation of RIPK1 | Regulation of cell viability | HCC | [58] | |
| p45-IKKα | Phosphorylation of SMRT and Histone H3 Regulation of anti-apoptotic and pro-metastatic genes | Tumour maintenance and apoptosis inhibition Tumour growth and metastasis | CRC CRC | [33] [34] |
| IKKβ | Phosphorylation of FOXO3a | Increased proliferation | Breast Cancer | [24] |
| Phosphorylation of RIPK1 | Regulation of cell viability | HCC | [58] | |
| Repression of MKK4/7-JNK signalling cascade | Tumour suppressor | HCC | [59] | |
| IκBα | Binding to HDACs and PRC2 | Regulation of HOX and IRX: keratinocyte differentiation | SCC | [44] |
| TAK1 | Suppression of specific NEMO function | Suppression of procarcinogenic and pronecrotic pathway | HCC | [60] |
| NEMO | NFκB activation | Tumour suppressor | HCC | [61][62] |
| Inhibition RIPK1 and Casp8 | Suppression of hepatocyte apoptosis | HCC | [62] | |
| HIFα stabilization | Cell survival | ccRCC | [66] | |
| Phosphorylation of CBP | Cell proliferation | Lung Cancer | [68] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colomer, C.; Marruecos, L.; Vert, A.; Bigas, A.; Espinosa, L. NF-κB Members Left Home: NF-κB-Independent Roles in Cancer. Biomedicines 2017, 5, 26. https://doi.org/10.3390/biomedicines5020026
Colomer C, Marruecos L, Vert A, Bigas A, Espinosa L. NF-κB Members Left Home: NF-κB-Independent Roles in Cancer. Biomedicines. 2017; 5(2):26. https://doi.org/10.3390/biomedicines5020026
Chicago/Turabian StyleColomer, Carlota, Laura Marruecos, Anna Vert, Anna Bigas, and Lluis Espinosa. 2017. "NF-κB Members Left Home: NF-κB-Independent Roles in Cancer" Biomedicines 5, no. 2: 26. https://doi.org/10.3390/biomedicines5020026
APA StyleColomer, C., Marruecos, L., Vert, A., Bigas, A., & Espinosa, L. (2017). NF-κB Members Left Home: NF-κB-Independent Roles in Cancer. Biomedicines, 5(2), 26. https://doi.org/10.3390/biomedicines5020026